
Neural Stem Cells as Potential Glioblastoma Cells of Origin
 , and
, and 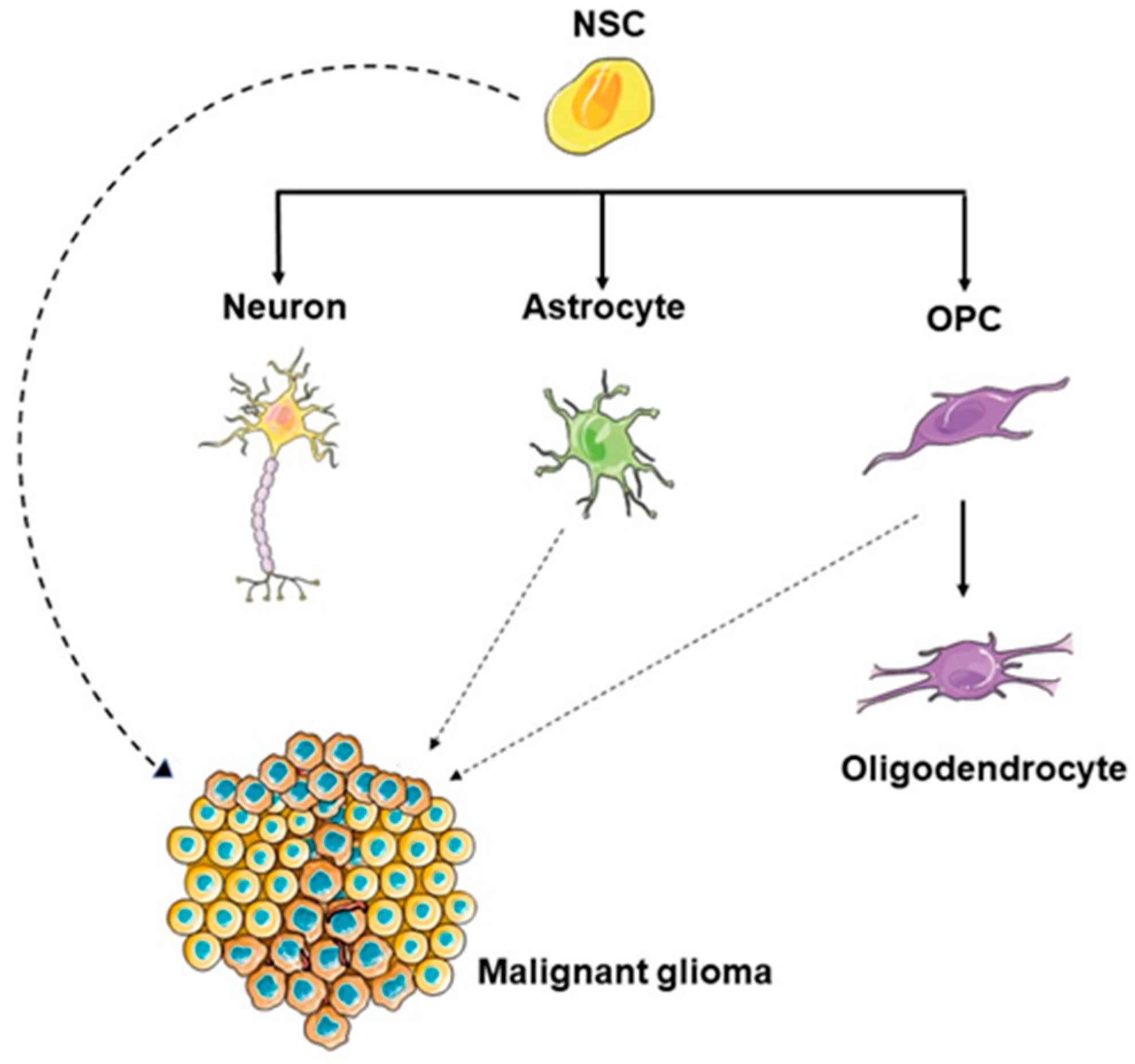{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Glioblastoma
1.2. The Potential GBM Cell of Origin
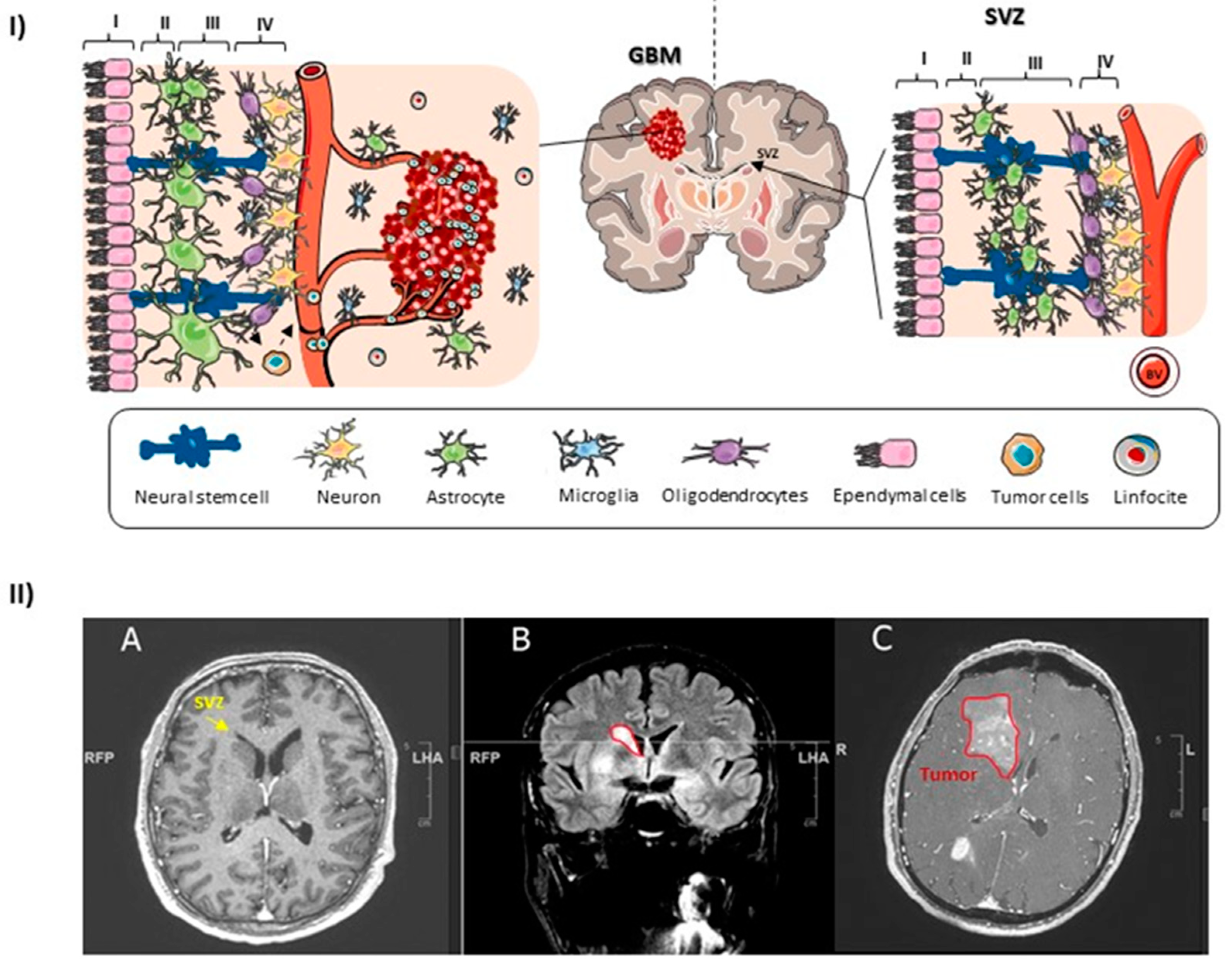
1.3. The Human SVZ
1.4. Similarities between NSCs and Glioma Stem Cells
1.5. NSCs of the SVZ as the GBM Cell of Origin
2. Materials and Methods
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and Molecular Prognostic Review of Glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma Stem Cells: Lessons from the Tumor Hierarchy in a Lethal Cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Hauser, A.; Dutta, S.W.; Showalter, T.N.; Sheehan, J.P.; Grover, S.; Trifiletti, D.M. Impact of Academic Facility Type and Volume on Post-Surgical Outcomes Following Diagnosis of Glioblastoma. J. Clin. Neurosci. 2018, 47, 103–110. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2006–2010. Neuro-Oncology 2013, 15, ii1–ii56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.; Karajannis, M.; Harter, D. Glioblastoma Multiforme: State of the Art and Future Therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar] [CrossRef]
- Cantrell, J.N.; Waddle, M.R.; Rotman, M.; Peterson, J.L.; Ruiz-Garcia, H.; Heckman, M.G.; Quiñones-Hinojosa, A.; Rosenfeld, S.S.; Brown, P.D.; Trifiletti, D.M. Progress Toward Long-Term Survivors of Glioblastoma. Mayo Clin. Proc. 2019, 94, 1278–1286. [Google Scholar] [CrossRef]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.-L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic Pathways to Glioblastoma. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [Green Version]
- Pan, I.-W.; Ferguson, S.D.; Lam, S. Patient and Treatment Factors Associated with Survival among Adult Glioblastoma Patients: A USA Population-Based Study from 2000–2010. J. Clin. Neurosci. 2015, 22, 1575–1581. [Google Scholar] [CrossRef]
- Franceschi, E.; Tosoni, A.; Minichillo, S.; Depenni, R.; Paccapelo, A.; Bartolini, S.; Michiara, M.; Pavesi, G.; Urbini, B.; Crisi, G.; et al. The Prognostic Roles of Gender and O6-Methylguanine-DNA Methyltransferase Methylation Status in Glioblastoma Patients: The Female Power. World Neurosurg. 2018, 112, e342–e347. [Google Scholar] [CrossRef]
- McCrea, H.J.; Bander, E.D.; Venn, R.A.; Reiner, A.S.; Iorgulescu, J.B.; Puchi, L.A.; Schaefer, P.M.; Cederquist, G.; Greenfield, J.P. Sex, Age, Anatomic Location, and Extent of Resection Influence Outcomes in Children With High-Grade Glioma. Neurosurgery 2015, 77, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 Mutations as Molecular Signature and Predictive Factor of Secondary Glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.-J.; Perry, S.R.; Tonon, G.; Chu, G.C.; Ding, Z.; et al. P53 and Pten Control Neural and Glioma Stem/Progenitor Cell Renewal and Differentiation. Nature 2008, 455, 1129–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT Promoter Mutations Occur Frequently in Gliomas and a Subset of Tumors Derived from Cells with Low Rates of Self-Renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, D.; Bender, S.; Jones, D.T.W.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and Adult Glioblastoma: Multiform (Epi)Genomic Culprits Emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [Green Version]
- Götz, M.; Barde, Y.-A. Radial Glial Cells. Neuron 2005, 46, 369–372. [Google Scholar] [CrossRef] [Green Version]
- Doetsch, F.; Caillé, I.; Lim, D.A.; García-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular Zone Astrocytes Are Neural Stem Cells in the Adult Mammalian Brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Gage, F.H. Mammalian Neural Stem Cells. Science 2000, 287, 1433–1438. [Google Scholar] [CrossRef]
- Ge, W.-P.; Miyawaki, A.; Gage, F.H.; Jan, Y.N.; Jan, L.Y. Local Generation of Glia Is a Major Astrocyte Source in Postnatal Cortex. Nature 2012, 484, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Gage, F.H.; Temple, S. Neural Stem Cells: Generating and Regenerating the Brain. Neuron 2013, 80, 588–601. [Google Scholar] [CrossRef] [Green Version]
- Rowitch, D.H. Glial Specification in the Vertebrate Neural Tube. Nat. Rev. Neurosci. 2004, 5, 409–419. [Google Scholar] [CrossRef]
- ffrench-Constant, C.; Raff, M.C. Proliferating Bipotential Glial Progenitor Cells in Adult Rat Optic Nerve. Nature 1986, 319, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, K.; Leblond, C.P. Radioautographic Investigation of Gliogenesis in the Corpus Callosum of Young Rats II. Origin of Microglial Cells. J. Comp. Neurol. 1978, 180, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Bardehle, S.; Krüger, M.; Buggenthin, F.; Schwausch, J.; Ninkovic, J.; Clevers, H.; Snippert, H.J.; Theis, F.J.; Meyer-Luehmann, M.; Bechmann, I.; et al. Live Imaging of Astrocyte Responses to Acute Injury Reveals Selective Juxtavascular Proliferation. Nat. Neurosci. 2013, 16, 580–586. [Google Scholar] [CrossRef]
- Kiaie, N.; Gorabi, A.M.; Loveless, R.; Teng, Y.; Jamialahmadi, T.; Sahebkar, A. The Regenerative Potential of Glial Progenitor Cells and Reactive Astrocytes in CNS Injuries. Neurosci. Biobehav. Rev. 2022, 140, 104794. [Google Scholar] [CrossRef]
- Garzón-Muvdi, T.; Quiñones-Hinojosa, A. Neural Stem Cell Niches and Homing: Recruitment and Integration into Functional Tissues. ILAR J. 2010, 51, 3–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human Glioblastoma Arises from Subventricular Zone Cells with Low-Level Driver Mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Amariglio, N.; Hirshberg, A.; Scheithauer, B.W.; Cohen, Y.; Loewenthal, R.; Trakhtenbrot, L.; Paz, N.; Koren-Michowitz, M.; Waldman, D.; Leider-Trejo, L.; et al. Donor-Derived Brain Tumor Following Neural Stem Cell Transplantation in an Ataxia Telangiectasia Patient. PLoS Med. 2009, 6, e1000029. [Google Scholar] [CrossRef]
- Sanai, N.; Tramontin, A.D.; Quiñones-Hinojosa, A.; Barbaro, N.M.; Gupta, N.; Kunwar, S.; Lawton, M.T.; McDermott, M.W.; Parsa, A.T.; Manuel-García Verdugo, J.; et al. Unique Astrocyte Ribbon in Adult Human Brain Contains Neural Stem Cells but Lacks Chain Migration. Nature 2004, 427, 740–744. [Google Scholar] [CrossRef]
- Doetsch, F.; García-Verdugo, J.M.; Alvarez-Buylla, A. Cellular Composition and Three-Dimensional Organization of the Subventricular Germinal Zone in the Adult Mammalian Brain. J. Neurosci. 1997, 17, 5046–5061. [Google Scholar] [CrossRef] [Green Version]
- Quiñones-Hinojosa, A.; Sanai, N.; Soriano-Navarro, M.; Gonzalez-Perez, O.; Mirzadeh, Z.; Gil-Perotin, S.; Romero-Rodriguez, R.; Berger, M.S.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Cellular Composition and Cytoarchitecture of the Adult Human Subventricular Zone: A Niche of Neural Stem Cells. J. Comp. Neurol. 2006, 494, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Ihrie, R.A.; Alvarez-Buylla, A. Cells in the Astroglial Lineage Are Neural Stem Cells. Cell Tissue Res. 2008, 331, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Lois, C.; Alvarez-Buylla, A. Proliferating Subventricular Zone Cells in the Adult Mammalian Forebrain Can Differentiate into Neurons and Glia. Proc. Natl. Acad. Sci. USA 1993, 90, 2074–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Cázares, H.; Gonzalez-Perez, O.; Soriano-Navarro, M.; Zamora-Berridi, G.; García-Verdugo, J.M.; Quinoñes-Hinojosa, A. Cytoarchitecture of the Lateral Ganglionic Eminence and Rostral Extension of the Lateral Ventricle in the Human Fetal Brain. J. Comp. Neurol. 2011, 519, 1165–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, M.A.; Kam, M.; Nannmark, U.; Anderson, M.F.; Axell, M.Z.; Wikkelso, C.; Holtås, S.; van Roon-Mom, W.M.C.; Björk-Eriksson, T.; Nordborg, C.; et al. Human Neuroblasts Migrate to the Olfactory Bulb via a Lateral Ventricular Extension. Science 2007, 315, 1243–1249. [Google Scholar] [CrossRef]
- Kam, M.; Curtis, M.A.; McGlashan, S.R.; Connor, B.; Nannmark, U.; Faull, R.L.M. The Cellular Composition and Morphological Organization of the Rostral Migratory Stream in the Adult Human Brain. J. Chem. Neuroanat. 2009, 37, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Sanai, N.; Nguyen, T.; Ihrie, R.A.; Mirzadeh, Z.; Tsai, H.-H.; Wong, M.; Gupta, N.; Berger, M.S.; Huang, E.; Garcia-Verdugo, J.-M.; et al. Corridors of Migrating Neurons in the Human Brain and Their Decline during Infancy. Nature 2011, 478, 382–386. [Google Scholar] [CrossRef] [Green Version]
- Paredes, M.F.; James, D.; Gil-Perotin, S.; Kim, H.; Cotter, J.A.; Ng, C.; Sandoval, K.; Rowitch, D.H.; Xu, D.; McQuillen, P.S.; et al. Extensive Migration of Young Neurons into the Infant Human Frontal Lobe. Science 2016, 354, aaf7073. [Google Scholar] [CrossRef] [Green Version]
- Ernst, A.; Alkass, K.; Bernard, S.; Salehpour, M.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; Frisén, J. Neurogenesis in the Striatum of the Adult Human Brain. Cell 2014, 156, 1072–1083. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e9. [Google Scholar] [CrossRef] [Green Version]
- Thirant, C.; Galan-Moya, E.; Dubois, L.G.; Pinte, S.; Chafey, P.; Broussard, C.; Varlet, P.; Devaux, B.; Soncin, F.; Gavard, J.; et al. Differential Proteomic Analysis of Human Glioblastoma and Neural Stem Cells Reveals HDGF as a Novel Angiogenic Secreted Factor. Stem Cells 2012, 30, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor Stem Cells Derived from Glioblastomas Cultured in BFGF and EGF More Closely Mirror the Phenotype and Genotype of Primary Tumors than Do Serum-Cultured Cell Lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Gangemi, R.M.R.; Griffero, F.; Marubbi, D.; Perera, M.; Capra, M.C.; Malatesta, P.; Ravetti, G.L.; Zona, G.L.; Daga, A.; Corte, G. SOX2 Silencing in Glioblastoma Tumor-Initiating Cells Causes Stop of Proliferation and Loss of Tumorigenicity. Stem Cells 2009, 27, 40–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.-N.; Borges, I.; Ruiz i Altaba, A. NANOG Regulates Glioma Stem Cells and Is Essential in Vivo Acting in a Cross-Functional Network with GLI1 and P53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 Signaling Regulates Human Glioma Growth, Cancer Stem Cell Self-Renewal, and Tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-Mediated Hedgehog Pathway Inhibition Depletes Stem-Like Cancer Cells in Glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Yuan, X.; Liu, G.; Black, K.L.; Yu, J.S. Hedgehog Signaling Regulates Brain Tumor-Initiating Cell Proliferation and Portends Shorter Survival for Patients with PTEN-Coexpressing Glioblastomas. Stem Cells 2008, 26, 3018–3026. [Google Scholar] [CrossRef] [Green Version]
- Fareh, M.; Turchi, L.; Virolle, V.; Debruyne, D.; Almairac, F.; de-la-Forest Divonne, S.; Paquis, P.; Preynat-Seauve, O.; Krause, K.-H.; Chneiweiss, H.; et al. The MiR 302-367 Cluster Drastically Affects Self-Renewal and Infiltration Properties of Glioma-Initiating Cells through CXCR4 Repression and Consequent Disruption of the SHH-GLI-NANOG Network. Cell Death Differ. 2012, 19, 232–244. [Google Scholar] [CrossRef]
- Rieske, P.; Golanska, E.; Zakrzewska, M.; Piaskowski, S.; Hulas-Bigoszewska, K.; Wolańczyk, M.; Szybka, M.; Witusik-Perkowska, M.; Jaskolski, D.J.; Zakrzewski, K.; et al. Arrested Neural and Advanced Mesenchymal Differentiation of Glioblastoma Cells-Comparative Study with Neural Progenitors. BMC Cancer 2009, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.-M.; Maciaczyk, J.; et al. NOTCH Pathway Blockade Depletes CD133-Positive Glioblastoma Cells and Inhibits Growth of Tumor Neurospheres and Xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Imayoshi, I.; Sakamoto, M.; Yamaguchi, M.; Mori, K.; Kageyama, R. Essential Roles of Notch Signaling in Maintenance of Neural Stem Cells in Developing and Adult Brains. J. Neurosci. 2010, 30, 3489–3498. [Google Scholar] [CrossRef] [Green Version]
- Panchision, D.M.; McKay, R.D.G. The Control of Neural Stem Cells by Morphogenic Signals. Curr. Opin. Genet. Dev. 2002, 12, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.K.; Miller, R.H. Emerging Roles for Bone Morphogenetic Proteins in Central Nervous System Glial Biology. J. Neurosci. Res. 2004, 76, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.A.; Tramontin, A.D.; Trevejo, J.M.; Herrera, D.G.; García-Verdugo, J.M.; Alvarez-Buylla, A. Noggin Antagonizes BMP Signaling to Create a Niche for Adult Neurogenesis. Neuron 2000, 28, 713–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Son, M.J.; Woolard, K.; Donin, N.M.; Li, A.; Cheng, C.H.; Kotliarova, S.; Kotliarov, Y.; Walling, J.; Ahn, S.; et al. Epigenetic-Mediated Dysfunction of the Bone Morphogenetic Protein Pathway Inhibits Differentiation of Glioblastoma-Initiating Cells. Cancer Cell 2008, 13, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, K.M.; Blin, C.; Alfazema, N.; Gangoso, E.; Pollard, S.M.; Marques-Torrejon, M.A. Lrig1 Regulates the Balance between Proliferation and Quiescence in Glioblastoma Stem Cells. Front. Cell Dev. Biol. 2022, 10, 983097. [Google Scholar] [CrossRef]
- Marqués-Torrejón, M.Á.; Williams, C.A.C.; Southgate, B.; Alfazema, N.; Clements, M.P.; Garcia-Diaz, C.; Blin, C.; Arranz-Emparan, N.; Fraser, J.; Gammoh, N.; et al. LRIG1 Is a Gatekeeper to Exit from Quiescence in Adult Neural Stem Cells. Nat. Commun. 2021, 12, 2594. [Google Scholar] [CrossRef]
- Kaur, N.; Chettiar, S.; Rathod, S.; Rath, P.; Muzumdar, D.; Shaikh, M.L.; Shiras, A. Wnt3a Mediated Activation of Wnt/β-Catenin Signaling Promotes Tumor Progression in Glioblastoma. Mol. Cell. Neurosci. 2013, 54, 44–57. [Google Scholar] [CrossRef]
- Ming, G.; Song, H. Adult Neurogenesis in the Mammalian Central Nervous System. Annu. Rev. Neurosci. 2005, 28, 223–250. [Google Scholar] [CrossRef]
- Ahn, S.; Joyner, A.L. In Vivo Analysis of Quiescent Adult Neural Stem Cells Responding to Sonic Hedgehog. Nature 2005, 437, 894–897. [Google Scholar] [CrossRef]
- de la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; DePinho, R.A.; Bonni, A. Identification of a PTEN-Regulated STAT3 Brain Tumor Suppressor Pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 Is Required for Proliferation and Maintenance of Multipotency in Glioblastoma Stem Cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef] [Green Version]
- Surena, A.-L.; de Faria, G.P.; Studler, J.-M.; Peiretti, F.; Pidoux, M.; Camonis, J.; Chneiweiss, H.; Formstecher, E.; Junier, M.-P. DLG1/SAP97 Modulates Transforming Growth Factor α Bioavailability. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2009, 1793, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoda, K.; Tanaka, K.; Nakagawa, S.; Thuy, D.H.D.; Ujifuku, K.; Kamada, K.; Hayashi, K.; Matsuo, T.; Nagata, I.; Niwa, M. Initial Contact of Glioblastoma Cells with Existing Normal Brain Endothelial Cells Strengthen the Barrier Function via Fibroblast Growth Factor 2 Secretion: A New In Vitro Blood–Brain Barrier Model. Cell. Mol. Neurobiol. 2013, 33, 489–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma Stem-like Cells Secrete the pro-Angiogenic VEGF-A Factor in Extracellular Vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Chen, Z.; Hu, Y.-D.; Wei, H.; Li, D.; Ji, H.; Wang, D.-L. Autocrine Factors Sustain Glioblastoma Stem Cell Self-Renewal. Oncol. Rep. 2009, 21, 419–424. [Google Scholar]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem Cell–like Glioma Cells Promote Tumor Angiogenesis through Vascular Endothelial Growth Factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [Green Version]
- El-Obeid, A.; Bongcam-Rudloff, E.; Sörby, M.; Ostman, A.; Nistér, M.; Westermark, B. Cell Scattering and Migration Induced by Autocrine Transforming Growth Factor Alpha in Human Glioma Cells in Vitro. Cancer Res. 1997, 57, 5598–5604. [Google Scholar]
- Lichti, C.F.; Liu, H.; Shavkunov, A.S.; Mostovenko, E.; Sulman, E.P.; Ezhilarasan, R.; Wang, Q.; Kroes, R.A.; Moskal, J.C.; Fenyö, D.; et al. Integrated Chromosome 19 Transcriptomic and Proteomic Data Sets Derived from Glioma Cancer Stem-Cell Lines. J. Proteome Res. 2014, 13, 191–199. [Google Scholar] [CrossRef]
- Mughal, A.A.; Zhang, L.; Fayzullin, A.; Server, A.; Li, Y.; Wu, Y.; Glass, R.; Meling, T.; Langmoen, I.A.; Leergaard, T.B.; et al. Patterns of Invasive Growth in Malignant Gliomas—The Hippocampus Emerges as an Invasion-Spared Brain Region. Neoplasia 2018, 20, 643–656. [Google Scholar] [CrossRef]
- Bergmann, O.; Liebl, J.; Bernard, S.; Alkass, K.; Yeung, M.S.Y.; Steier, P.; Kutschera, W.; Johnson, L.; Landén, M.; Druid, H.; et al. The Age of Olfactory Bulb Neurons in Humans. Neuron 2012, 74, 634–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, E.Y.; Cooper, D.D.; Abbott, K.L.; Lennon, J.; Nagaraja, S.; Mackay, A.; Jones, C.; Vogel, H.; Jackson, P.K.; Monje, M. Neural Precursor-Derived Pleiotrophin Mediates Subventricular Zone Invasion by Glioma. Cell 2017, 170, 845–859.e19. [Google Scholar] [CrossRef] [PubMed]
- Zappaterra, M.W.; Lehtinen, M.K. The Cerebrospinal Fluid: Regulator of Neurogenesis, Behavior, and Beyond. Cell. Mol. Life Sci. 2012, 69, 2863–2878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrano, A.; Zarco, N.; Phillipps, J.; Lara-Velazquez, M.; Suarez-Meade, P.; Norton, E.S.; Chaichana, K.L.; Quiñones-Hinojosa, A.; Asmann, Y.W.; Guerrero-Cázares, H. Human Cerebrospinal Fluid Modulates Pathways Promoting Glioblastoma Malignancy. Front. Oncol. 2021, 11, 624145. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Takeda, N.; Jain, R.; Manderfield, L.J.; Liu, F.; Li, L.; Anderson, S.A.; Epstein, J.A. Hopx Distinguishes Hippocampal from Lateral Ventricle Neural Stem Cells. Stem Cell Res. 2015, 15, 522–529. [Google Scholar] [CrossRef] [Green Version]
- Er, M.; Am, P. Neural Stem Cells of the Subventricular Zone as the Origin of Human Glioblastoma Stem Cells. Therapeutic Implications. Front. Oncol. 2019, 9, 779. [Google Scholar] [CrossRef] [Green Version]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and Characterization of Tumorigenic, Stem-like Neural Precursors from Human Glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Guignard, F.; Zhao, D.; Liu, L.; Burns, D.K.; Mason, R.P.; Messing, A.; Parada, L.F. Early Inactivation of P53 Tumor Suppressor Gene Cooperating with NF1 Loss Induces Malignant Astrocytoma. Cancer Cell 2005, 8, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcantara Llaguno, S.R.; Xie, X.; Parada, L.F. Cell of Origin and Cancer Stem Cells in Tumor Suppressor Mouse Models of Glioblastoma. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Kim, Y.-S.; Lee, K.; Kang, M.; Shin, H.; Oh, J.-W.; Koo, H.; Kim, D.; Kim, Y.; Kong, D.-S.; et al. Novel Semi-Replicative Retroviral Vector Mediated Double Suicide Gene Transfer Enhances Antitumor Effects in Patient-Derived Glioblastoma Models. Cancers 2019, 11, 1090. [Google Scholar] [CrossRef] [Green Version]
- Holland, E.C.; Celestino, J.; Dai, C.; Schaefer, L.; Sawaya, R.E.; Fuller, G.N. Combined Activation of Ras and Akt in Neural Progenitors Induces Glioblastoma Formation in Mice. Nat. Genet. 2000, 25, 55–57. [Google Scholar] [CrossRef]
- Jacques, T.S.; Swales, A.; Brzozowski, M.J.; Henriquez, N.V.; Linehan, J.M.; Mirzadeh, Z.; O’ Malley, C.; Naumann, H.; Alvarez-Buylla, A.; Brandner, S. Combinations of Genetic Mutations in the Adult Neural Stem Cell Compartment Determine Brain Tumour Phenotypes. EMBO J. 2010, 29, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Alcantara Llaguno, S.; Chen, J.; Kwon, C.-H.; Jackson, E.L.; Li, Y.; Burns, D.K.; Alvarez-Buylla, A.; Parada, L.F. Malignant Astrocytomas Originate from Neural Stem/Progenitor Cells in a Somatic Tumor Suppressor Mouse Model. Cancer Cell 2009, 15, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human Cortical Glial Tumors Contain Neural Stem-like Cells Expressing Astroglial and Neuronal Markers in Vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef]
- Jafri, N.F.; Clarke, J.L.; Weinberg, V.; Barani, I.J.; Cha, S. Relationship of Glioblastoma Multiforme to the Subventricular Zone Is Associated with Survival. Neuro-Oncology 2013, 15, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Adeberg, S.; Bostel, T.; König, L.; Welzel, T.; Debus, J.; Combs, S.E. A Comparison of Long-Term Survivors and Short-Term Survivors with Glioblastoma, Subventricular Zone Involvement: A Predictive Factor for Survival? Radiat. Oncol. 2014, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, D.A.; Cha, S.; Mayo, M.C.; Chen, M.-H.; Keles, E.; VandenBerg, S.; Berger, M.S. Relationship of Glioblastoma Multiforme to Neural Stem Cell Regions Predicts Invasive and Multifocal Tumor Phenotype. Neuro-Oncology 2007, 9, 424–429. [Google Scholar] [CrossRef]
- Berger, F.; Gay, E.; Pelletier, L.; Tropel, P.; Wion, D. Development of Gliomas: Potential Role of Asymmetrical Cell Division of Neural Stem Cells. Lancet Oncol. 2004, 5, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Vescovi, A.L.; Galli, R.; Reynolds, B.A. Brain Tumour Stem Cells. Nat. Rev. Cancer 2006, 6, 425–436. [Google Scholar] [CrossRef]
- Quiñones-Hinojosa, A.; Chaichana, K. The Human Subventricular Zone: A Source of New Cells and a Potential Source of Brain Tumors. Exp. Neurol. 2007, 205, 313–324. [Google Scholar] [CrossRef]
- Lai, A.; Kharbanda, S.; Pope, W.B.; Tran, A.; Solis, O.E.; Peale, F.; Forrest, W.F.; Pujara, K.; Carrillo, J.A.; Pandita, A.; et al. Evidence for Sequenced Molecular Evolution of IDH1 Mutant Glioblastoma From a Distinct Cell of Origin. J. Clin. Oncol. 2011, 29, 4482–4490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canoll, P.; Goldman, J.E. The Interface between Glial Progenitors and Gliomas. Acta Neuropathol. 2008, 116, 465–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, L.C.; Veeravagu, A.; Hsu, A.R.; Tse, V.C.K. Recurrent Glioblastoma Multiforme: A Review of Natural History and Management Options. Neurosurg. Focus 2006, 20, E3. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Kohn, E.C. The Microenvironment of the Tumour–Host Interface. Nature 2001, 411, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Aboody, K.S.; Brown, A.; Rainov, N.G.; Bower, K.A.; Liu, S.; Yang, W.; Small, J.E.; Herrlinger, U.; Ourednik, V.; Black, P.M.; et al. Neural Stem Cells Display Extensive Tropism for Pathology in Adult Brain: Evidence from Intracranial Gliomas. Proc. Natl. Acad. Sci. USA 2000, 97, 12846–12851. [Google Scholar] [CrossRef] [Green Version]
- Assanah, M.; Lochhead, R.; Ogden, A.; Bruce, J.; Goldman, J.; Canoll, P. Glial Progenitors in Adult White Matter Are Driven to Form Malignant Gliomas by Platelet-Derived Growth Factor-Expressing Retroviruses. J. Neurosci. 2006, 26, 6781–6790. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Gao, Y.; Tokuyama, T.; Yamamoto, J.; Yokota, N.; Yamamoto, S.; Terakawa, S.; Kitagawa, M.; Namba, H. Genetically Engineered Neural Stem Cells Migrate and Suppress Glioma Cell Growth at Distant Intracranial Sites. Cancer Lett. 2007, 251, 220–227. [Google Scholar] [CrossRef]
- Kim, S.-K.; Kim, S.U.; Park, I.H.; Bang, J.H.; Aboody, K.S.; Wang, K.-C.; Cho, B.-K.; Kim, M.; Menon, L.G.; Black, P.M.; et al. Human Neural Stem Cells Target Experimental Intracranial Medulloblastoma and Deliver a Therapeutic Gene Leading to Tumor Regression. Clin. Cancer Res. 2006, 12, 5550–5556. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zong, H. Developmental Origins of Brain Tumors. Curr. Opin. Neurobiol. 2012, 22, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A Transcriptome Database for Astrocytes, Neurons, and Oligodendrocytes: A New Resource for Understanding Brain Development and Function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Kupp, R.; Shtayer, L.; Tien, A.-C.; Szeto, E.; Sanai, N.; Rowitch, D.H.; Mehta, S. Lineage-Restricted OLIG2-RTK Signaling Governs the Molecular Subtype of Glioma Stem-like Cells. Cell Rep. 2016, 16, 2838–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carén, H.; Stricker, S.H.; Bulstrode, H.; Gagrica, S.; Johnstone, E.; Bartlett, T.E.; Feber, A.; Wilson, G.; Teschendorff, A.E.; Bertone, P.; et al. Glioblastoma Stem Cells Respond to Differentiation Cues but Fail to Undergo Commitment and Terminal Cell-Cycle Arrest. Stem Cell Rep. 2015, 5, 829–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, N.I.; Guilhamon, P.; Desai, K.; McAdam, R.F.; Langille, E.; O’Connor, M.; Lan, X.; Whetstone, H.; Coutinho, F.J.; Vanner, R.J.; et al. ASCL1 Reorganizes Chromatin to Direct Neuronal Fate and Suppress Tumorigenicity of Glioblastoma Stem Cells. Cell Stem Cell 2017, 21, 209–224.e7. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Verhaak, R.G.; Canoll, P. The Cellular Origin for Malignant Glioma and Prospects for Clinical Advancements. Expert Rev. Mol. Diagn. 2012, 12, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, D.; Funk, C.C.; Caballero, J.; Shah, N.; Rouleau, K.; Earls, J.C.; Soroceanu, L.; Foltz, G.; Cobbs, C.S.; Price, N.D.; et al. A Cell-Surface Membrane Protein Signature for Glioblastoma. Cell Syst. 2017, 4, 516–529.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loras, A.; Gonzalez-Bonet, L.G.; Gutierrez-Arroyo, J.L.; Martinez-Cadenas, C.; Marques-Torrejon, M.A. Neural Stem Cells as Potential Glioblastoma Cells of Origin. Life 2023, 13, 905. https://doi.org/10.3390/life13040905
Loras A, Gonzalez-Bonet LG, Gutierrez-Arroyo JL, Martinez-Cadenas C, Marques-Torrejon MA. Neural Stem Cells as Potential Glioblastoma Cells of Origin. Life. 2023; 13(4):905. https://doi.org/10.3390/life13040905
Chicago/Turabian StyleLoras, Alba, Luis G. Gonzalez-Bonet, Julia L. Gutierrez-Arroyo, Conrado Martinez-Cadenas, and Maria Angeles Marques-Torrejon. 2023. "Neural Stem Cells as Potential Glioblastoma Cells of Origin" Life 13, no. 4: 905. https://doi.org/10.3390/life13040905