
Pathophysiological Effects of Contemporary Lifestyle on Evolutionary-Conserved Survival Mechanisms in Polycystic Ovary Syndrome


Abstract
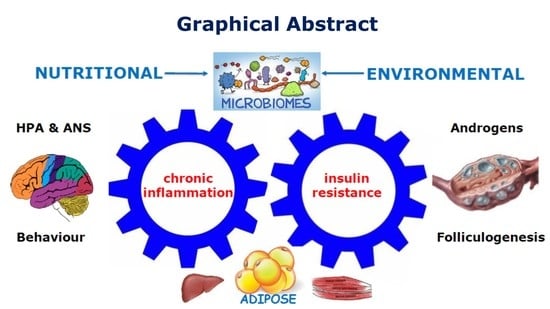
:
1. Introduction
2. Materials and Methods
3. Chronic Systemic Inflammation
3.1. Evolution and the Advantages of a Proinflammatory Design
3.2. Overview of the Inflammatory Response
3.2.1. Oxidative Stress in PCOS
3.2.2. Advanced Glycation End Products and PCOS
3.2.3. Pattern Recognition Receptors and the Innate Immune System
3.2.4. Inflammasomes in PCOS
3.2.5. Adaptive Immune Response in PCOS
3.3. Neuroimmunomodulation and the Link between the Nervous System and PCOS
3.3.1. Anatomy of Neuroendocrine-Immune Connections

3.3.2. Neuroimmunomodulation in PCOS
3.4. Hyperandrogenism and Chronic Inflammation
3.5. Summary of the Role of Inflammation in PCOS from an Evolutionary Perspective
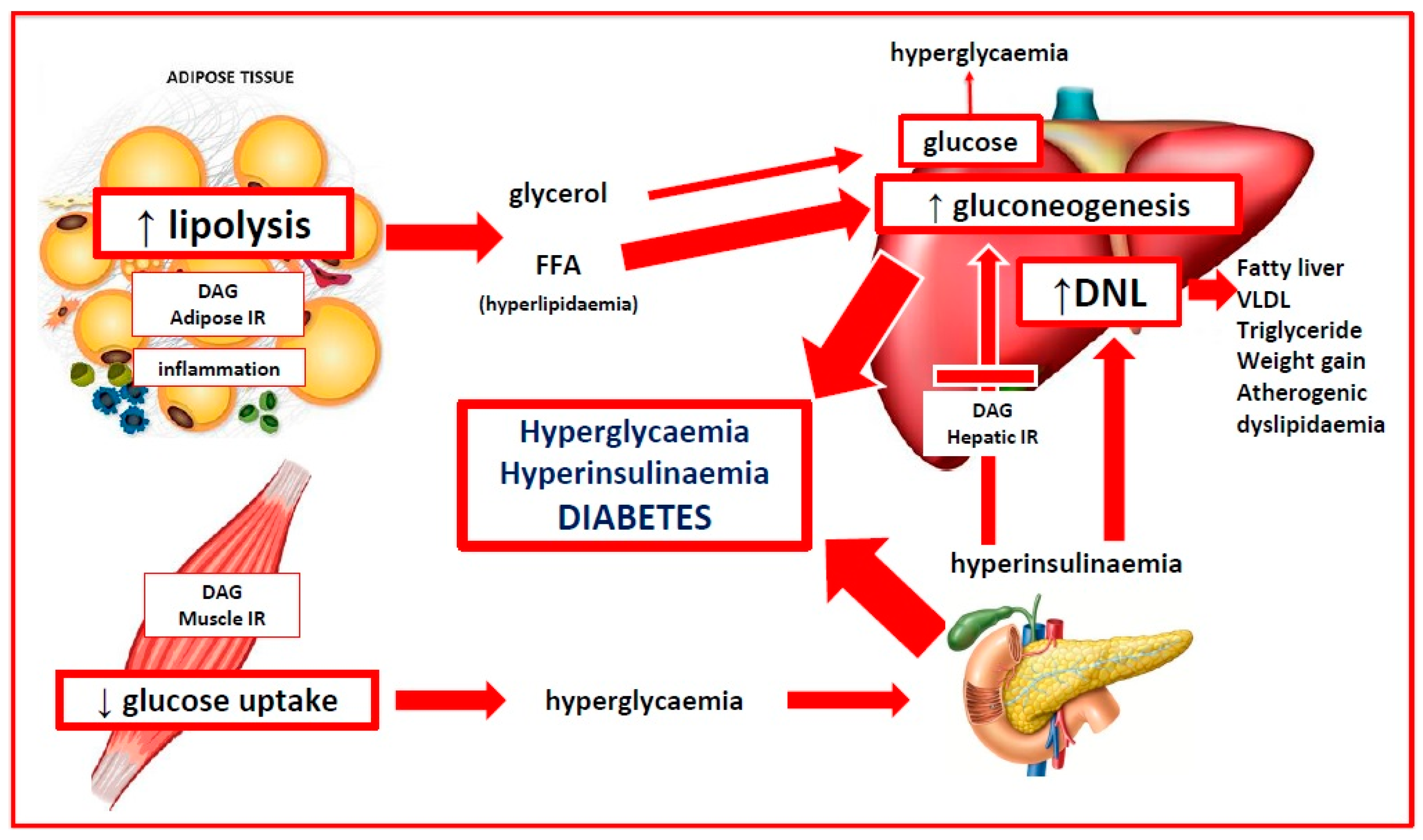
4. Insulin Resistance and Reduced Insulin Sensitivity
4.1. Physiological Actions of Insulin
4.2. Reduced Insulin Sensitivity versus Insulin Resistance in PCOS
4.3. Mechanisms of Insulin Resistance in PCOS
4.4. Evolutionary Adaptive Role of Insulin Resistance in PCOS
4.4.1. Insulin Resistance and Infection
4.4.2. Insulin Resistance, Starvation and Dehydration
4.4.3. Insulin Resistance and Pregnancy
4.5. Insulin Resistance and Hyperandrogenism
5. Evolutionary Significance of Adipose Tissue in PCOS
6. Central Role of the Microbiome in the Pathogenesis of PCOS
7. Environmental and Endocrine Disrupting Chemicals in PCOS
8. Evolutionary Model of PCOS and the Hallmarks of Health
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTH | Adrenocorticotropic Hormone |
| AGE | Advanced Glycation End-Products |
| ANS | Autonomic Nervous System |
| AT | Adipose Tissue |
| BAT | Brown Adipose Tissue |
| CDR | Cell Danger Response |
| DAMPS | Danger Associated Molecular Patterns |
| DNA | Deoxyribose Nucleic Acid |
| EDC | Endocrine Disrupting Chemical |
| GDM | Gestational Diabetes |
| GI | Gastrointestinal |
| GLUT | Glucose Transporter |
| HPA | Hypothalamic-Pituitary-Adrenal |
| IR | Insulin Resistance |
| LH | Luteinizing Hormone |
| LPS | Lipopolysaccharide |
| PAMPS | Pathogen Associated Molecular Patterns |
| PCOS | Polycystic Ovary Syndrome |
| PNS | Peripheral Nervous System |
| PRR | Pattern Recognition Receptors |
| RAGE | Receptor of Advanced Glycation End-Products |
| RNA | Ribose Nucleic Acid |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| SAT | Subcutaneous Adipose Tissue |
| SNS | Sympathetic Nervous System |
| TLR | Toll-like Receptor |
| TNF | Tumour Necrosis Factor |
| T2DM | Type 2 Diabetes Mellitis |
| WAT | White Adipose Tissue |
| VAT | Visceral Adipose Tissue |
References
- Parker, J.; O’brien, C.; Hawrelak, J.; Gersh, F.L. Polycystic Ovary Syndrome: An Evolutionary Adaptation to Lifestyle and the Environment. Int. J. Environ. Res. Public Health 2022, 19, 1336. [Google Scholar] [CrossRef] [PubMed]
- Teede, H.J.; Misso, M.L.; Costello, M.F.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R.J.; Andersen, M.; Azziz, R.; et al. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Fertil. Steril. 2018, 110, 364–379. [Google Scholar] [CrossRef]
- Cowan, S.; Lim, S.; Alycia, C.; Pirotta, S.; Thomson, R.; Gibson-Helm, M.; Blackmore, R.; Naderpoor, N.; Bennett, C.; Ee, C.; et al. Lifestyle management in polycystic ovary syndrome—Beyond diet and physical activity. BMC Endocr. Disord. 2023, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Lu, W.; Feng, Y.; Xu, C.; Hsueh, A.J.W. Out of step societal and Darwinian adaptation during evolution is the cause of multiple women’s health issues. Hum. Reprod. 2022, 37, 1959–1969. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, D.A.; Padmanabhan, V.; Chazenbalk, G.D.; Abbott, D.H. Polycystic ovary syndrome as a plausible evolutionary outcome of metabolic adaptation. Reprod. Biol. Endocrinol. 2022, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Pathak, G.; Nichter, M. Polycystic ovary syndrome in globalizing India: An ecosocial perspective on an emerging lifestyle disease. Soc. Sci. Med. 2015, 146, 21–28. [Google Scholar] [CrossRef]
- Parker, J.; O’Brien, C. Evolutionary and genetic antecedents to the pathogenesis of polycystic ovary syndrome (PCOS). J. ACNEM 2021, 40, 12–20. [Google Scholar]
- Aboeldalyl, S.; James, C.; Seyam, E.; Ibrahim, E.M.; Shawki, H.E.D.; Amer, S. The role of chronic inflammation in polycystic ovarian syndrome—A systematic review and meta-analysis. Int. J. Mol. Sci. 2021, 22, 2734. [Google Scholar] [CrossRef] [PubMed]
- Giampaolino, P.; Foreste, V.; Di Filippo, C.; Gallo, A.; Mercorio, A.; Serafino, P.; Improda, F.P.; Verrazzo, P.; Zara, G.; Buonfantino, C.; et al. Microbiome and PCOS: State-of-art and future aspects. Int. J. Mol. Sci. 2021, 22, 2048. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, D.; Guo, H.; Li, M. Hyperandrogenemia and insulin resistance: The chief culprit of polycystic ovary syndrome. Life Sci. 2019, 236, 116940. [Google Scholar] [CrossRef] [PubMed]
- Palomba, S.; De Wilde, M.A.; Falbo, A.; Koster, M.P.H.; La Sala, G.B.; Fauser, B.C.J.M. Pregnancy complications in women with polycystic ovary syndrome. Hum. Reprod. Update 2015, 21, 575–592. [Google Scholar] [CrossRef]
- Brutocao, C.; Zaiem, F.; Alsawas, M.; Morrow, A.S.; Murad, M.H.; Javed, A. Psychiatric disorders in women with polycystic ovary syndrome: A systematic review and meta-analysis. Endocrine 2018, 62, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Zore, T.; Joshi, N.V.; Lizneva, D.; Azziz, R. Polycystic Ovarian Syndrome: Long-Term Health Consequences. Semin. Reprod. Med. 2017, 35, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yao, X.Y.; Shi, R.X.; Liu, S.F.; Wang, X.Y. A potential link between polycystic ovary syndrome and non-alcoholic fatty liver disease: An update meta-analysis. Reprod. Health 2018, 15, 77. [Google Scholar] [CrossRef]
- Yumiceba, V.; Lopez-Cortes, A.; Perez-Villa, A. Oncology and Pharmacogenomics Insights in Polycystic Ovary Syndrome: An Integrative Analysis. Front. Endocrinol. 2020, 11, 5130. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Li, F.; Li, S.; Ding, L.; Liu, M. Causal relationship between polycystic ovary syndrome and chronic kidney disease: A Mendelian randomization study. Front. Endocrinol. 2023, 14, 119. [Google Scholar] [CrossRef]
- Rodgers, R.J.; Avery, J.C.; Moore, V.M.; Davies, M.J.; Azziz, R.; Stener-Victorin, E.; Moran, L.J.; Robertson, S.A.; Stepto, N.K.; Norman, R.J.; et al. Complex diseases and co-morbidities: Polycystic ovary syndrome and type 2 diabetes mellitus. Endocr. Connect. 2019, 8, R71–R75. [Google Scholar] [CrossRef]
- Parker, J. NEM: A New Paradigm for Understanding the Common Origins of the Chronic Disease Epidemic. ACNEM J. 2018, 37, 6–11. [Google Scholar]
- Dunaif, A. Perspectives in polycystic ovary syndrome: From hair to eternity. J. Clin. Endocrinol. Metab. 2016, 101, 759–768. [Google Scholar] [CrossRef]
- Day, F.; Karaderi, T.; Jones, M.R.; Meun, C.; He, C.; Drong, A.; Kraft, P.; Lin, N.; Huang, H.; Broer, L.; et al. Large-scale genome-wide meta-analysis of polycystic ovary syndrome suggests shared genetic architecture for different diagnosis criteria. PLoS Genet. 2018, 14, e1007813. [Google Scholar] [CrossRef]
- Vink, J.M.; Sadrzadeh, S.; Lambalk, C.B.; Boomsma, D.I. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J. Clin. Endocrinol. Metab. 2006, 91, 2100–2104. [Google Scholar] [CrossRef] [PubMed]
- Kahsar-Miller, M.D.; Nixon, C.; Boots, L.R.; Go, R.C.; Azziz, R. Prevalence of polycystic ovary syndrome (PCOS) in first-degree relatives of patients with PCOS. Fertil. Steril. 2001, 75, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Gao, Y.; Yang, J.; Lu, J.; Feng, W.; Yang, W. Mendelian Randomization Analysis Identified Potential Genes Pleiotropically Associated with Polycystic Ovary Syndrome. Reprod. Sci. 2021, 29, 1028–1037. [Google Scholar] [CrossRef]
- Zhu, T.; Goodarzi, M. Causes and consequences of polycystic ovary syndrome: Insights from Mendelian Randomization. J. Clin. Endocrinol. Metab. 2021, 107, e899–e911. [Google Scholar] [CrossRef]
- Lyle, S.M.; Ahmed, S.; Elliott, J.E.; Stener-Victorin, E.; Nachtigal, M.W.; Drögemöller, B.I. Transcriptome-wide association analyses identify an association between ARL14EP and polycystic ovary syndrome. J. Hum. Genet. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Charifson, M.A.; Trumble, B.C. Evolutionary origins of polycystic ovary syndrome: An environmental mismatch disorder. Evol. Med. Public Health 2019, 2019, 50–63. [Google Scholar] [CrossRef]
- Shaw, L.M.A.; Elton, S. Polycystic ovary syndrome: A transgenerational evolutionary adaptation. BJOG An Int. J. Obstet. Gynaecol. 2008, 115, 144–148. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Piperi, C. Genetics of polycystic ovary syndrome: Searching for the way out of the labyrinth. Hum. Reprod. Update 2005, 11, 631–643. [Google Scholar] [CrossRef]
- Khan, M.J.; Ullah, A.; Basit, S. Genetic basis of polycystic ovary syndrome (PCOS): Current perspectives. Appl. Clin. Genet. 2019, 12, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Chodasewicz, K. Evolution, reproduction and definition of life. Theory Biosci. 2014, 133, 39–45. [Google Scholar] [CrossRef]
- Corbett, S.; Morin-Papunen, L. The Polycystic Ovary Syndrome and recent human evolution. Mol. Cell. Endocrinol. 2013, 373, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Benton, M.L. The influence of evolutionary history on human health and disease. Nat. Rev. Genet. 2021, 22, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.P.; Bolnick, D.I.; Barrett, R.D.H.; Chapman, L.; Crispo, E.; Derry, A.M.; Eckert, C.G.; Fraser, D.J.; Fussmann, G.F.; Gonzalez, A.; et al. Understanding maladaptation by uniting ecological and evolutionary perspectives. Am. Nat. 2019, 194, 495–515. [Google Scholar] [CrossRef] [PubMed]
- Parker, J. Glucose metabolism, energy production and regulation of cellular and whole-body metabolism. ACNEM J. 2020, 39, 29–33. [Google Scholar]
- Chantranupong, L.; Wolfson, R.L.; Sabatini, D.M. Nutrient-sensing mechanisms across evolution. Cell 2015, 161, 67–83. [Google Scholar] [CrossRef]
- Holly, J.M.P.; Biernacka, K.; Perks, C.M. Systemic metabolism, its regulators, and cancer: Past mistakes and future potential. Front. Endocrinol. 2019, 10, 65. [Google Scholar] [CrossRef]
- Norman, R.; Teede, H. A new evidence-based guideline for assessment and management of polycystic ovary syndrome. Med. J. Aust. 2018, 209, 299–300. [Google Scholar] [CrossRef]
- Casarini, L.; Simoni, M.; Brigante, G. Is polycystic ovary syndrome a sexual conflict? A review. Reprod. Biomed. Online 2016, 32, 350–361. [Google Scholar] [CrossRef]
- Parker, J. Emerging Concepts in the Pathogenesis and Treatment of Polycystic Ovary Syndrome. Curr. Womens. Health Rev. 2016, 10, 107–112. [Google Scholar] [CrossRef]
- Azziz, R.; Dumesic, D.A.; Goodarzi, M.O. Polycystic ovary syndrome: An ancient disorder? Fertil. Steril. 2011, 95, 1544–1548. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Low, F.M.; Hanson, M.A. Anthropocene-related disease. Evol. Med. Public Health 2020, 2020, 304–310. [Google Scholar] [CrossRef]
- Rubio-Ruiz, M.E.; Peredo-Escárcega, A.E.; Cano-Martínez, A.; Guarner-Lans, V. An Evolutionary Perspective of Nutrition and Inflammation as Mechanisms of Cardiovascular Disease. Int. J. Evol. Biol. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Watve, M.G.; Yajnik, C.S. Evolutionary origins of insulin resistance: A behavioral switch hypothesis. BMC Evol. Biol. 2007, 7, 61. [Google Scholar] [CrossRef]
- López-Otín, C.; Kroemer, G. Hallmarks of Health. Cell 2021, 184, 33–63. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Rönn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef]
- Szukiewicz, D.; Trojanowski, S.; Kociszewska, A.; Szewczyk, G. Modulation of the Inflammatory Response in Polycystic Ovary Syndrome (PCOS)—Searching for Epigenetic Factors. Int. J. Mol. Sci. 2022, 23, 14663. [Google Scholar] [CrossRef] [PubMed]
- Shorakae, S.; Ranasinha, S.; Abell, S.; Lambert, G.; Lambert, E.; de Courten, B.; Teede, H. Inter-related effects of insulin resistance, hyperandrogenism, sympathetic dysfunction and chronic inflammation in PCOS. Clin. Endocrinol. 2018, 89, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Barlampa, D.; Bompoula, M.S.; Bargiota, A.; Kalantaridou, S.; Mastorakos, G.; Valsamakis, G. Hypothalamic inflammation as a potential pathophysiologic basis for the heterogeneity of clinical, hormonal, and metabolic presentation in pcos. Nutrients 2021, 13, 520. [Google Scholar] [CrossRef] [PubMed]
- Okin, D.; Medzhitov, R. Evolution of inflammatory diseases. Curr. Biol. 2012, 22, R733–R740. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K. Metabolic features and regulation of the healing cycle—A new model for chronic disease pathogenesis and treatment. Mitochondrion 2019, 46, 278–297. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.Y.; Li, X.L. Pyroptosis and inflammasomes in obstetrical and gynecological diseases. Gynecol. Endocrinol. 2021, 37, 385–391. [Google Scholar] [CrossRef]
- Katayama, P.L.; Leirão, I.P.; Kanashiro, A.; Luiz, J.P.M.; Cunha, F.Q.; Navegantes, L.C.C.; Menani, J.V.; Zoccal, D.B.; Colombari, D.S.A.; Colombari, E. The carotid body detects circulating tumor necrosis factor-alpha to activate a sympathetic anti-inflammatory reflex. Brain Behav. Immun. 2022, 102, 370–386. [Google Scholar] [CrossRef] [PubMed]
- Tremellen, K.; Pearce, K. Dysbiosis of Gut Microbiota (DOGMA)—A novel theory for the development of polycystic ovarian syndrome. Med. Hypotheses 2012, 79, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Xiong, P.; Zhang, F.; Liu, F.; Zhao, J.; Huang, X. Metaflammation in glucolipid metabolic disorders: Pathogenesis and treatment. Biomed. Pharmacother. 2023, 161, 114545. [Google Scholar] [CrossRef]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, J.; Gong, Z.; Zhu, J.K. Abiotic stress responses in plants. Nat. Rev. Genet. 2022, 23, 104–119. [Google Scholar] [CrossRef]
- Khan, R.N.; Hay, D.P. A clear and present danger: Inflammasomes DAMPing down disorders of pregnancy. Hum. Reprod. Update 2015, 21, 388–405. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern recognition receptors and the host cell death molecular machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. Cell evolution and Earth history: Stasis and revolution. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 969–1006. [Google Scholar] [CrossRef]
- Tang, K.H.; Blankenship, R.E. Both forward and reverse TCA cycles operate in green sulfur bacteria. J. Biol. Chem. 2010, 285, 35848–35854. [Google Scholar] [CrossRef]
- Naviaux, R.K. Oxidative shielding or oxidative stress? J. Pharmacol. Exp. Ther. 2012, 342, 608–618. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Nikiforov, N.G.; Eid, A.H.; Nedosugova, L.V.; Starodubova, A.V.; Popkova, T.V.; Bezsonov, E.E.; Orekhov, A.N. Mitochondrial dysfunction and chronic inflammation in polycystic ovary syndrome. Int. J. Mol. Sci. 2021, 22, 3923. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhao, Y.; Li, T.; Li, M.; Li, J.; Li, R.; Liu, P.; Yu, Y.; Qiao, J. Metabolism alteration in follicular niche: The nexus among intermediary metabolism, mitochondrial function, and classic polycystic ovary syndrome. Free Radic. Biol. Med. 2015, 86, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M. Oxidative Stress and Polycystic Ovary Syndrome: A Brief Review. Int. J. Prev. Med. 2017, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Zhu, M.; Xu, W. Roles of oxidative stress in polycystic ovary syndrome and cancers. Oxid. Med. Cell. Longev. 2016, 2016, 8589318. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Herman, R.; Jensterle, M.; Janež, A.; Goričar, K.; Dolžan, V. Genetic variability in antioxidative and inflammatory pathways modifies the risk for PCOs and influences metabolic profile of the syndrome. Metabolites 2020, 10, 439. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Dunaif, A. Insulin resistance and the polycystic ovary syndrome revisited: An update on mechanisms and implications. Endocr. Rev. 2012, 33, 981–1030. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, B.; Haghollahi, F.; Shariat, M.; Zayerii, F. The Effects of Calcium-Vitamin D and Metformin on Polycystic Ovary Syndrome: A Pilot Study. Taiwan. J. Obstet. Gynecol. 2009, 48, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Kaya, C.; Erkan, A.F.; Cengiz, S.D.; Dünder, I.; Demirel, Ö.E.; Bilgihan, A. Advanced oxidation protein products are increased in women with polycystic ovary syndrome: Relationship with traditional and nontraditional cardiovascular risk factors in patients with polycystic ovary syndrome. Fertil. Steril. 2009, 92, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Enechukwu, C.I.; Onuegbu, A.J.; Olisekodiaka, M.J.; Eleje, G.U.; Ikechebelu, J.I.; Ugboaja, J.O.; Amah, U.K.; Okwara, J.E.; Igwegbe, A.O. Oxidative stress markers and lipid profiles of patients with polycystic ovary syndrome in a Nigerian tertiary hospital. Obstet. Gynecol. Sci. 2019, 62, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Dincer, Y.; Akcay, T.; Erdem, T.; Ilker Saygili, E.; Gundogdu, S. DNA damage, DNA susceptibility to oxidation and glutathione level in women with polycystic ovary syndrome. Scand. J. Clin. Lab. Investig. 2005, 65, 721–728. [Google Scholar] [CrossRef]
- Noormohammadi, M.; Eslamian, G.; Malek, S.; Shoaibinobarian, N.; Mirmohammadali, S.N. The association between fertility diet score and polycystic ovary syndrome: A Case-Control study. Health Care Women Int. 2022, 43, 70–84. [Google Scholar] [CrossRef]
- Shoaibinobarian, N.; Eslamian, G.; Noormohammadi, M.; Malek, S.; Rouhani, S.; Mirmohammadali, S.N. Dietary Total Antioxidant Capacity and Risk of Polycystic Ovary Syndrome: A Case-Control Study. Int. J. Fertil. Steril. 2022, 16, 200–205. [Google Scholar] [CrossRef]
- Parker, J.; Hawrelak, J.; Gersh, F. Nutritional Role of Polyphenols as a Component of a Wholefood Diet in the Management of Polycystic Ovary Syndrome. Australas. Coll. Nutr. Environ. Med. J. 2021, 40, 6–12. Available online: https://search.ebscohost.com/login.aspx?direct=true&db=rzh&AN=154032844&lang=ja&site=ehost-live (accessed on 21 June 2021).
- Piperi, C.; Adamopoulos, C.; Dalagiorgou, G.; Diamanti-Kandarakis, E.; Papavassiliou, A.G. Crosstalk between advanced glycation and endoplasmic reticulum stress: Emerging therapeutic targeting for metabolic diseases. J. Clin. Endocrinol. Metab. 2012, 97, 2231–2242. [Google Scholar] [CrossRef]
- Inagi, R. Inhibitors of advanced glycation and endoplasmic reticulum stress. In Methods in Enzymology, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011; Volume 491, pp. 361–380. [Google Scholar] [CrossRef]
- Goldberg, T.; Cai, W.; Peppa, M.; Dardaine, V.; Baliga, B.S.; Uribarri, J.; Vlassara, H. Advanced glycoxidation end products in commonly consumed foods. J. Am. Diet. Assoc. 2004, 104, 1287–1291. [Google Scholar] [CrossRef]
- Garg, D.; Merhi, Z. Advanced glycation end products: Link between diet and ovulatory dysfunction in PCOS? Nutrients 2015, 7, 10129–10144. [Google Scholar] [CrossRef] [PubMed]
- Basta, G. Receptor for advanced glycation endproducts and atherosclerosis: From basic mechanisms to clinical implications. Atherosclerosis 2008, 196, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Kathryn, C.; Tan, B.; Sammy, W.; Shiu, M.; Wong, Y. Xystus Tam Serum advanced glycation end products (AGEs) are associated with insulin resistance. Diabetes. Metab. Res. Rev. 2011, 27, 1488–1492. [Google Scholar] [CrossRef]
- Palimeri, S.; Palioura, E.; Diamanti-Kandarakis, E. Current perspectives on the health risks associated with the consumption of advanced glycation end products: Recommendations for dietary management. Diabetes Metab. Syndr. Obes. Targets Ther. 2015, 8, 415–426. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Piperi, C.; Patsouris, E.; Korkolopoulou, P.; Panidis, D.; Pawelczyk, L.; Papavassiliou, A.G.; Duleba, A.J. Immunohistochemical localization of advanced glycation end-products (AGEs) and their receptor (RAGE) in polycystic and normal ovaries. Histochem. Cell Biol. 2007, 127, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Azhary, J.M.K.; Harada, M.; Kunitomi, C.; Kusamoto, A.; Takahashi, N.; Nose, E.; Oi, N.; Wada-Hiraike, O.; Urata, Y.; Hirata, T.; et al. Androgens Increase Accumulation of Advanced Glycation End Products in Granulosa Cells by Activating ER Stress in PCOS. Endocrinology 2020, 161, 1–13. [Google Scholar] [CrossRef]
- Tatone, C.; Di Emidio, G.; Placidi, M.; Rossi, G.; Ruggieri, S.; Taccaliti, C.; D’Alfonso, A.; Amicarelli, F.; Guido, M. AGEs-related dysfunctions in PCOS: Evidence from animal and clinical research. J. Endocrinol. 2021, 251, R1–R9. [Google Scholar] [CrossRef]
- Garg, D.; Merhi, Z. Relationship between Advanced Glycation End Products and Steroidogenesis in PCOS. Reprod. Biol. Endocrinol. 2016, 14, 1–13. [Google Scholar] [CrossRef]
- Van Der Lugt, T.; Weseler, A.R.; Gebbink, W.A.; Vrolijk, M.F.; Opperhuizen, A.; Bast, A. Dietary advanced glycation endproducts induce an inflammatory response in human macrophages in vitro. Nutrients 2018, 10, 1868. [Google Scholar] [CrossRef]
- Uribarri, J.; del Castillo, M.D.; de la Maza, M.P.; Filip, R.; Gugliucci, A.; Luevano-Contreras, C.; Macías-Cervantes, M.H.; Markowicz Bastos, D.H.; Medrano, A.; Menini, T.; et al. Dietary advanced glycation end products and their role in health and disease. Adv. Nutr. 2015, 6, 461–473. [Google Scholar] [CrossRef]
- Tantalaki, E.; Piperi, C.; Livadas, S.; Kollias, A.; Adamopoulos, C.; Koulouri, A.; Christakou, C.; Diamanti-Kandarakis, E. Impact of dietary modification of advanced glycation end products (AGEs) on the hormonal and metabolic profile of women with polycystic ovary syndrome (PCOS). Hormones 2014, 13, 65–73. [Google Scholar] [CrossRef]
- Vlassara, H.; Striker, G.E. AGE restriction in diabetes mellitus: A paradigm shift. Nat. Rev. Endocrinol. 2011, 7, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K. Metabolic features of the cell danger response. Mitochondrion 2014, 16, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Moretti, J.; Blander, J.M. Insights into phagocytosis-coupled activation of pattern recognition receptors and inflammasomes. Curr. Opin. Immunol. 2014, 26, 100–110. [Google Scholar] [CrossRef]
- Zhou, F.; Li, C.; Zhang, S.Y. NLRP3 inflammasome: A new therapeutic target for high-risk reproductive disorders? Chin. Med. J. 2020, 134, 20–27. [Google Scholar] [CrossRef]
- Moretti, J.; Blander, J.M. Cell-autonomous stress responses in innate immunity. J. Leukoc. Biol. 2017, 101, 77–86. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Parker, J.; O’Brien, C.; Hawrelak, J. A narrative review of the role of gastrointestinal dysbiosis in the pathogenesis of polycystic ovary syndrome. Obstet. Gynecol. Sci. 2021, 65, 14–28. [Google Scholar] [CrossRef]
- Murthi, P.; Pinar, A.A.; Dimitriadis, E.; Samuel, C.S. Inflammasomes—A molecular link for altered immunoregulation and inflammation mediated vascular dysfunction in preeclampsia. Int. J. Mol. Sci. 2020, 21, 1406. [Google Scholar] [CrossRef]
- Rostamtabar, M.; Esmaeilzadeh, M.; Tourani, M.; Rahmani, A.; Baee, M.; Shirafkan, F.; Saleki, K.; Mirzababayi, S.; Ebrahimpour, S.; Nouri, H. Pathophysiological roles of chronic low-grade inflammation in polycystic ovary syndrome. J. Cell. Physiol. 2020, 236, 824–838. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S. Purinergic Signaling during Inflammation. N. Engl. J. Med. 2013, 368, 1260. [Google Scholar] [CrossRef] [PubMed]
- Rostamtabar, M.; Esmaeilzadeh, S.; Karkhah, A.; Amiri, M.; Rahmani, A.; Bakouei, F.; Nouri, H.R. Elevated expression of IL-18 but not IL-1β gene is associated with NALP3 and AIM2 inflammasome in Polycystic Ovary Syndrome. Gene 2020, 731, 144352. [Google Scholar] [CrossRef]
- Guo, Q.J.; Shan, J.; Xu, Y.F.; Hu, Y.Y.; Huo, C.L.; Song, J.Y.; Wang, C.Q.; Zhou, H.; Yu, C.Q.; Huang, Q. Pioglitazone Metformin Complex Improves Polycystic Ovary Syndrome Comorbid Psychological Distress via Inhibiting NLRP3 Inflammasome Activation: A Prospective Clinical Study. Mediat. Inflamm. 2020, 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Pang, B.; Ma, Z.; Yi, H. Immunophenotypic profiles in polycystic ovary syndrome. Mediat. Inflamm. 2020, 2020, 5894768. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Vita, R.; Shackelford, D.; Lane, J.; Schulten, V.; Zarebski, L.; Jespersen, M.C.; Marcatili, P.; Nielsen, M.; Sette, A.; et al. Epitope specific antibodies and T cell receptors in the immune epitope database. Front. Immunol. 2018, 9, 2688. [Google Scholar] [CrossRef] [PubMed]
- Rees, A.R. Understanding the human antibody repertoire. MAbs 2020, 12, 1729683. [Google Scholar] [CrossRef]
- Alberto Kölliker Frers, R.; Otero-Losada, M.; Inés Herrera, M.; Porta, S.; Cosentino, V.; Kerzberg, E.; Udovin, L.; Capani, F. Immune-Mediated Inflammation: Human T CD4 Helper Lymphocyte Diversity and Plasticity in Health and Disease. In Cells of the Immune System, 1st ed.; Ota Fuchs, Q., Athari, S.S., Eds.; Intechopen: London, UK, 2020; pp. 1–14. Available online: https://www.intechopen.com/chapters/69166 (accessed on 17 February 2023).
- Sbierski-Kind, J.; Goldeck, D.; Buchmann, N.; Spranger, J.; Volk, H.D.; Steinhagen-Thiessen, E.; Pawelec, G.; Demuth, I.; Spira, D. T cell phenotypes associated with insulin resistance: Results from the Berlin Aging Study II. Immun. Ageing 2020, 17, 1–11. [Google Scholar] [CrossRef]
- Wu, R.; Fujii, S.; Ryan, N.K.; Van der Hoek, K.H.; Jasper, M.J.; Sini, I.; Robertson, S.A.; Robker, R.L.; Norman, R.J. Ovarian leukocyte distribution and cytokine/chemokine mRNA expression in follicular fluid cells in women with polycystic ovary syndrome. Hum. Reprod. 2007, 22, 527–535. [Google Scholar] [CrossRef]
- Krishna, M.B.; Joseph, A.; Subramaniam, A.G.; Gupta, A.; Pillai, S.M.; Laloraya, M. Reduced tregs in peripheral blood of PCOS patients—A consequence of aberrant Il2 signaling. J. Clin. Endocrinol. Metab. 2015, 100, 282–292. [Google Scholar] [CrossRef]
- Ma, M.; Wang, M.; Xu, F.; Hao, S. The Imbalance in Th17 and Treg Cells in Polycystic Ovarian Syndrome Patients with Autoimmune Thyroiditis. Immunol. Investig. 2022, 51, 1170–1181. [Google Scholar] [CrossRef]
- Luan, Y.-Y.; Zhang, L.; Peng, Y.-Q.; Li, Y.-Y.; Liu, R.-X.; Yin, C.-H. Immune regulation in polycystic ovary syndrome. Clin. Chim. Acta 2022, 531, 265–272. [Google Scholar] [CrossRef]
- Salvador, A.F.; de Lima, K.A.; Kipnis, J. Neuromodulation by the immune system: A focus on cytokines. Nat. Rev. Immunol. 2021, 21, 526–541. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Ueda, M.; Suzuki, M.; Kohmura-Kobayashi, Y. Developmental Origins of Metaflammation; A Bridge to the Future Between the DOHaD Theory and Evolutionary Biology. Front. Endocrinol. 2022, 13, 9436. [Google Scholar] [CrossRef] [PubMed]
- Patel, S. Polycystic ovary syndrome (PCOS), an inflammatory, systemic, lifestyle endocrinopathy. J. Steroid Biochem. Mol. Biol. 2018, 182, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Wang, H.; Czura, C.J.; Friedman, S.G.; Tracey, K.J. The Cholinergic Anti-inflammatory Pathway: A Missing Link in Neuroimmunomodulation. Mol. Med. 2003, 9, 125–134. [Google Scholar] [CrossRef]
- Bellocchi, C.; Carandina, A.; Montinaro, B.; Targetti, E.; Furlan, L.; Rodrigues, G.D.; Tobaldini, E.; Montano, N. The Interplay between Autonomic Nervous System and Inflammation across Systemic Autoimmune Diseases. Int. J. Mol. Sci. 2022, 23, 2449. [Google Scholar] [CrossRef]
- Ingegnoli, F.; Buoli, M.; Antonucci, F.; Coletto, L.A.; Esposito, C.M.; Caporali, R. The Link Between Autonomic Nervous System and Rheumatoid Arthritis: From Bench to Bedside. Front. Med. 2020, 7, 589079. [Google Scholar] [CrossRef]
- del Rey, A.; Besedovsky, H.O.; Sorkin, E.; da Prada, M.; Arrenbrecht, S. Immunoregulation mediated by the sympathetic nervous system, II. Cell. Immunol. 1981, 63, 329–334. [Google Scholar] [CrossRef]
- Martelli, D. The inflammatory reflex reloaded. Brain Behav. Immun. 2022, 104, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Zangeneh, F.Z.; Bagheri, M.; Naghizadeh, M.M. Hyponeurotrophinemia in Serum of Women with Polycystic Ovary Syndrome as a Low Grade Chronic Inflammation. Open J. Obstet. Gynecol. 2015, 05, 459–469. [Google Scholar] [CrossRef]
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Wexler, B.C.; Dolgin, A.E.; Tryczynski, E.W. Effects of a bacterial polysaccharide (piromen) on the pituitary-adrenal axis: Adrenal ascorbic acid, cholesterol and histologic alterations. Endocrinology 1957, 61, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Broadwell, R. Passage of cytokines across the blood-brain-barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef]
- Buller, K.M. Role of circumventricular organs in pro-inflammatory cytokine-induced activation of the hypothalamic-pituitary-adrenal axis. Clin. Exp. Pharmacol. Physiol. 2001, 28, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, H.R.; Neuhuber, W.L. Functional and chemical anatomy of the afferent vagal system. Auton. Neurosci. Basic Clin. 2000, 85, 1–17. [Google Scholar] [CrossRef]
- Dag, Z.O.; Alpua, M.; Turkel, Y.; Isik, Y. Autonomic dysfunction in patients with polycystic ovary syndrome. Taiwan. J. Obstet. Gynecol. 2015, 54, 381–384. [Google Scholar] [CrossRef]
- Webster, J.I.; Tonelli, L.; Sternberg, E.M. Neuroendocrine regulation of immunity. Annu. Rev. Immunol. 2002, 20, 125–163. [Google Scholar] [CrossRef] [PubMed]
- Kox, M.; van Eijk, L.T.; Zwaag, J.; van den Wildenberg, J.; Sweep, F.C.G.J.; van der Hoeven, J.G.; Pickkers, P. Voluntary activation of the sympathetic nervous system and attenuation of the innate immune response in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7379–7384. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Liu, D.G.; Ye, X.M. Nicotinic Acetylcholine Receptor α7 Subunit Is an Essential Regulator of Seizure Susceptibility. Front. Neurol. 2021, 12, 6752. [Google Scholar] [CrossRef]
- Shaikh, S.; Verma, H.; Yadav, N.; Jauhari, M.; Bullangowda, J. Applications of Steroid in Clinical Practice: A Review. ISRN Anesthesiol. 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Lansdown, A.; Rees, D.A. The sympathetic nervous system in polycystic ovary syndrome: A novel therapeutic target? Clin. Endocrinol. 2012, 77, 791–801. [Google Scholar] [CrossRef]
- Chen, M.J.; Yang, W.S.; Yang, J.H.; Chen, C.L.; Ho, H.N.; Yang, Y.S. Relationship between androgen levels and blood pressure in young women with polycystic ovary syndrome. Hypertension 2007, 49, 1442–1447. [Google Scholar] [CrossRef]
- Perciaccante, A.; Fiorentini, A.; Valente, R.; Tubani, L. Polycystic ovary syndrome: Androgens, autonomic nervous system, and hypertension. Hypertension 2007, 50, 91710. [Google Scholar] [CrossRef]
- Giallauria, F.; Palomba, S.; Maresca, L.; Vuolo, L.; Tafuri, D.; Lombardi, G.; Colao, A.; Vigorito, C.; Orio, F. Exercise training improves autonomic function and inflammatory pattern in women with polycystic ovary syndrome (PCOS). Clin. Endocrinol. 2008, 69, 792–798. [Google Scholar] [CrossRef]
- Trigunaite, A.; Dimo, J.; Jørgensen, T.N. Suppressive effects of androgens on the immune system. Cell. Immunol. 2015, 294, 87–94. [Google Scholar] [CrossRef]
- González, F. Inflammation in Polycystic Ovary Syndrome: Underpinning of insulin resistance and ovarian dysfunction. Steroids 2012, 77, 300–305. [Google Scholar] [CrossRef]
- Wagner, I.V.; Savchuk, I.; Sahlin, L.; Kulle, A.; Klöting, N.; Dietrich, A.; Holterhus, P.M.; Dötsch, J.; Blüher, M.; Söder, O. De Novo and Depot-Specific Androgen Production in Human Adipose Tissue: A Source of Hyperandrogenism in Women with Obesity. Obes. Facts 2022, 15, 281–291. [Google Scholar] [CrossRef]
- Velez, L.M.; Seldin, M.; Motta, A.B. Inflammation and reproductive function in women with polycystic ovary syndrome. Biol. Reprod. 2021, 104, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.L.; Liang, X.Y.; Yang, X.; Li, Y.; Wei, L.N. Low-grade chronic inflammation in the peripheral blood and ovaries of women with polycystic ovarian syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011, 159, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Alexandraki, K.; Piperi, C.; Protogerou, A.; Katsikis, I.; Paterakis, T.; Lekakis, J.; Panidis, D. Inflammatory and endothelial markers in women with polycystic ovary syndrome. Eur. J. Clin. Investig. 2006, 36, 691–697. [Google Scholar] [CrossRef]
- Escobar-Morreale, H.F.; Luque-Ramírez, M.; González, F. Circulating inflammatory markers in polycystic ovary syndrome: A systematic review and metaanalysis. Fertil. Steril. 2011, 95, 1048–1058.e2. [Google Scholar] [CrossRef]
- Azhary, J.M.K.; Harada, M.; Takahashi, N.; Nose, E.; Kunitomi, C.; Koike, H.; Hirata, T.; Hirota, Y.; Koga, K.; Wada-Hiraike, O.; et al. Endoplasmic reticulum stress activated by androgen enhances apoptosis of granulosa cells via induction of death receptor 5 in PCOS. Endocrinology 2019, 160, 119–132. [Google Scholar] [CrossRef]
- Koike, H.; Harada, M.; Kusamoto, A.; Xu, Z.; Tanaka, T.; Sakaguchi, N.; Kunitomi, C.; Azhary, J.M.K.; Takahashi, N.; Urata, Y.; et al. Roles of endoplasmic reticulum stress in the pathophysiology of polycystic ovary syndrome. Front. Endocrinol. 2023, 4, 1124405. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, Q.; Sun, D.; Cui, X.; Chen, S.; Bulbul, A.; Liu, S.; Yan, Q. Dehydroepiandrosterone stimulates inflammation and impairs ovarian functions of polycystic ovary syndrome. J. Cell. Physiol. 2019, 234, 7435–7447. [Google Scholar] [CrossRef]
- Zhai, Y.; Pang, Y. Systemic and ovarian inflammation in women with polycystic ovary syndrome. J. Reprod. Immunol. 2022, 151, 103628. [Google Scholar] [CrossRef]
- Rahman, M.S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Pang, M.G. Role of insulin in health and disease: An update. Int. J. Mol. Sci. 2021, 22, 6403. [Google Scholar] [CrossRef]
- Watanabe, M.; Hayasaki, H.; Tamayama, T.; Shimada, M. Histologic distribution of insulin and glucagon receptors. Braz. J. Med. Biol. Res. 1998, 31, 243–256. [Google Scholar] [CrossRef]
- Khalid, M.; Alkaabi, J.; Khan, M.A.B.; Adem, A. Insulin signal transduction perturbations in insulin resistance. Int. J. Mol. Sci. 2021, 22, 8590. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.S.; Wang, A.; Yu, H. Link between insulin resistance and hypertension: What is the evidence from evolutionary biology? Diabetol. Metab. Syndr. 2014, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Li, J.; Gao, F. New insights into insulin: The anti-inflammatory effect and its clinical relevance. World J. Diabetes 2014, 5, 89. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.W.; Hung, L.C.; Chen, Y.C.; Wang, W.H.; Lin, C.Y.; Tzeng, H.H.; Suen, J.L.; Chen, Y.H. Insulin Reduces Inflammation by Regulating the Activation of the NLRP3 Inflammasome. Front. Immunol. 2021, 11, 7229. [Google Scholar] [CrossRef]
- Fernández-Real, J.M.; Ricart, W. Insulin resistance and inflammation in an evolutionary perspective: The contribution of cytokine genotype/phenotype to thriftiness. Diabetologia 1999, 42, 1367–1374. [Google Scholar] [CrossRef]
- Wan, Y.Y. Regulatory T cells: Immune suppression and beyond. Cell. Mol. Immunol. 2010, 7, 204–210. [Google Scholar] [CrossRef]
- Jacobse, J.; Li, J.; Rings, E.H.H.M.; Samsom, J.N.; Goettel, J.A. Intestinal Regulatory T Cells as Specialized Tissue-Restricted Immune Cells in Intestinal Immune Homeostasis and Disease. Front. Immunol. 2021, 12, 6499. [Google Scholar] [CrossRef]
- Makhijani, P.; Basso, P.J.; Chan, Y.T.; Chen, N.; Baechle, J.; Khan, S.; Furman, D.; Tsai, S.; Winer, D. Regulation of the immune system by the insulin receptor in health and disease. Front. Endocrinol. 2023, 14, 1128622. [Google Scholar] [CrossRef]
- Cassar, S.; Misso, M.L.; Hopkins, W.G.; Shaw, C.S.; Teede, H.J.; Stepto, N.K. Insulin resistance in polycystic ovary syndrome: A systematic review and meta-analysis of euglycaemic-hyperinsulinaemic clamp studies. Hum. Reprod. 2016, 31, 2619–2631. [Google Scholar] [CrossRef]
- Stepto, N.K.; Cassar, S.; Joham, A.E.; Hutchison, S.K.; Harrison, C.L.; Goldstein, R.F.; Teede, H.J. Women with polycystic ovary syndrome have intrinsic insulin resistance on euglycaemic-hyperinsulaemic clamp. Hum. Reprod. 2013, 28, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Katic, M.; Kahn, C.R. The role of insulin and IGF-1 signaling in longevity. Cell. Mol. Life Sci. 2005, 62, 320–343. [Google Scholar] [CrossRef]
- Cibula, D. Is insulin resistance an essential component of PCOS? The influence of confounding factors. Hum. Reprod. 2004, 19, 757–759. [Google Scholar] [CrossRef]
- Plomin, R.; Haworth, C.M.A.; Davis, O.S.P. Common disorders are quantitative traits. Nat. Rev. Genet. 2009, 10, 872–878. [Google Scholar] [CrossRef]
- Tam, C.S.; Xie, W.; Johnson, W.D.; Cefalu, W.T.; Redman, L.M.; Ravussin, E. Defining insulin resistance from hyperinsulinemic-euglycemic clamps. Diabetes Care 2012, 35, 1605–1610. [Google Scholar] [CrossRef]
- Dunaif, A.; Segal, K.R.; Futterweit, W.; Dobrjansky, A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 1989, 38, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Teede, H.J.; Hutchison, S.K.; Zoungas, S. The management of insulin resistance in polycystic ovary syndrome. Trends Endocrinol. Metab. 2007, 18, 273–279. [Google Scholar] [CrossRef]
- Toosy, S.; Sodi, R.; Pappachan, J.M. Lean polycystic ovary syndrome (PCOS): An evidence-based practical approach. J. Diabetes Metab. Disord. 2018, 17, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Morciano, A.; Romani, F.; Sagnella, F.; Scarinci, E.; Palla, C.; Moro, F.; Tropea, A.; Policola, C.; Della Casa, S.; Guido, M.; et al. Assessment of insulin resistance in lean women with polycystic ovary syndrome. Fertil. Steril. 2014, 102, 250–256.e3. [Google Scholar] [CrossRef] [PubMed]
- Gorjão, R.; Takahashi, H.K.; Pan, J.A.; Massao Hirabara, S. Molecular mechanisms involved in inflammation and insulin resistance in chronic diseases and possible interventions. J. Biomed. Biotechnol. 2012, 2012, 2012–2014. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Murillo, A. Dietary Treatments to Reduce Insulin Resistance and Inflammation in Type-2 Diabetic Patients. Med. Res. Arch. 2022, 10, 1–20. [Google Scholar] [CrossRef]
- Tsatsoulis, A.; Mantzaris, M.D.; Sofia, B.; Andrikoula, M. Insulin resistance: An adaptive mechanism becomes maladaptive in the current environment—An evolutionary perspective. Metabolism 2012, 62, 622–633. [Google Scholar] [CrossRef]
- Wensveen, F.M.; Šestan, M.; Turk Wensveen, T.; Polić, B. ‘Beauty and the beast’ in infection: How immune–endocrine interactions regulate systemic metabolism in the context of infection. Eur. J. Immunol. 2019, 49, 982–995. [Google Scholar] [CrossRef]
- Wang, P.; Mariman, E.C.M. Insulin resistance in an energy-centered perspective. Physiol. Behav. 2008, 94, 198–205. [Google Scholar] [CrossRef]
- Kampmann, U.; Knorr, S.; Fuglsang, J.; Ovesen, P. Determinants of Maternal Insulin Resistance during Pregnancy: An Updated Overview. J. Diabetes Res. 2019, 2019, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sprague, J.E.; Gandica, R.; Kelsey, M.M. Insulin Resistance in Puberty; Humana Press Inc.: Totowa, NJ, USA, 2020; ISBN 9783030250553. [Google Scholar]
- Straub, R.H.; Cutolo, M.; Buttgereit, F.; Pongratz, G. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J. Intern. Med. 2010, 267, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Rabasa, C.; Dickson, S.L. Impact of stress on metabolism and energy balance. Curr. Opin. Behav. Sci. 2016, 9, 71–77. [Google Scholar] [CrossRef]
- Ieronymaki, E.; Daskalaki, M.G.; Lyroni, K.; Tsatsanis, C. Insulin signaling and insulin resistance facilitate trained immunity in macrophages through metabolic and epigenetic changes. Front. Immunol. 2019, 10, 1330. [Google Scholar] [CrossRef]
- Chaudhari, A.P.; Mazumdar, K.; Deepak, P. Anxiety, Depression, and Quality of Life in Women with Polycystic Ovarian Syndrome. Indian J. Psychol. Med. 2018, 40, 239–246. [Google Scholar] [CrossRef]
- Soeters, M.R.; Soeters, P.B. The evolutionary benefit of insulin resistance. Clin. Nutr. 2012, 31, 1002–1007. [Google Scholar] [CrossRef]
- Soeters, M.R.; Sauerwein, H.P.; Dubbelhuis, P.F.; Groener, J.E.; Ackermans, M.T.; Fliers, E.; Aerts, J.M.; Serlie, M.J. Muscle adaptation to short-term fasting in healthy lean humans. J. Clin. Endocrinol. Metab. 2008, 93, 2900–2903. [Google Scholar] [CrossRef]
- Horita, S.; Seki, G.; Yamada, H.; Suzuki, M.; Koike, K.; Fujita, T. Insulin resistance, obesity, hypertension, and renal sodium transport. Int. J. Hypertens. 2011, 2011, 1–8. [Google Scholar] [CrossRef]
- Velloso, L.A.; Folli, F.; Perego, L.; Saad, M.J.A. The multi-faceted cross-talk between the insulin and angiotensin II signaling systems. Diabetes. Metab. Res. Rev. 2006, 22, 98–107. [Google Scholar] [CrossRef]
- Zhou, M.S.; Schulman, I.H.; Raij, L. Vascular inflammation, insulin resistance, and endothelial dysfunction in salt-sensitive hypertension: Role of nuclear factor kappa B activation. J. Hypertens. 2010, 28, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Ehrmann, D.A.; Liljenquist, D.R.; Kasza, K.; Azziz, R.; Legro, R.S.; Ghazzi, M.N.; Aronoff, S.; Bernstein, R.; Bodenner, D.; Braithwaite, S.; et al. Prevalence and predictors of the metabolic syndrome in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 48–53. [Google Scholar] [CrossRef]
- Sonagra, A.D. Normal Pregnancy- A State of Insulin Resistance. J. Clin. Diagnostic Res. 2014, 8, CC01–CC03. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; Huston, L.; Amini, S.B.; Kalhan, S.C. Longitudinal changes in glucose metabolism during pregnancy in obese women with normal glucose tolerance and gestational diabetes mellitus. Am. J. Obstet. Gynecol. 1999, 180, 903–916. [Google Scholar] [CrossRef]
- Jayabalan, N.; Nair, S.; Nuzhat, Z.; Rice, G.E.; Zuñiga, F.A.; Sobrevia, L.; Leiva, A.; Sanhueza, C.; Gutiérrez, J.A.; Lappas, M.; et al. Cross talk between adipose tissue and placenta in obese and gestational diabetes mellitus pregnancies via exosomes. Front. Endocrinol. 2017, 8, 239. [Google Scholar] [CrossRef]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Kling Bäckhed, H.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, X.; Zuo, Y.; Gao, J.; Liu, Y.; Zheng, W. The risk factors of gestational diabetes mellitus in patients with polycystic ovary syndrome: What should we care. Medicine 2021, 100, E26521. [Google Scholar] [CrossRef]
- Yan, Q.; Qiu, D.; Liu, X.; Xing, Q.; Liu, R.; Hu, Y. The incidence of gestational diabetes mellitus among women with polycystic ovary syndrome: A meta-analysis of longitudinal studies. BMC Pregnancy Childbirth 2022, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Noctor, E. Type 2 diabetes after gestational diabetes: The influence of changing diagnostic criteria. World J. Diabetes 2015, 6, 234. [Google Scholar] [CrossRef] [PubMed]
- Vounzoulaki, E.; Khunti, K.; Abner, S.C.; Tan, B.K.; Davies, M.J.; Gillies, C.L. Progression to type 2 diabetes in women with a known history of gestational diabetes: Systematic review and meta-analysis. BMJ 2020, 369, m1361. [Google Scholar] [CrossRef]
- Bonora, E.; Trombetta, M.; Dauriz, M.; Travia, D.; Cacciatori, V.; Brangani, C.; Negri, C.; Perrone, F.; Pichiri, I.; Stoico, V.; et al. Chronic complications in patients with newly diagnosed type 2 diabetes: Prevalence and related metabolic and clinical features: The Verona Newly Diagnosed Type 2 Diabetes Study (VNDS) 9. BMJ Open Diabetes Res. Care 2020, 8, e001549. [Google Scholar] [CrossRef]
- Bolton, A. (Ed.) International Diabetes Federation Diabetes Atlas, 10th ed.; Diabetes Research and Clinical Practice; The IDF Diabetes Atlas: Brussels, Belgium, 2021; Volume 102, Available online: www.diabetesatlas.org (accessed on 15 December 2021).
- Yang, J.; Qian, F.; Chavarro, J.E.; Ley, S.H.; Tobias, D.K.; Yeung, E.; Hinkle, S.N.; Bao, W.; Li, M.; Liu, A.; et al. Modifiable risk factors and long term risk of type 2 diabetes among individuals with a history of gestational diabetes mellitus: Prospective cohort study. BMJ 2022, 378, e070312. [Google Scholar] [CrossRef]
- Patel, S. Disruption of aromatase homeostasis as the cause of a multiplicity of ailments: A comprehensive review. J. Steroid Biochem. Mol. Biol. 2017, 168, 19–25. [Google Scholar] [CrossRef]
- Evans, S.F.; Hull, M.L.; Hutchinson, M.R.; Rolan, P.E. Androgens, Endometriosis and Pain. Front. Reprod. Health 2021, 3, 2920. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.P.; Grainger, D.; Diamond, M.C.; Sherwin, R.S.; Defronzo, R.A. Effects of methyltestosterone on insulin secretion and sensitivity in women. J. Clin. Endocrinol. Metab. 1998, 83, 4420–4425. [Google Scholar] [CrossRef]
- Polderman, K.H.; Gooren, L.J.; Asscheman, H. Induction of Insulin Resistance by Androgens and Estrogens. J. Clin. Endocrinol. Metab. 1994, 79, 265–271. Available online: https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jcem/79/1/10.1210_jcem.79.1.8027240/1/jcem0265.pdf?Expires=1498610821&Signature=TRE3mYh6fQ2FNOn4dry9Y~-Y-oZVmlGINOvJhlM7Lrm2J-l-9VG1X9UeJK1XUU9wSsmEDz4kiZRNOXaeefJFQDNsFOPEIfUp~vqLk91tWtQ (accessed on 19 February 2023). [PubMed]
- Skinner, M.K.; Schmidt, M.; Savenkova, M.I.; Sadler-Riggleman, I.; Nilsson, E.E. Regulation of granulosa and theca cell transcriptomes during ovarian antral follicle development. Mol. Reprod. Dev. 2008, 75, 1457–1472. [Google Scholar] [CrossRef] [PubMed]
- Clegg, D.J. Minireview: The year in review of estrogen regulation of metabolism. Mol. Endocrinol. 2012, 26, 1957–1960. [Google Scholar] [CrossRef]
- Stocco, C. Aromatase expression in the ovary: Hormonal and molecular regulation. Steroids 2008, 73, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Armanini, D.; Boscaro, M.; Bordin, L.; Sabbadin, C. Controversies in the Pathogenesis, Diagnosis and Treatment of PCOS: Focus on Insulin Resistance, Inflammation, and Hyperandrogenism. Int. J. Mol. Sci. 2022, 23, 4110. [Google Scholar] [CrossRef]
- Di Nardo, G.; Zhang, C.; Marcelli, A.G.; Gilardi, G. Molecular and structural evolution of cytochrome p450 aromatase. Int. J. Mol. Sci. 2021, 22, 631. [Google Scholar] [CrossRef] [PubMed]
- Simeon, S.; Spjuth, O.; Lapins, M.; Nabu, S.; Anuwongcharoen, N.; Prachayasittikul, V.; Wikberg, J.E.S.; Nantasenamat, C. Origin of aromatase inhibitory activity via proteochemometric modeling. PeerJ 2016, 2016, e1979. [Google Scholar] [CrossRef]
- Conley, A.; Mapes, S.; Jo Corbin, C.; Greger, D.; Graham, S. Structural determinants of aromatase cytochrome p450 inhibition in substrate recognition site-1. Mol. Endocrinol. 2002, 16, 1456–1468. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Choi, Y.J.; Heo, K.; Park, S.J. Melatonin as an oncostatic molecule based on its anti-aromatase role in breast cancer. Int. J. Mol. Sci. 2021, 22, 438. [Google Scholar] [CrossRef]
- Cheng, J.C.; Fang, L.; Li, Y.; Wang, S.; Li, Y.; Yan, Y.; Jia, Q.; Wu, Z.; Wang, Z.; Han, X.; et al. Melatonin stimulates aromatase expression and estradiol production in human granulosa-lutein cells: Relevance for high serum estradiol levels in patients with ovarian hyperstimulation syndrome. Exp. Mol. Med. 2020, 52, 1341–1350. [Google Scholar] [CrossRef]
- Kim, J.Y.; Han, E.H.; Kim, H.G.; Oh, K.N.; Kim, S.K.; Lee, K.Y.; Jeong, H.G. Bisphenol A-induced aromatase activation is mediated by cyclooxygenase-2 up-regulation in rat testicular Leydig cells. Toxicol. Lett. 2010, 193, 200–208. [Google Scholar] [CrossRef]
- Maia, H.; Haddad, C.; Pinheiro, N.; Casoy, J. The effect of oral contraceptives on aromatase and Cox-2 expression in the endometrium of patients with idiopathic menorrhagia or adenomyosis. Int. J. Womens. Health 2013, 5, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mäkelä, T.; Hase, T.; Adlercreutz, H.; Kurzer, M.S. Lignans and flavonoids inhibit aromatase enzyme in human preadipocytes. J. Steroid Biochem. Mol. Biol. 1994, 50, 205–212. [Google Scholar] [CrossRef]
- Singh, S.; Awasthi, M.; Pandey, V.P.; Dwivedi, U.N. Plant derived anti-cancerous secondary metabolites as multipronged inhibitor of COX, Topo, and aromatase: Molecular modeling and dynamics simulation analyses. J. Biomol. Struct. Dyn. 2017, 35, 3082–3097. [Google Scholar] [CrossRef]
- Richard, S.; Moslemi, S.; Sipahutar, H.; Benachour, N.; Seralini, G.E. Differential effects of glyphosate and roundup on human placental cells and aromatase. Environ. Health Perspect. 2005, 113, 716–720. [Google Scholar] [CrossRef]
- Zarn, J.A.; Brüschweiler, B.J.; Schlatter, J.R. Azole fungicides affect mammalian steroidogenesis by inhibiting sterol 14α-demethylase and aromatase. Environ. Health Perspect. 2003, 111, 255–261. [Google Scholar] [CrossRef]
- Rojas, J.; Chávez-Castillo, M.; Torres, W.; Arraiz, N.; Cabrera, M. Metformin in Cancer: Chemical Pathways for Tumoral Control Independent of Amp-Dependent Kinase. J. Endocrinol. Diabetes Obes. 2014, 2, 1036. [Google Scholar]
- Biegon, A.; Alia-Klein, N.; Fowler, J.S. Potential contribution of aromatase inhibition to the effects of nicotine and related compounds on the brain. Front. Pharmacol. 2012, 3, 185. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Baillargeon, J.P.; Nestler, J.E. Commentary: Polycystic ovary syndrome: A syndrome of ovarian hypersensitivity to insulin? J. Clin. Endocrinol. Metab. 2006, 91, 22–24. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, J.; Cheng, X.; Nie, X.; He, B. Insulin resistance in polycystic ovary syndrome across various tissues: An updated review of pathogenesis, evaluation, and treatment. J. Ovarian Res. 2023, 16, 1–17. [Google Scholar] [CrossRef]
- Bremer, A.A.; Miller, W.L. The serine phosphorylation hypothesis of polycystic ovary syndrome: A unifying mechanism for hyperandrogenemia and insulin resistance. Fertil. Steril. 2008, 89, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, L.; Potau, N.; Zampolli, M.; Riqué, S.; Saenger, P.; Carrascosa, A. Hyperinsulinemia and decreased insulin-like growth factor-binding protein-1 are common features in prepubertal and pubertal girls with a history of premature pubarche. J. Clin. Endocrinol. Metab. 1997, 82, 2283–2288. [Google Scholar] [CrossRef]
- Soldani, R.; Cagnacci, A.; Yen, S.S.C. Insulin insulin-like growth factor I (IGF-I) and IGF-II enhance basal and gonadotrophin-releasing hormone-stimulated luteinizing hormone release from rat anterior pituitary cells in vitro. Eur. J. Endocrinol. 1994, 131, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Nestler, J.E.; Powers, L.P.; Matt, D.W.; Steingold, K.A.; Plymate, S.R.; Rittmaster, R.S.; Clore, J.N.; Blackard, W.G. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 1991, 72, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.; Inouye, K.; Akirav, E.; Park, E.; Riddell, M.C.; Vranic, M.; Matthews, S.G. Insulin alone increases hypothalamo-pituitary-adrenal activity, and diabetes lowers peak stress responses. Endocrinology 2005, 146, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Kanbour, S.A.; Dobs, A.S. Hyperandrogenism in Women with Polycystic Ovarian Syndrome: Pathophysiology and Controversies. Androgens 2022, 3, 22–30. [Google Scholar] [CrossRef]
- Pateguana, N.B.; Janes, A. The contribution of hyperinsulinemia to the hyperandrogenism of polycystic ovary syndrome. J. Insul. Resist. 2019, 4, 3. [Google Scholar] [CrossRef]
- Vyrides, A.A.; El Mahdi, E.; Giannakou, K. Ovulation induction techniques in women with polycystic ovary syndrome. Front. Med. 2022, 9, 2230. [Google Scholar] [CrossRef]
- Panth, N.; Gavarkovs, A.; Tamez, M.; Mattei, J. The Influence of Diet on Fertility and the Implications for Public Health Nutrition in the United States. Front. Public Health 2018, 6, 211. [Google Scholar] [CrossRef]
- Elnashar, A.M. The role of metformin in ovulation induction: Current status. Middle East Fertil. Soc. J. 2011, 16, 175–181. [Google Scholar] [CrossRef]
- Nehir Aytan, A.; Bastu, E.; Demiral, I.; Bulut, H.; Dogan, M.; Buyru, F. Relationship between hyperandrogenism, obesity, inflammation and polycystic ovary syndrome. Gynecol. Endocrinol. 2016, 32, 709–713. [Google Scholar] [CrossRef]
- West-Eberhard, M.J. Nutrition, the visceral immune system, and the evolutionary origins of pathogenic obesity. Proc. Natl. Acad. Sci. USA 2019, 116, 723–731. [Google Scholar] [CrossRef]
- Ottaviani, E.; Malagoli, D.; Franceschi, C. The evolution of the adipose tissue: A neglected enigma. Gen. Comp. Endocrinol. 2011, 174, 1–4. [Google Scholar] [CrossRef]
- Kuzawa, C.W. Adipose Tissue in Human Infancy and Childhood: An Evolutionary Perspective. Yearb. Phys. Anthropol. 1998, 41, 177–209. [Google Scholar] [CrossRef]
- Parra-Peralbo, E.; Talamillo, A.; Barrio, R. Origin and Development of the Adipose Tissue, a Key Organ in Physiology and Disease. Front. Cell Dev. Biol. 2021, 9, 786129. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Spritzer, P.M.; Lecke, S.B.; Satler, F.; Morsch, D.M. Adipose tissue dysfunction, adipokines, and low-grade chronic inflammation in polycystic ovary syndrome. Reproduction 2015, 149, R219–R227. [Google Scholar] [CrossRef]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef]
- Pawłowski, B.; Żelaźniewicz, A. The evolution of perennially enlarged breasts in women: A critical review and a novel hypothesis. Biol. Rev. 2021, 2798, 2794–2809. [Google Scholar] [CrossRef]
- Tan, C.Y.; Vidal-Puig, A. Adipose tissue expandability: The metabolic problems of obesity may arise from the inability to become more obese. Biochem. Soc. Trans. 2008, 36, 935–940. [Google Scholar] [CrossRef]
- Villa, J.; Pratley, R.E. Adipose tissue dysfunction in polycystic ovary syndrome. Curr. Diab. Rep. 2011, 11, 179–184. [Google Scholar] [CrossRef]
- Echiburú, B.; Pérez-Bravo, F.; Galgani, J.E.; Sandoval, D.; Saldías, C.; Crisosto, N.; Maliqueo, M.; Sir-Petermann, T. Enlarged adipocytes in subcutaneous adipose tissue associated to hyperandrogenism and visceral adipose tissue volume in women with polycystic ovary syndrome. Steroids 2018, 130, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Arpaci, D.; Gurkan Tocoglu, A.; Yilmaz, S.; Ergenc, H.; Tamer, A.; Keser, N.; Gunduz, H. The relationship between epicardial fat tissue thickness and visceral adipose tissue in lean patients with polycystic ovary syndrome. J. Ovarian Res. 2015, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Glueck, C.J.; Goldenberg, N. Characteristics of obesity in polycystic ovary syndrome: Etiology, treatment, and genetics. Metabolism 2019, 92, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Kałużna, M.; Czlapka-Matyasik, M.; Bykowska-Derda, A.; Moczko, J.; Ruchala, M.; Ziemnicka, K. Indirect predictors of visceral adipose tissue in women with polycystic ovary syndrome: A comparison of methods. Nutrients 2021, 13, 2494. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.W.; Hill, D.G.; Jones, S.A. Understanding immune cells in tertiary lymphoid organ development: It is all starting to come together. Front. Immunol. 2016, 7, 401. [Google Scholar] [CrossRef]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar] [CrossRef]
- Parker, J.; O’Brien, C.; Gersh, F.L. Developmental origins and transgenerational inheritance of polycystic ovary syndrome. Aust. N. Z. J. Obstet. Gynaecol. 2021, 61, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.R.; Sanders, J.G.; Song, S.J.; Amato, K.R.; Clark, A.G.; Knight, R. The human microbiome in evolution. BMC Biol. 2017, 15, 1–12. [Google Scholar] [CrossRef]
- Rizk, M.G.; Thackray, V.G. Intersection of Polycystic Ovary Syndrome and the Gut Microbiome. J. Endocr. Soc. 2021, 5, bvaa177. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, 36–44. [Google Scholar] [CrossRef]
- Martinez, J.E.; Kahana, D.D.; Ghuman, S.; Wilson, H.P.; Wilson, J.; Kim, S.C.J.; Lagishetty, V.; Jacobs, J.P.; Sinha-Hikim, A.P.; Friedman, T.C. Unhealthy Lifestyle and Gut Dysbiosis: A Better Understanding of the Effects of Poor Diet and Nicotine on the Intestinal Microbiome. Front. Endocrinol. 2021, 12, 7066. [Google Scholar] [CrossRef] [PubMed]
- Degruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current understanding of dysbiosis in disease in human and animal models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef]
- Shahi, S.K.; Ghimire, S.; Lehman, P.; Mangalam, A.K. Obesity induced gut dysbiosis contributes to disease severity in an animal model of multiple sclerosis. Front. Immunol. 2022, 13, 6417. [Google Scholar] [CrossRef] [PubMed]
- Moya, A.; Ferrer, M. Functional Redundancy-Induced Stability of Gut Microbiota Subjected to Disturbance. Trends Microbiol. 2016, 24, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Jiang, Y.; Xi, H.; Chen, L.; Feng, X. Exploration of the Relationship between Gut Microbiota and Polycystic Ovary Syndrome (PCOS): A Review. Geburtshilfe Frauenheilkd. 2020, 80, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.; Terekeci, H.; Sandal, S.; Kelestimur, F. Endocrine disrupting chemicals: Exposure, effects on human health, mechanism of action, models for testing and strategies for prevention. Rev. Endocr. Metab. Disord. 2020, 21, 127–147. [Google Scholar] [CrossRef]
- Fabozzi, G.; Rebuzzini, P.; Cimadomo, D.; Allori, M.; Franzago, M.; Stuppia, L.; Garagna, S.; Ubaldi, F.M.; Zuccotti, M.; Rienzi, L. Endocrine-Disrupting Chemicals, Gut Microbiota, and Human (In) Fertility—It Is Time to Consider the Triad. Cells 2022, 11, 3335. [Google Scholar] [CrossRef]
- Bellingham, M.; Sharpe, R. Chemical Exposures During Pregnancy: Dealing with Potential, but Unproven, Risks to Child Health. R. Coll. Obstet. Gynaecol. 2013, 1–7. [Google Scholar]
- Di Renzo, G.C.; Conry, J.A.; Blake, J.; Defrancesco, M.S.; Denicola, N.; Martin, J.N.; McCue, K.A.; Richmond, D.; Shah, A.; Sutton, P.; et al. International Federation of Gynecology and Obstetrics opinion on reproductive health impacts of exposure to toxic environmental chemicals. Int. J. Gynecol. Obstet. 2015, 131, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lu, Y.; Zhong, K.; Wang, C.; Xu, X. The associations between endocrine disrupting chemicals and markers of inflammation and immune responses: A systematic review and meta-analysis. Ecotoxicol. Environ. Saf. 2022, 234, 113382. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Magdalena, P.; Morimoto, S.; Ripoll, C.; Fuentes, E.; Nadal, A. The estrogenic effect of bisphenol a disrupts pancreatic β-cell function in vivo and induces insulin resistance. Environ. Health Perspect. 2006, 114, 106–112. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, Q.; Dang, X.; He, Y.; Li, X.; Sun, Y. Local effect of bisphenol A on the estradiol synthesis of ovarian granulosa cells from PCOS. Gynecol. Endocrinol. 2017, 33, 21–25. [Google Scholar] [CrossRef]
- Sun, Y.; Gao, S.; Ye, C.; Zhao, W. Gut microbiota dysbiosis in polycystic ovary syndrome: Mechanisms of progression and clinical applications. Front. Cell. Infect. Microbiol. 2023, 13, 1142041. [Google Scholar] [CrossRef]
- Ananthasubramanian, P.; Ananth, S.; Kumaraguru, S.; Barathi, S.; Santosh, W.; Vasantharekha, R. Associated Effects of Endocrine Disrupting Chemicals (EDCs) on Neuroendocrine Axes and Neurotransmitter Profile in Polycystic Ovarian Syndrome Condition. Proc. Zool. Soc. 2021, 74, 378–386. [Google Scholar] [CrossRef]
- Gupta, R.; Kumar, P.; Fahmi, N.; Garg, B.; Dutta, S.; Sachar, S.; Matharu, A.S.; Vimaleswaran, K.S. Endocrine disruption and obesity: A current review on environmental obesogens. Curr. Res. Green Sustain. Chem. 2020, 3, 100009. [Google Scholar] [CrossRef]
- Aydemir, D.; Ulusu, N.N. The possible role of the endocrine disrupting chemicals on the premature and early menopause associated with the altered oxidative stress metabolism. Front. Endocrinol. 2023, 14, 1081704. [Google Scholar] [CrossRef]
- Ravichandran, G.; Lakshmanan, D.K.; Raju, K.; Elangovan, A.; Nambirajan, G.; Devanesan, A.A.; Thilagar, S. Food advanced glycation end products as potential endocrine disruptors: An emerging threat to contemporary and future generation. Environ. Int. 2019, 123, 486–500. [Google Scholar] [CrossRef]
- Bansal, A.; Henao-Mejia, J.; Simmons, R.A. Immune system: An emerging player in mediating effects of endocrine disruptors on metabolic health. Endocrinology 2018, 159, 32–45. [Google Scholar] [CrossRef]
- Tudurí, E.; Marroqui, L.; Dos Santos, R.S.; Quesada, I.; Fuentes, E.; Alonso-Magdalena, P. Timing of exposure and Bisphenol-A: Implications for diabetes development. Front. Endocrinol. 2018, 9, 648. [Google Scholar] [CrossRef] [PubMed]
- Resnik, D.B. The precautionary principle and medical decision making. J. Med. Philos. 2004, 29, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Johnson, A.F.; Birnbaum, L.S.; Colborn, T.; Guillette, L.J.; Crews, D.P.; Collins, T.; Soto, A.M.; Vom Saal, F.S.; McLachlan, J.A.; et al. Minireview: Endocrine disruptors: Past lessons and future directions. Mol. Endocrinol. 2016, 30, 833–847. [Google Scholar] [CrossRef] [PubMed]
- More, S.; Bampidis, V.; Benford, D.; Bragard, C.; Halldorsson, T.; Hernández-Jerez, A.; Hougaard Bennekou, S.; Koutsoumanis, K.; Lambré, C.; Machera, K.; et al. Guidance on risk assessment of nanomaterials to be applied in the food and feed chain: Human and animal health. EFSA J. 2021, 19, e06768. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska, A.Z.; Diamanti-Kandarakis, E. Polycystic ovary syndrome and environmental toxins. Fertil. Steril. 2016, 106, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Jala, A.; Varghese, B.; Kaur, G.; Rajendiran, K.; Dutta, R.; Adela, R.; Borkar, R.M. Implications of endocrine-disrupting chemicals on polycystic ovarian syndrome: A comprehensive review. Environ. Sci. Pollut. Res. 2022, 29, 58484–58513. [Google Scholar] [CrossRef]
- Jozkowiak, M.; Piotrowska-Kempisty, H.; Kobylarek, D.; Gorska, N.; Mozdziak, P.; Kempisty, B.; Rachon, D.; Spaczynski, R.Z. Endocrine Disrupting Chemicals in Polycystic Ovary Syndrome: The Relevant Role of the Theca and Granulosa Cells in the Pathogenesis of the Ovarian Dysfunction. Cells 2023, 12, 174. [Google Scholar] [CrossRef]
- Parker, J. A new hypothesis for the mechanism of glyphosate induced intestinal permeability in the pathogenesis of polycystic ovary syndrome. J. Australas. Coll. Nutr. Environ. Med. 2015, 34, 3. [Google Scholar]
- Kumar, M.; Sarma, D.K.; Shubham, S.; Kumawat, M.; Verma, V.; Prakash, A.; Tiwari, R. Environmental Endocrine-Disrupting Chemical Exposure: Role in Non-Communicable Diseases. Front. Public Health 2020, 8, 3850. [Google Scholar] [CrossRef]
- Mitro, S.D.; Johnson, T.; Zota, A.R. Cumulative Chemical Exposures During Pregnancy and Early Development. Curr. Environ. Health Rep. 2015, 2, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Starling, A.P.; Adgate, J.L.; Hamman, R.F.; Kechris, K.; Calafat, A.M.; Ye, X.; Dabelea, D. Perfluoroalkyl substances during pregnancy and offspring weight and adiposity at birth: Examining mediation by maternal fasting glucose in the healthy start study. Environ. Health Perspect. 2017, 125, 067016. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, C.S. Transcriptomics and Other Omics Approaches to Investigate Effects of Xenobiotics on the Placenta. Front. Cell Dev. Biol. 2021, 9, 3656. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, M.; Chow, E.; Aschengrau, A.; Mahalingaiah, S. Prenatal Exposure to Endocrine Disruptors: A Developmental Etiology for Polycystic Ovary Syndrome. Reprod. Sci. 2017, 24, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Corbett, G.A.; Lee, S.; Woodruff, T.J.; Hanson, M.; Hod, M.; Charlesworth, A.M.; Giudice, L.; Conry, J.; McAuliffe, F.M. Nutritional interventions to ameliorate the effect of endocrine disruptors on human reproductive health: A semi-structured review from FIGO. Int. J. Gynecol. Obstet. 2022, 157, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Piazza, M.J.; Urbanetz, A.A. Environmental toxins and the impact of other endocrine disrupting chemicals in women’s reproductive health. J. Bras. Reprod. Assist. 2019, 23, 154–164. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194. [Google Scholar] [CrossRef]



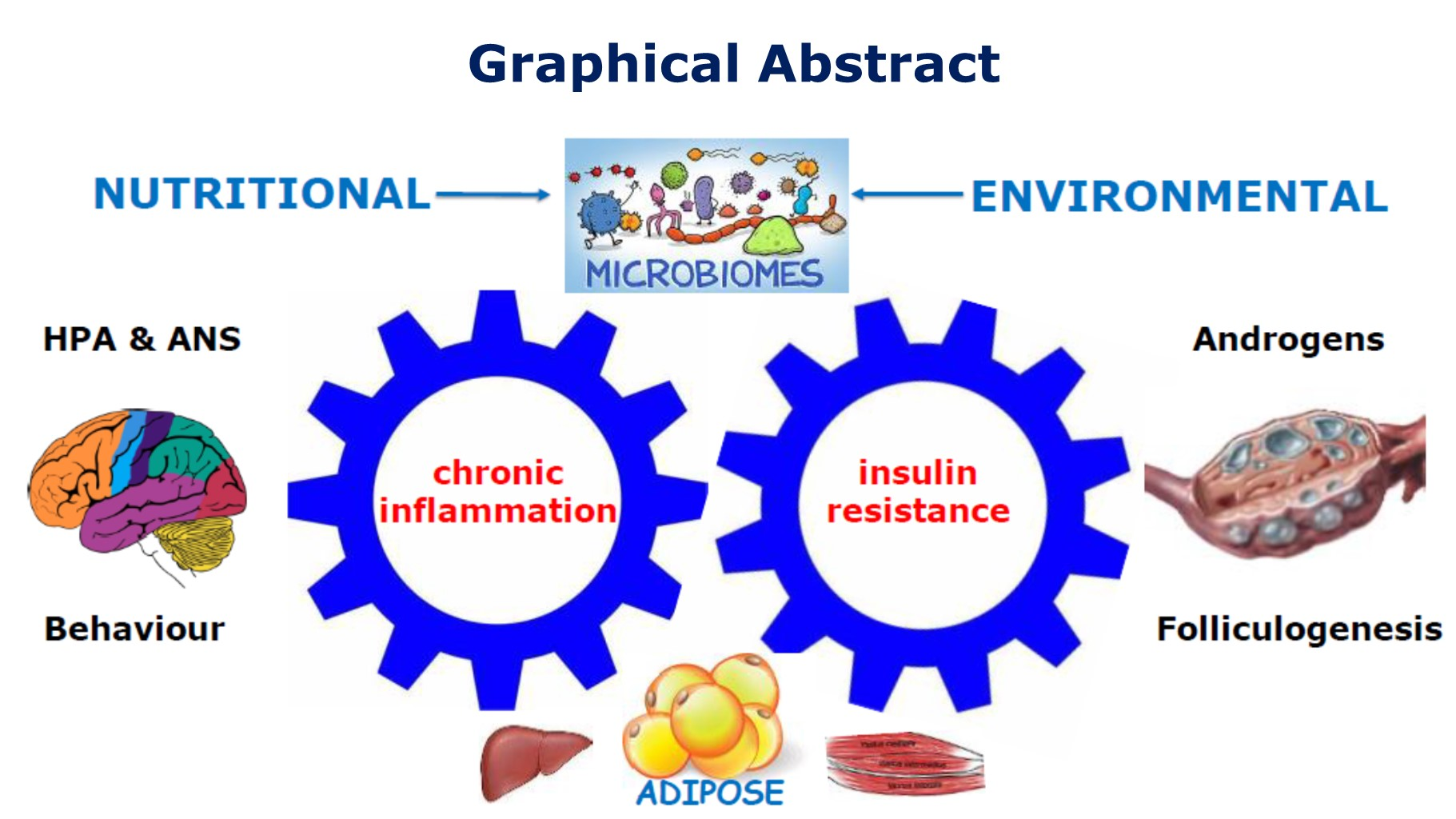{kind=link}
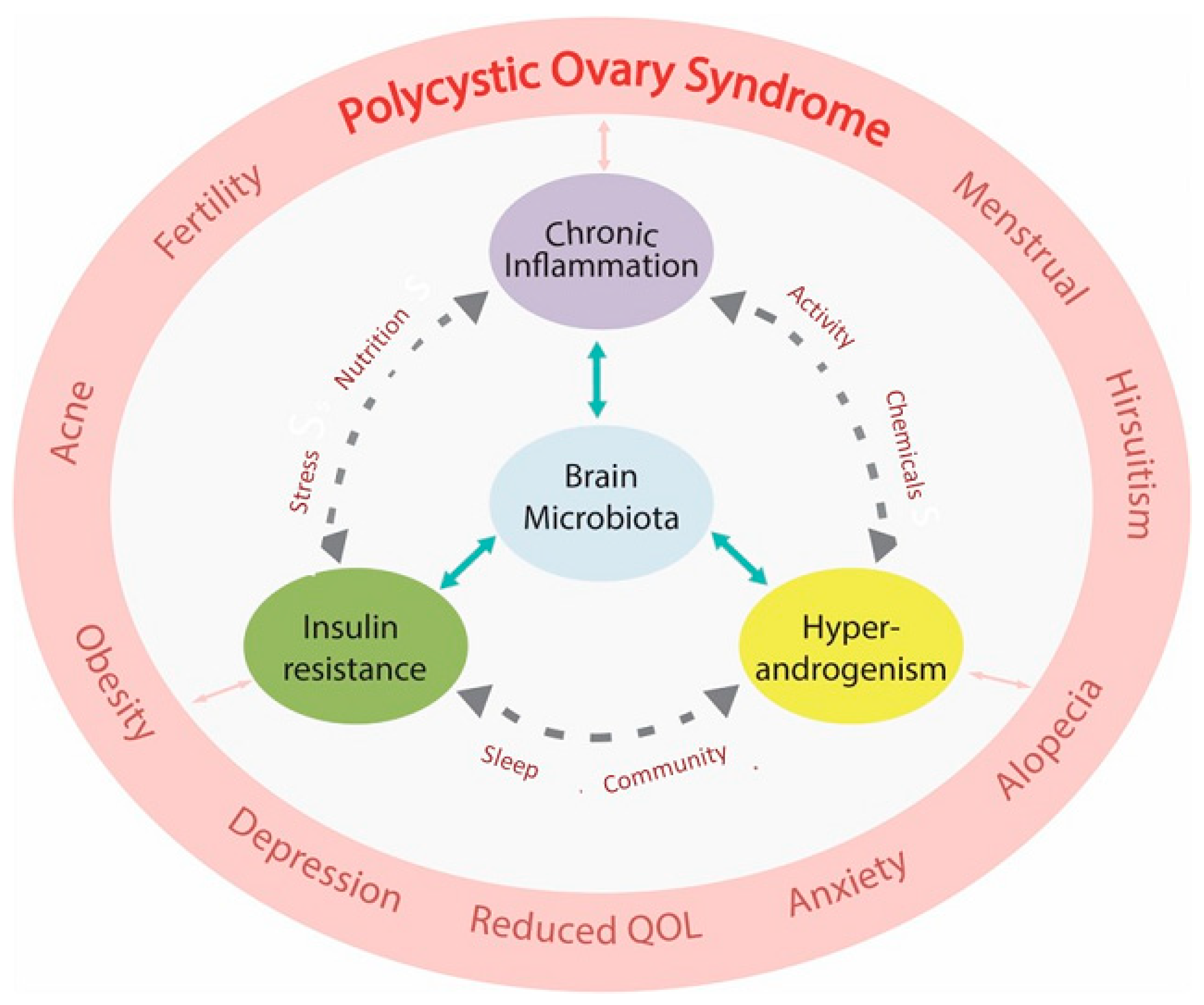{kind=link}
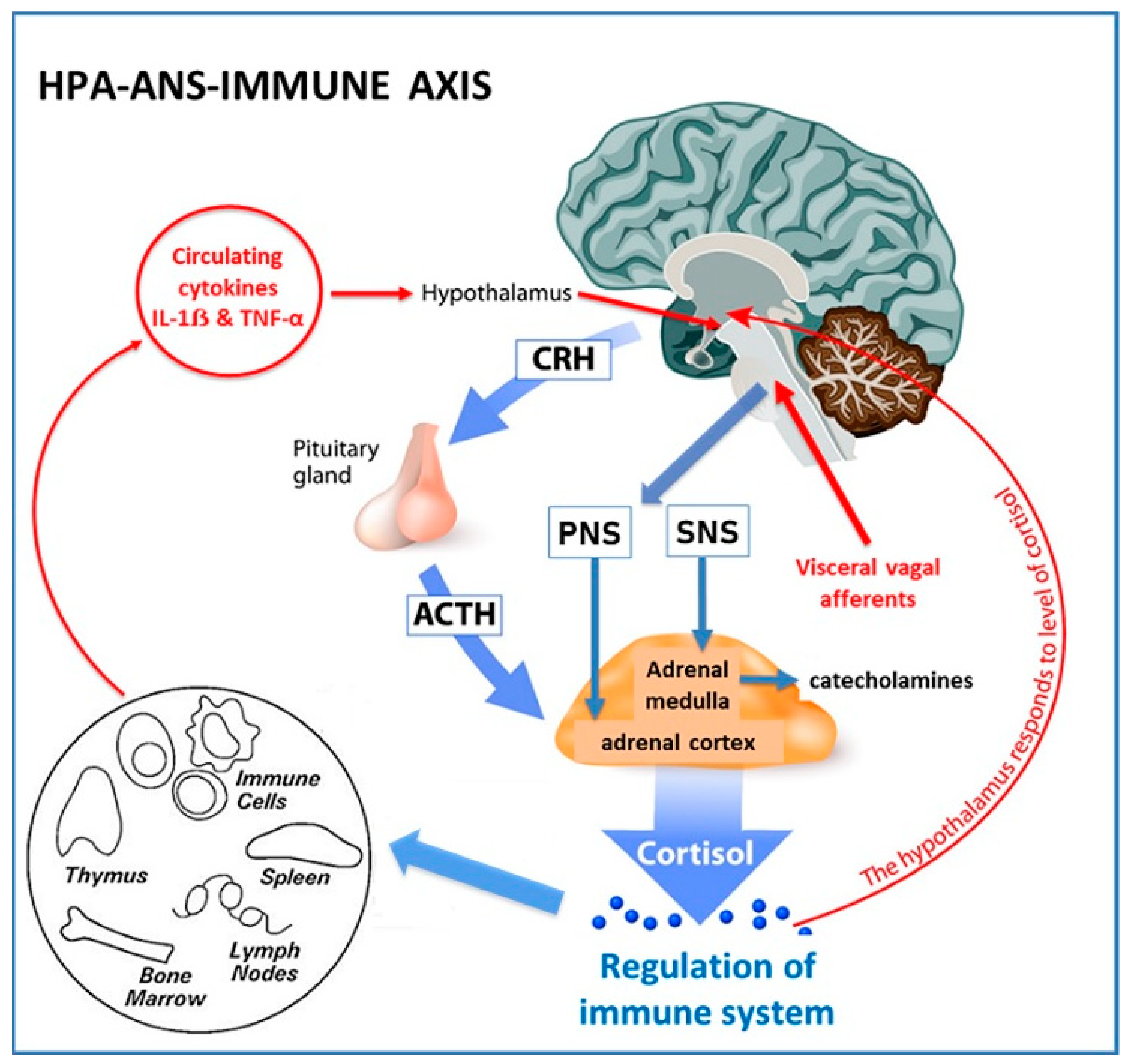{kind=link}
{kind=link}
| Hallmark | Polycystic Ovary Syndrome |
|---|---|
| Barrier integrity | Gastrointestinal permeability Respiratory mucosa |
| Containment of local perturbations | Mucosal and adaptive immunity ROS, AGE, DAMPS, PAMPS |
| Recycling and turnover | Autophagy, apoptosis, pyroptosis GI stem cell turnover |
| Integration of circuitries | Metabolic, reproductive neurological, circadian |
| Rhythmic oscillations | Menstrual cycle, circadian GI peristalsis |
| Homeostatic resilience | Endocrine, ANS metabolic, immune, microbiome |
| Hormetic regulation | oxidative stress, inactivity xenohormesis # |
| Repair and regeneration | Endoplasmic reticular stress mitochondrial stress |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parker, J. Pathophysiological Effects of Contemporary Lifestyle on Evolutionary-Conserved Survival Mechanisms in Polycystic Ovary Syndrome. Life 2023, 13, 1056. https://doi.org/10.3390/life13041056
Parker J. Pathophysiological Effects of Contemporary Lifestyle on Evolutionary-Conserved Survival Mechanisms in Polycystic Ovary Syndrome. Life. 2023; 13(4):1056. https://doi.org/10.3390/life13041056
Chicago/Turabian StyleParker, Jim. 2023. "Pathophysiological Effects of Contemporary Lifestyle on Evolutionary-Conserved Survival Mechanisms in Polycystic Ovary Syndrome" Life 13, no. 4: 1056. https://doi.org/10.3390/life13041056