
Docking and Molecular Dynamics Study to Identify Novel Phytobiologics from Dracaena trifasciata against Metabolic Reprogramming in Rheumatoid Arthritis



Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection and Identification
2.2. Extract Preparation
2.3. Biochemical Testing of Extracts
2.4. Gas Chromatography–Mass Spectrometry Analysis
2.5. Drug like Properties
2.6. Structure Retrieval
2.7. Preparation of Targets
2.8. Active Site Prediction
2.9. Docking and Protein Ligand Interaction Profiling
2.10. Pose Analysis
2.11. MD Simulations
3. Results
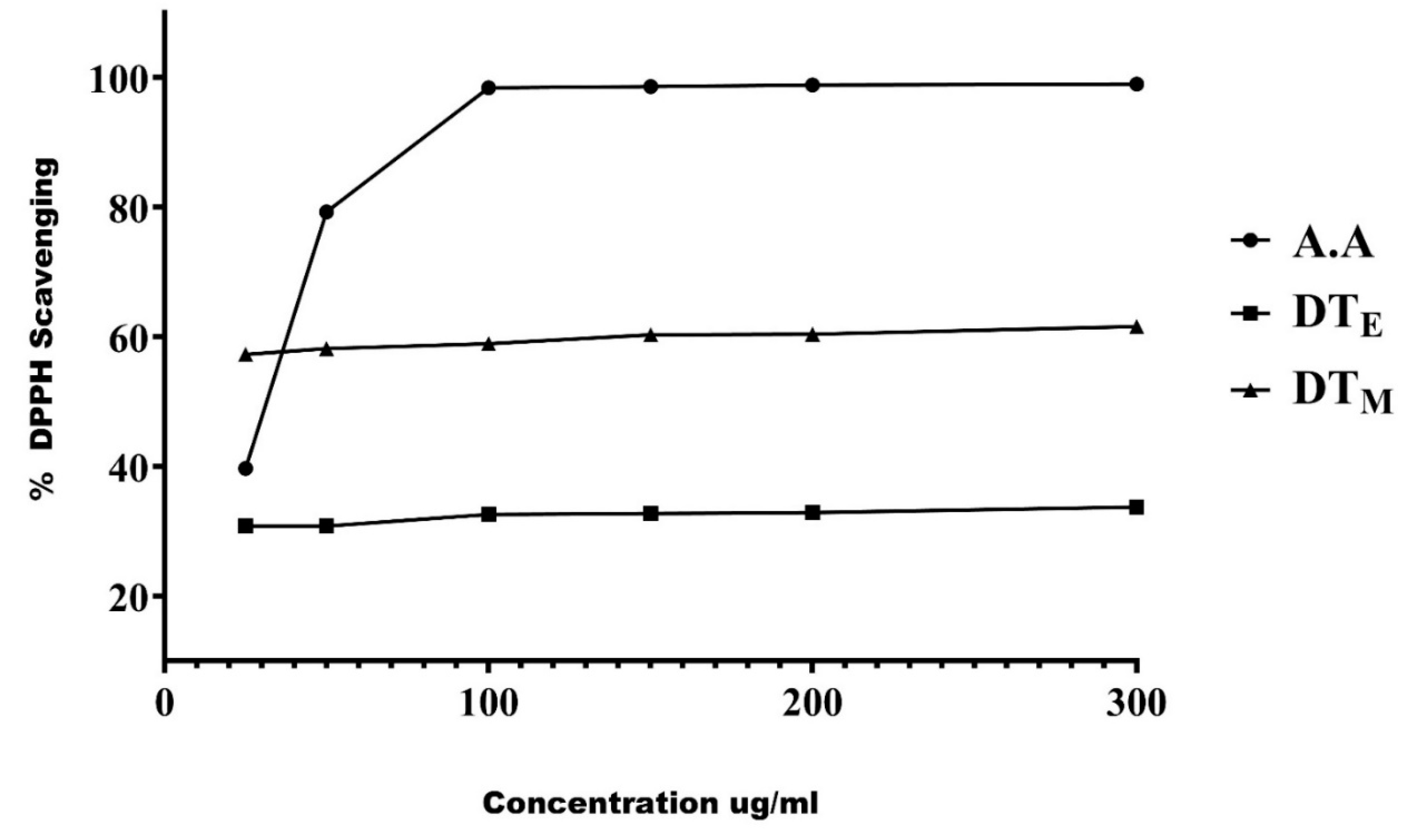
3.1. Phytobiological Screening and Radical Scavenging Activity of the Extracts
3.2. Library Generation and Drug Likeness Analysis
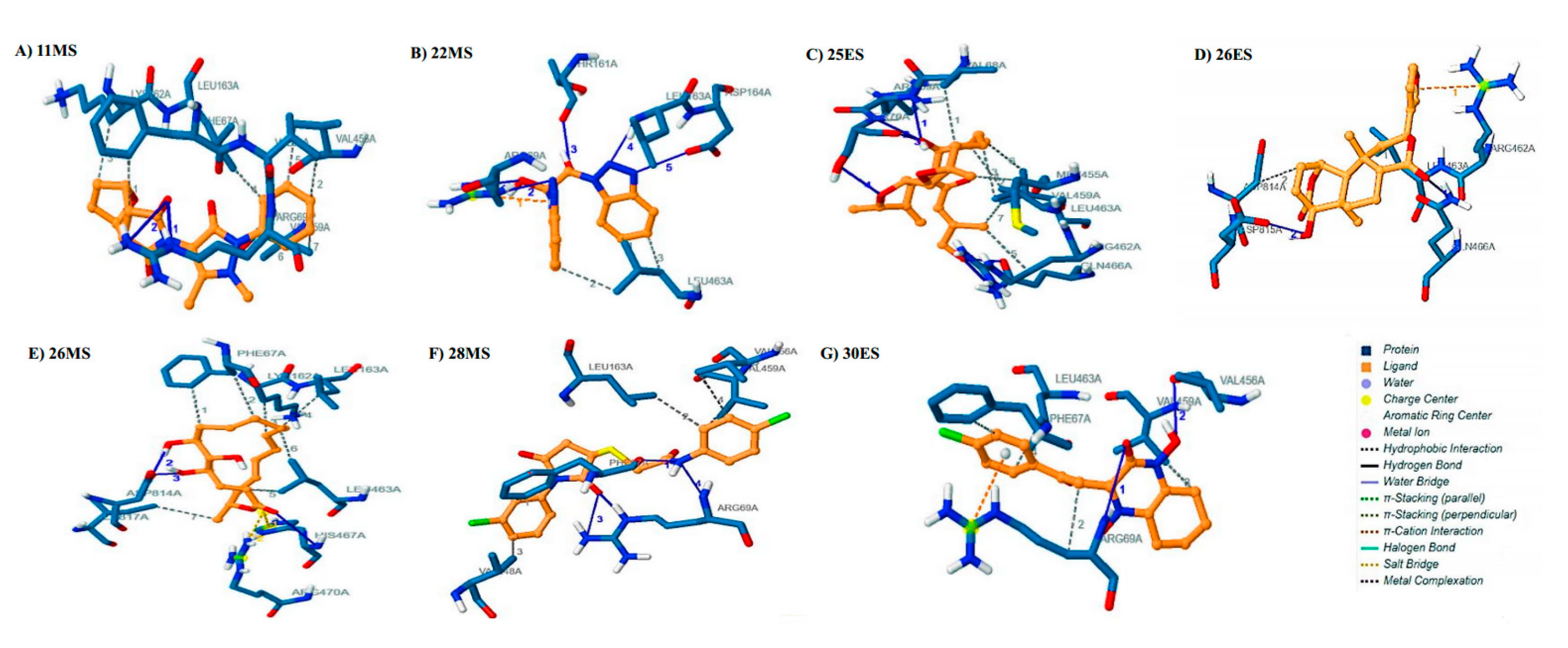
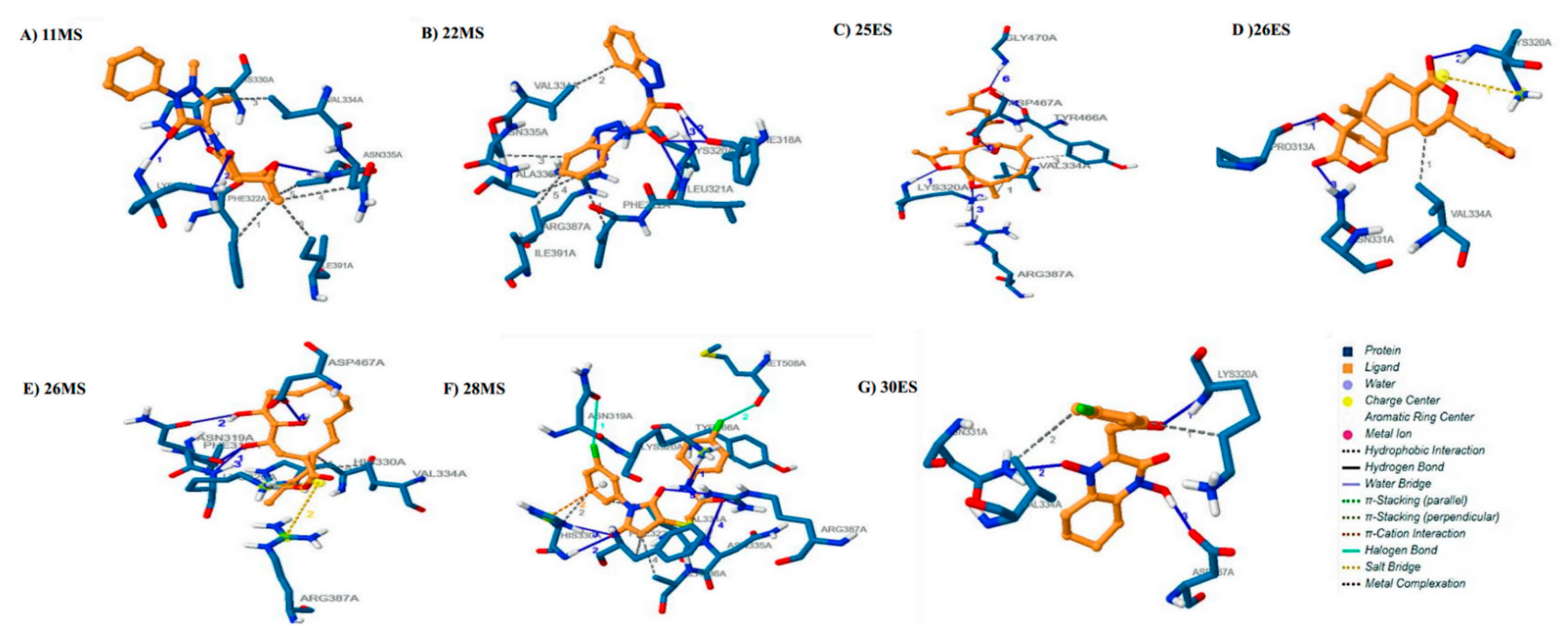
3.3. Molecular Docking
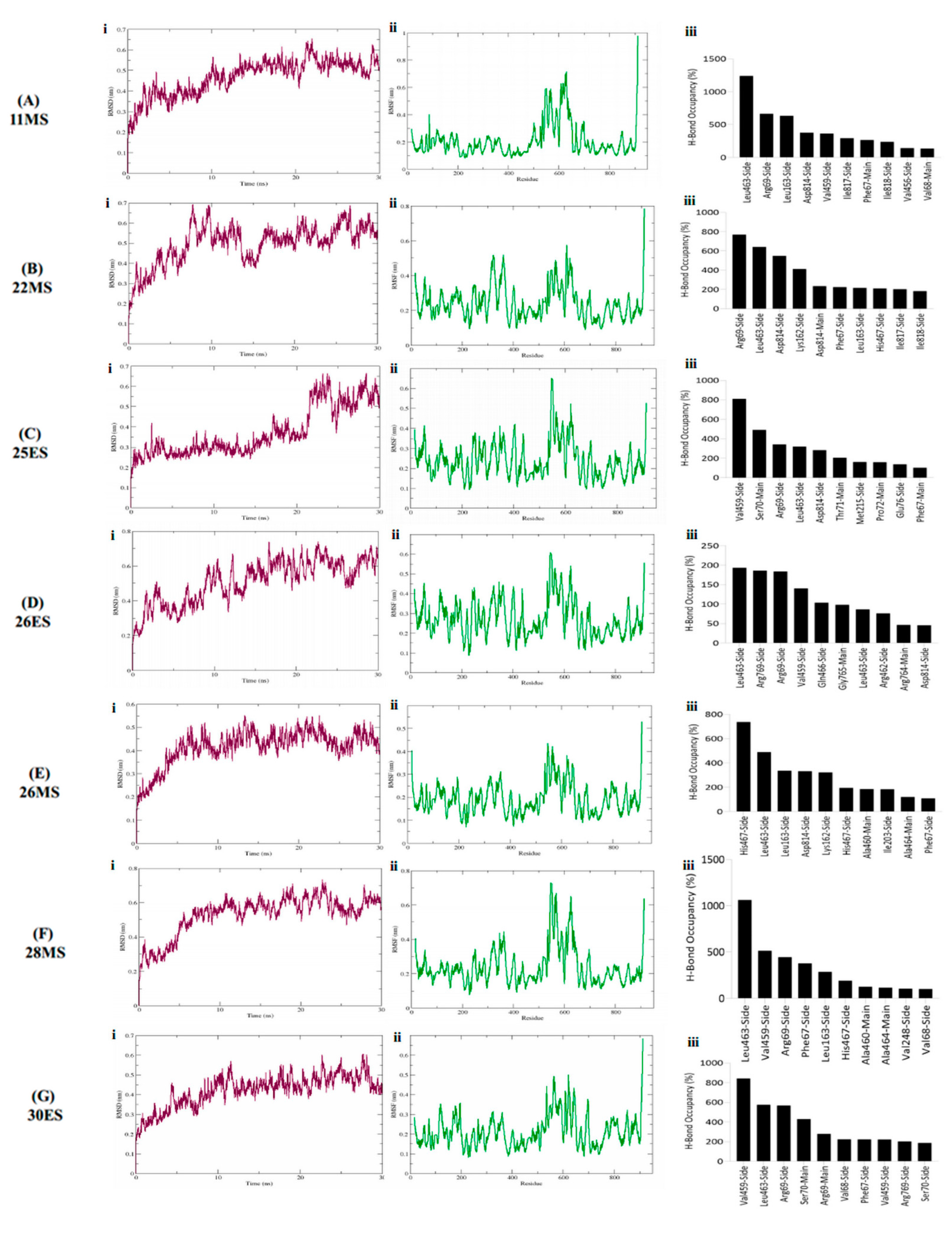
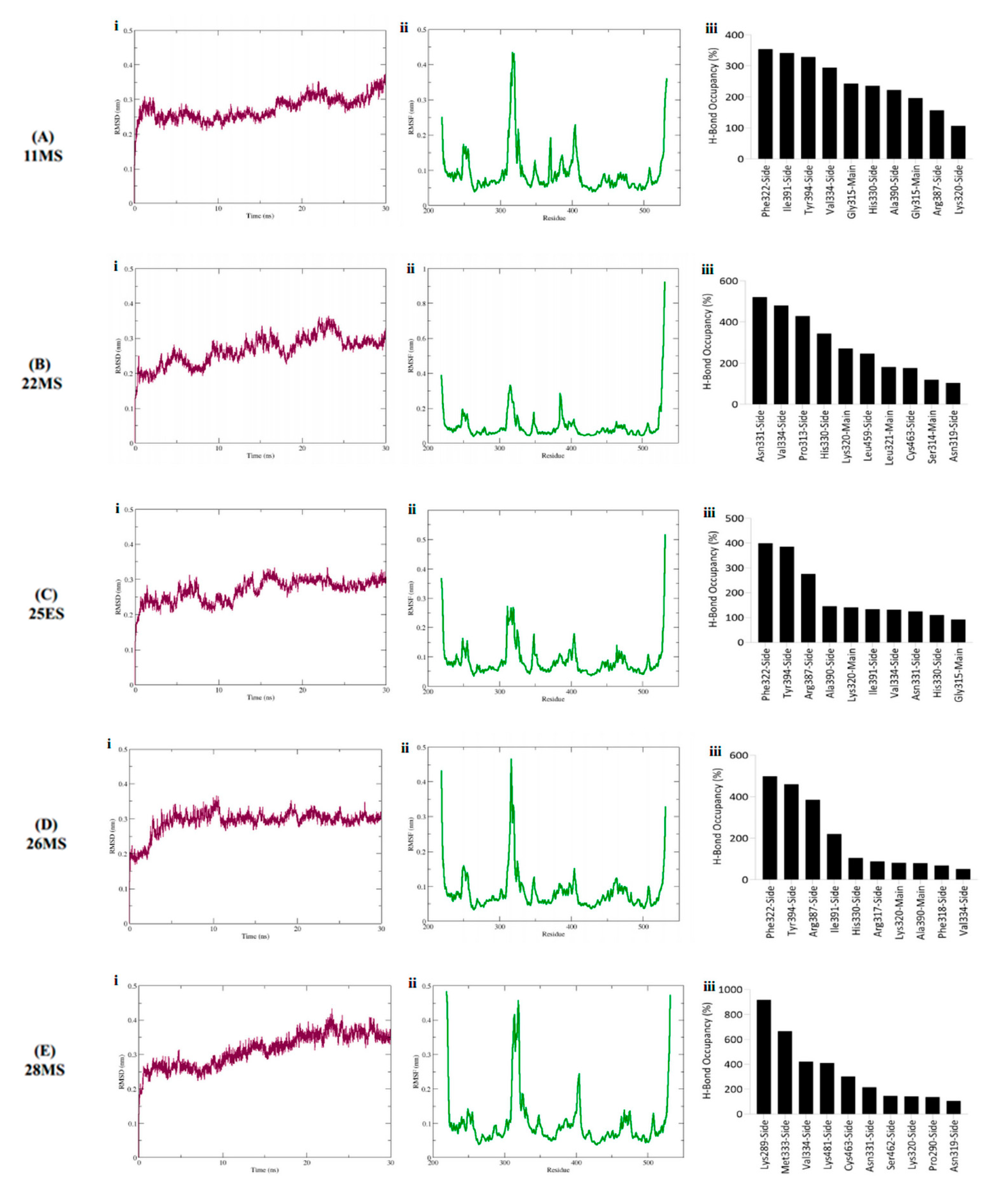
3.4. Molecular Dynamics Simulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| RA | Rheumatoid Arthritis |
| RASF | Rheumatoid Arthritis Synovial FIbroblasts |
| HK2 | Hexokinase 2 |
| GLS1 | Glutaminase 1 |
| 2DG | 2 Deoxyglucose |
| 3-BrPA | 3 Bromopyruvate |
| IL-6 | Interleukin-6 |
| CADD | Computer Aided Drug Designing |
| DTM | Dried Methanolic Extracts |
| DTE | Dried Ethanolic Extracts |
| DPPH | Diphenyl-1-picrylhydrazyl |
| PDB | Protein Data Bank |
| PLIP | Protein Ligand Interaction Profiling |
| MD Simulations | Molecular Dynamics Simulations |
| GROMACS | Groningen Machine for Chemical Simulations |
| CHARMM | Chemistry at Harvard Macromolecular Mechanics |
References
- Hansildaar, R.; Vedder, D.; Baniaamam, M.; Tausche, A.-K.; Gerritsen, M.; Nurmohamed, M.T. Cardiovascular risk in inflammatory arthritis: Rheumatoid arthritis and gout. Lancet Rheumatol. 2021, 3, e58–e70. [Google Scholar]
- Leclair, N.; Knopf, J.; Baldwin, M.; Forouhar, F.; Onyiuke, H. Rheumatoid pannus presenting as a large epidural mass in the subaxial cervical spine: A case report. Neurochirurgie 2022, 68, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Aghakhani, S.; Zerrouk, N.; Niarakis, A. Metabolic reprogramming of fibroblasts as therapeutic target in rheumatoid arthritis and cancer: Deciphering key mechanisms using computational systems biology approaches. Cancers 2021, 13, 35. [Google Scholar] [CrossRef]
- Falconer, J.; Murphy, A.N.; Young, S.P.; Clark, A.R.; Tiziani, S.; Guma, M.; Buckley, C.D. Synovial cell metabolism and chronic inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2018, 70, 984–999. [Google Scholar] [PubMed]
- Bustamante, M.F.; Garcia-Carbonell, R.; Whisenant, K.D.; Guma, M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 110. [Google Scholar] [PubMed]
- Sanchez-Lopez, E.; Cheng, A.; Guma, M. Can metabolic pathways be therapeutic targets in rheumatoid arthritis? J. Clin. Med. 2019, 8, 753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biniecka, M.; Canavan, M.; McGarry, T.; Gao, W.; McCormick, J.; Cregan, S.; Gallagher, L.; Smith, T.; Phelan, J.; Ryan, J. Dysregulated bioenergetics: A key regulator of joint inflammation. Ann. Rheum. Dis. 2016, 75, 2192–2200. [Google Scholar] [CrossRef]
- Araujo, L.; Khim, P.; Mkhikian, H.; Mortales, C.-L.; Demetriou, M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. Elife 2017, 6, e21330. [Google Scholar]
- Garcia-Carbonell, R.; Divakaruni, A.S.; Lodi, A.; Vicente-Suarez, I.; Saha, A.; Cheroutre, H.; Boss, G.R.; Tiziani, S.; Murphy, A.N.; Guma, M. Critical role of glucose metabolism in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol. 2016, 68, 1614–1626. [Google Scholar]
- Bustamante, M.F.; Oliveira, P.G.; Garcia-Carbonell, R.; Croft, A.P.; Smith, J.M.; Serrano, R.L.; Sanchez-Lopez, E.; Liu, X.; Kisseleva, T.; Hay, N. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann. Rheum. Dis. 2018, 77, 1636–1643. [Google Scholar]
- Oliveira, P.G.; Farinon, M.; Sanchez-Lopez, E.; Miyamoto, S.; Guma, M. Fibroblast-like synoviocytes glucose metabolism as a therapeutic target in rheumatoid arthritis. Front. Immunol. 2019, 10, 1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, A.; Pedersen, B.; Cobo, I.; Ai, R.; Coras, R.; Murillo-Saich, J.; Nygaard, G.; Sanchez-Lopez, E.; Murphy, A.; Wang, W. Epigenetic Regulation of Nutrient Transporters in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. Arthritis Rheumatol. 2022, 74, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Saegusa, J.; Sendo, S.; Okano, T.; Akashi, K.; Irino, Y.; Morinobu, A. Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flood, S.; Duance, V.; Mason, D. The role of glutamate signalling in rheumatoid arthritis. Int. J. Exp. Pathol. 2004, 85, A19–A20. [Google Scholar] [CrossRef]
- Anada, M.; Tohyama, M.; Oda, Y.; Kamoshima, Y.; Amino, I.; Nakano, F.; Miyazaki, Y.; Akimoto, S.; Minami, N.; Kikuchi, S. Progressive multifocal leukoencephalopathy during tocilizumab treatment for rheumatoid arthritis. Intern. Med. 2020, 59, 2053–2059. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Anzaghe, M.; Schülke, S. Update on the pathomechanism, diagnosis, and treatment options for rheumatoid arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef] [Green Version]
- Mewar, D.; Wilson, A.G. Treatment of rheumatoid arthritis with tumour necrosis factor inhibitors. Br. J. Pharmacol. 2011, 162, 785–791. [Google Scholar]
- Shareef, H.; Naeem, S.; Zaheer, E. Comparative Analgesic Activity of Selected Medicinal Plants from Pakistan: Analgesics from plants. Proc. Pak. Acad. Sci. B Life Environ. Sci. 2019, 56, 57–67. [Google Scholar]
- Malik, K.; Ahmad, M.; Zhang, G.; Rashid, N.; Zafar, M.; Sultana, S.; Shah, S.N. Traditional plant based medicines used to treat musculoskeletal disorders in Northern Pakistan. Eur. J. Integr. Med. 2018, 19, 17–64. [Google Scholar] [CrossRef]
- Abdul-Hafeez, E.Y.; Orabi, M.A.; Ibrahim, O.H.; Ilinskaya, O.; Karamova, N.S. In vitro cytotoxic activity of certain succulent plants against human colon, breast and liver cancer cell lines. S. Afr. J. Bot. 2020, 131, 295–301. [Google Scholar] [CrossRef]
- Andhare, R.N.; Raut, M.K.; Naik, S.R. Evaluation of antiallergic and anti-anaphylactic activity of ethanolic extract of Sanseveiria trifasciata leaves (EEST) in rodents. J. Ethnopharmacol. 2012, 142, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Pinky, S.S.; Monira, S.; Hossain, M.A.; Hossain, A. Antioxidant, Anti-inflammatory, Cytotoxic and Analgesic Activities of Sensevieria trifasciata. Bangladesh Pharm. J. 2020, 23, 195–200. [Google Scholar] [CrossRef]
- Dewatisari, W.; Nugroho, L.H.; Retnaningrum, E.; Purwestri, Y.A. The potency of Sansevieria trifasciata and S. cylindrica leaves extracts as an antibacterial against Pseudomonas aeruginosa. Biodivers. J. Biol. Divers. 2021, 22. [Google Scholar] [CrossRef]
- Harborne, J.; Greenham, J.; Williams, C. Phytochemical Analysis; Chapman and Hall Company Ltd.: London, UK, 1973; Volume 1, pp. 5–6. [Google Scholar]
- Kodangala, C.; Saha, S.; Kodangala, P. Phytochemical studies of aerial parts of the plant Leucas lavandulaefolia. Pharma Chem. 2010, 2, 434–437. [Google Scholar]
- Njoku, V.O.; Obi, C. Phytochemical constituents of some selected medicinal plants. Afr. J. Pure Appl. Chem. 2009, 3, 228–233. [Google Scholar] [CrossRef]
- Trease, G.; Gajendiran, K.; Evans, W. Phytochemical screaning of Citrus. In Pharmacognosy, 11th ed.; Bailliere Tindall: London, UK, 1989; Volume 25, pp. 45–50. [Google Scholar]
- Hassan, M.; Baig, A.A.; Attique, S.A.; Abbas, S.; Khan, F.; Zahid, S.; Ain, Q.U.; Usman, M.; Simbak, N.B.; Kamal, M.A. Molecular docking of alpha-enolase to elucidate the promising candidates against Streptococcus pneumoniae infection. DARU J. Pharm. Sci. 2021, 29, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 11. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cousins, K.R. Computer review of ChemDraw ultra 12.0. J. Am. Chem. Soc. 2011, 133, 8388. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Chem. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, F.; Yang, K.; Wu, C.; Gao, S.; Wang, P.; Chen, L.; Li, H. New natural inhibitors of hexokinase 2 (HK2): Steroids from Ganoderma sinense. Fitoterapia 2018, 125, 123–129. [Google Scholar] [CrossRef]
- Cheng, L.; Wu, C.-R.; Zhu, L.-H.; Li, H.; Chen, L.-X. Physapubescin, a natural withanolide as a kidney-type glutaminase (KGA) inhibitor. Bioorg. Med. Chem. Lett. 2017, 27, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [PubMed]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Lemkul, J.A. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package, v1.0. Living J. Comp. Mol. Sci. 2018, 1, 5068. [Google Scholar]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM force field in GROMACS: Analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Yu, M. Computational Modeling of Protein Dynamics with GROMACS and Java. Master’s Thesis, San Jose State University, San Jose, CA, USA, 2012. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dudics, S.; Langan, D.; Meka, R.R.; Venkatesha, S.H.; Berman, B.M.; Che, C.-T.; Moudgil, K.D. Natural products for the treatment of autoimmune arthritis: Their mechanisms of action, targeted delivery, and interplay with the host microbiome. Int. J. Mol. Sci. 2018, 19, 2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Williams, O.; Inderjeeth, C.A.; Almutairi, K.B.; Keen, H.; Preen, D.B.; Nossent, J.C. Total Hip Replacement in Patients with Rheumatoid Arthritis: Trends in Incidence and Complication Rates over 35 Years. Rheumatol. Ther. 2022, 9, 565–580. [Google Scholar] [CrossRef]
- Jamari, J.; Ammarullah, M.I.; Santoso, G.; Sugiharto, S.; Supriyono, T.; Prakoso, A.T.; Basri, H.; van der Heide, E. Computational contact pressure prediction of CoCrMo, SS 316L and Ti6Al4V femoral head against UHMWPE acetabular cup under gait cycle. J. Funct. Biomater. 2022, 13, 64. [Google Scholar] [CrossRef]
- Cai, W.-W.; Yu, Y.; Zong, S.-Y.; Wei, F. Metabolic reprogramming as a key regulator in the pathogenesis of rheumatoid arthritis. Inflamm. Res. 2020, 69, 1087–1101. [Google Scholar] [CrossRef]
- Augustin, R.C.; Delgoffe, G.M.; Najjar, Y.G. Characteristics of the Tumor Microenvironment That Influence Immune Cell Functions: Hypoxia, Oxidative Stress, Metabolic Alterations. Cancers 2020, 12, 3802. [Google Scholar] [CrossRef]
- Harnanik, T.; Soeroso, J.; Suryokusumo, M.G.; Juliandhy, T. Effects of Hyperbaric Oxygen on T helper 17/regulatory T Polarization in Antigen and Collagen-induced Arthritis: Hypoxia-inducible Factor-1α as a Target. Oman Med. J. 2020, 35, e90. [Google Scholar] [CrossRef] [PubMed]
- Aravilli, R.K.; Vikram, S.L.; Kohila, V. Phytochemicals as potential antidotes for targeting NF-κB in rheumatoid arthritis. 3 Biotech 2017, 7, 253. [Google Scholar] [CrossRef] [PubMed]
- Chelliah, V.; Blundell, T.L.; Fernández-Recio, J. Efficient restraints for protein–protein docking by comparison of observed amino acid substitution patterns with those predicted from local environment. J. Mol. Biol. 2006, 357, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Pandey, S.K.; Singh, V.K.; Goel, Y.; Kumar, A.; Singh, S.M. Molecular docking studies of 3-bromopyruvate and its derivatives to metabolic regulatory enzymes: Implication in designing of novel anticancer therapeutic strategies. PLoS ONE 2017, 12, e0176403. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Meng, Y.; Li, L.; Xu, P.; Wang, J.; Li, Z.; Bian, J. Overview of the development of glutaminase inhibitors: Achievements and future directions. J. Med. 2018, 62, 1096–1115. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (DTM) | (DTE) | ||
|---|---|---|---|
| 1 | Alkaloids | + | + |
| 2 | Phenols | − | + |
| 3 | Anthraquinones | − | − |
| 4 | Flavonoids | + | + |
| 5 | Anthocyanins | − | − |
| 6 | Leucoanthocyanins | − | + |
| 7 | Tannins | + | + |
| 8 | Phlobatannins | − | − |
| 9 | Coumarins | + | − |
| 10 | Terpenoids | − | − |
| 11 | Diterpenes | − | + |
| 12 | Triterpenes | + | + |
| 13 | Steroids | + | + |
| 14 | Sterols | + | + |
| 15 | Saponins | − | − |
| 16 | Resins | − | − |
| 17 | Emodins | − | − |
| 18 | Glycosides | + | + |
| 19 | Cardiac glycosides | + | − |
| Compound Name | Target 1 (HK2) | Target 2 (GLS1) | ||
|---|---|---|---|---|
| Binding Energy (kcal/mol) | Inhibition Constant (Ki) | Binding Energy (kcal/mol) | Inhibition Constant (Ki) | |
| 11MS | −8.34 | 776.71 nM | −7.81 | 1.90 μM |
| 22MS | −7.06 | 6.64 μM | −7.79 | 1.94 μM |
| 25ES | −7.85 | 1.77 μM | −9.04 | 237.37 nM |
| 26ES | −7.47 | 3.33 μM | −8.75 | 383.40 nM |
| 26MS | −7.05 | 6.84 μM | −8.01 | 1.35 μM |
| 28MS | −8.19 | 998.63 nM | −8.99 | 257.91 nM |
| 30ES | −7.86 | 1.74 μM | −9.47 | 113.85 nM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, S.; John, P.; Paracha, R.Z.; Bhatti, A.; Guma, M. Docking and Molecular Dynamics Study to Identify Novel Phytobiologics from Dracaena trifasciata against Metabolic Reprogramming in Rheumatoid Arthritis. Life 2022, 12, 1148. https://doi.org/10.3390/life12081148
Ahmed S, John P, Paracha RZ, Bhatti A, Guma M. Docking and Molecular Dynamics Study to Identify Novel Phytobiologics from Dracaena trifasciata against Metabolic Reprogramming in Rheumatoid Arthritis. Life. 2022; 12(8):1148. https://doi.org/10.3390/life12081148
Chicago/Turabian StyleAhmed, Shanzay, Peter John, Rehan Zafar Paracha, Attya Bhatti, and Monica Guma. 2022. "Docking and Molecular Dynamics Study to Identify Novel Phytobiologics from Dracaena trifasciata against Metabolic Reprogramming in Rheumatoid Arthritis" Life 12, no. 8: 1148. https://doi.org/10.3390/life12081148