
Pathogenesis of Distal Symmetrical Polyneuropathy in Diabetes

 , , and
, , and 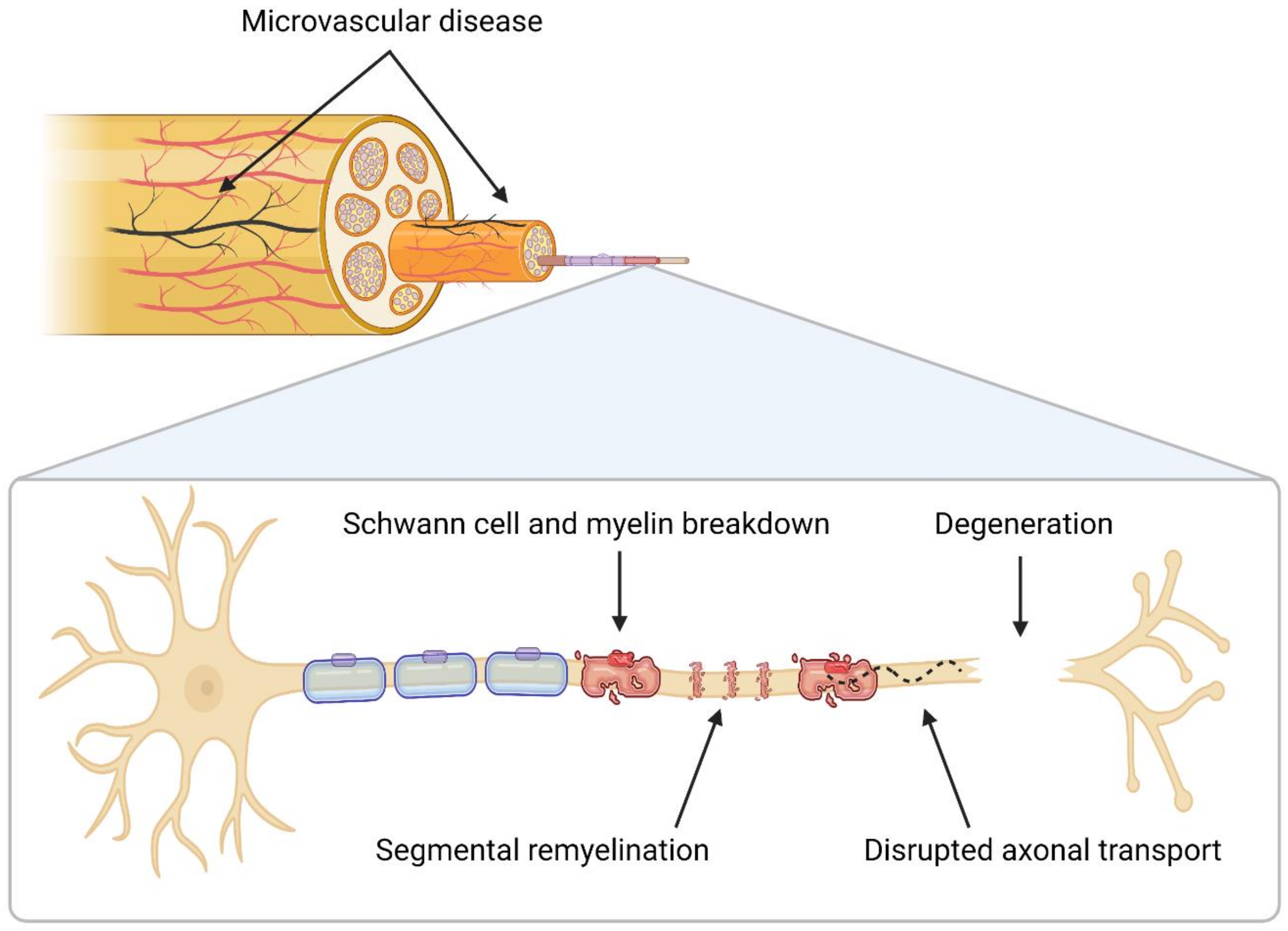{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neuropathology
3. Metabolic Mechanisms
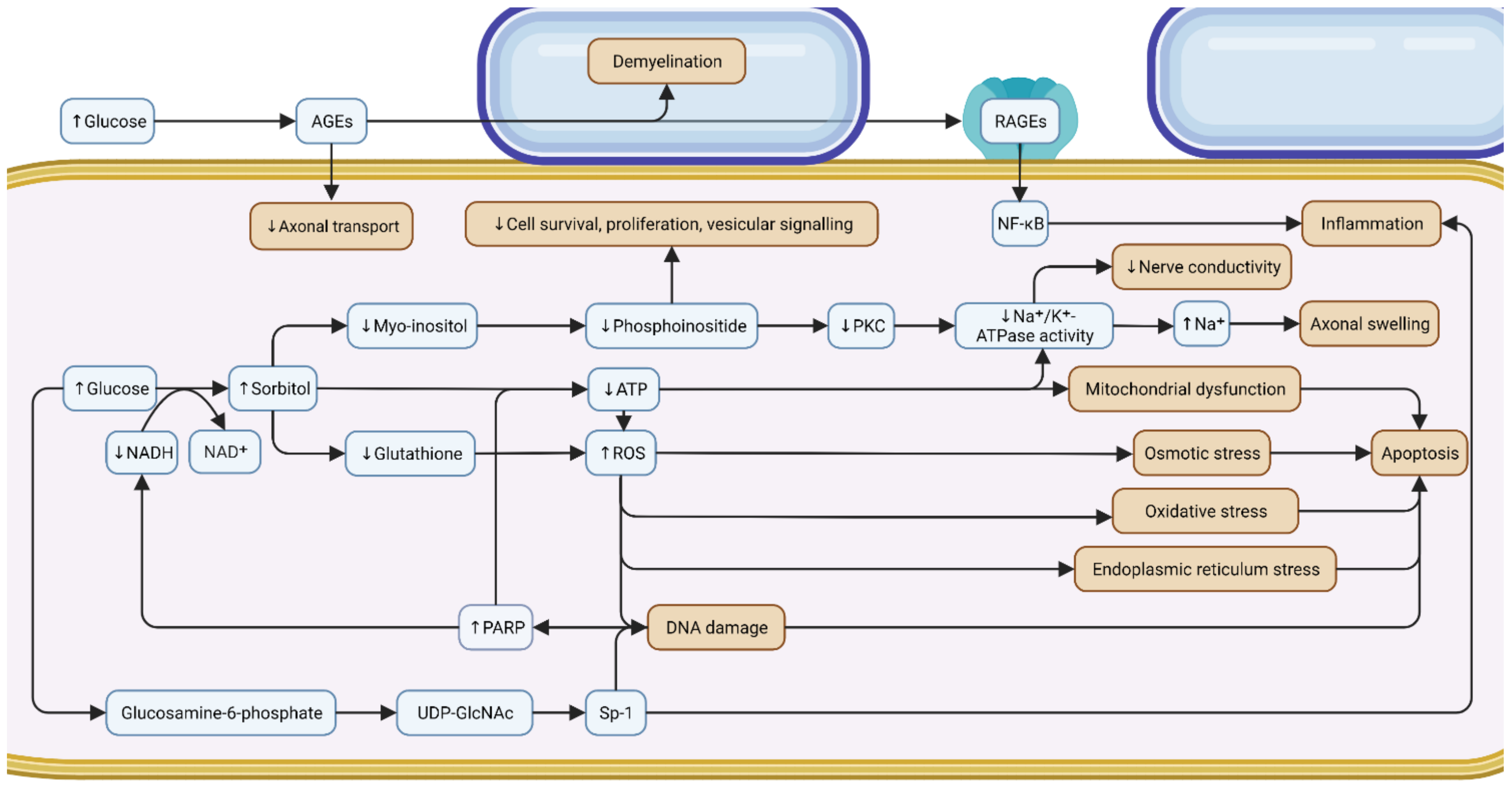
3.1. Hyperglycemia
3.1.1. Polyol
3.1.2. Glycation
3.1.3. Protein Kinase C (PKC)
3.1.4. Poly (ADP-Ribose) Polymerase (PARP)
3.1.5. Hexosamine
3.2. Hyperlipidemia
3.3. Impaired Insulin Signaling
4. Central Nervous System (CNS) Changes
4.1. Spinal Cord
4.2. Brainstem
4.3. Thalamus
4.4. Somatosensory Cortex
4.5. Motor and Somatosensory Tracts
5. Current Therapies and Future Recommendations
5.1. Glycemic Control
5.2. Pain Management
5.3. Spinal Cord Stimulation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pop-Busui, R.; Ang, L.; Boulton, A.J.M.; Feldman, E.L.; Marcus, R.L.; Mizokami-Stout, K.; Singleton, J.R.; Ziegler, D. Diagnosis and Treatment of Painful Diabetic Peripheral Neuropathy. ADA Clin. Compend. 2022, 2022, 1–32. [Google Scholar] [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and Regional Diabetes Prevalence Estimates for 2019 and Projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th Edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pop-Busui, R.; Boulton, A.J.M.; Feldman, E.L.; Bril, V.; Freeman, R.; Malik, R.A.; Sosenko, J.M.; Ziegler, D. Diabetic Neuropathy: A Position Statement by the American Diabetes Association. Diabetes Care 2017, 40, 136–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albers, J.W.; Pop-Busui, R. Diabetic Neuropathy: Mechanisms, Emerging Treatments and Subtypes. Curr. Neurol. Neurosci. Rep. 2014, 14, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, G.; Selvarajah, D.; Tesfaye, S. Pathogenesis, Diagnosis and Clinical Management of Diabetic Sensorimotor Peripheral Neuropathy. Nat. Rev. Endocrinol. 2021, 17, 400–420. [Google Scholar] [CrossRef] [PubMed]
- Kempler, P.; Amarenco, G.; Freeman, R.; Frontoni, S.; Horowitz, M.; Stevens, M.; Low, P.; Pop-Busui, R.; Tahrani, A.A.; Tesfaye, S.; et al. Management Strategies for Gastrointestinal, Erectile, Bladder, and Sudomotor Dysfunction in Patients with Diabetes. Diabetes Metab. Res. Rev. 2011, 27, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burakgazi, A.Z.; Alsowaity, B.; Burakgazi, Z.A.; Unal, D.; Kelly, J.J. Bladder Dysfunction in Peripheral Neuropathies. Muscle Nerve 2012, 45, 2–8. [Google Scholar] [CrossRef]
- Freeman, R. Chapter 6-Diabetic Autonomic Neuropathy. In Handbook of Clinical Neurology; Zochodne, D.W., Malik, R.A., Eds.; Diabetes and the Nervous System; Elsevier: Amsterdam, The Netherlands, 2014; Volume 126, pp. 63–79. [Google Scholar]
- Jin, H.Y.; Baek, H.S.; Park, T.S. Morphologic Changes in Autonomic Nerves in Diabetic Autonomic Neuropathy. Diabetes Metab. J. 2015, 39, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Spallone, V. Update on the Impact, Diagnosis and Management of Cardiovascular Autonomic Neuropathy in Diabetes: What Is Defined, What Is New, and What Is Unmet. Diabetes Metab. J. 2019, 43, 3–30. [Google Scholar] [CrossRef]
- Bharucha, A.E.; Kudva, Y.C.; Prichard, D.O. Diabetic Gastroparesis. Endocr. Rev. 2019, 40, 1318–1352. [Google Scholar] [CrossRef]
- Marathe, C.S.; Jones, K.L.; Wu, T.; Rayner, C.K.; Horowitz, M. Gastrointestinal Autonomic Neuropathy in Diabetes. Auton. Neurosci. 2020, 229, 102718. [Google Scholar] [CrossRef]
- Körei, A.E.; Istenes, I.; Papanas, N.; Kempler, P. Small-Fiber Neuropathy: A Diabetic Microvascular Complication of Special Clinical, Diagnostic, and Prognostic Importance. Angiology 2016, 67, 49–57. [Google Scholar] [CrossRef]
- Sène, D. Small Fiber Neuropathy: Diagnosis, Causes, and Treatment. Jt. Bone Spine 2018, 85, 553–559. [Google Scholar] [CrossRef]
- Laughlin, R.S.; Dyck, P.J.B. Chapter 4-Diabetic Radiculoplexus Neuropathies. In Handbook of Clinical Neurology; Zochodne, D.W., Malik, R.A., Eds.; Diabetes and the Nervous System; Elsevier: Amsterdam, The Netherlands, 2014; Volume 126, pp. 45–52. [Google Scholar]
- Glenn, M.D.; Jabari, D. Diabetic Lumbosacral Radiculoplexus Neuropathy (Diabetic Amyotrophy). Neurol. Clin. 2020, 38, 553–564. [Google Scholar] [CrossRef]
- Vinik, A.; Mehrabyan, A.; Colen, L.; Boulton, A. Focal Entrapment Neuropathies in Diabetes. Diabetes Care 2004, 27, 1783–1788. [Google Scholar] [CrossRef] [Green Version]
- Duby, J.J.; Campbell, R.K.; Setter, S.M.; White, J.R.; Rasmussen, K.A. Diabetic Neuropathy: An Intensive Review. Am. J. Health Syst. Pharm. 2004, 61, 160–173. [Google Scholar] [CrossRef]
- Boulton, A.J.M.; Malik, R.A. Chapter 53-Diabetes Mellitus: Neuropathy. In Endocrinology: Adult and Pediatric, 7th ed.; Jameson, J.L., de Groot, L.J., de Kretser, D.M., Giudice, L.C., Grossman, A.B., Melmed, S., Potts, J.T., Jr., Weir, G.C., Eds.; Saunders: Philadelphia, PA, USA, 2016; pp. 920–933. [Google Scholar]
- Samakidou, G.; Eleftheriadou, I.; Tentolouris, A.; Papanas, N.; Tentolouris, N. Rare Diabetic Neuropathies: It Is Not Only Distal Symmetrical Polyneuropathy. Diabetes Res. Clin. Pract. 2021, 177, 108932. [Google Scholar] [CrossRef]
- Reeves, N.D.; Orlando, G.; Brown, S.J. Sensory-Motor Mechanisms Increasing Falls Risk in Diabetic Peripheral Neuropathy. Medicina 2021, 57, 457. [Google Scholar] [CrossRef]
- Vileikyte, L.; Gonzalez, J.S. Chapter 14-Recognition and Management of Psychosocial Issues in Diabetic Neuropathy. In Handbook of Clinical Neurology; Zochodne, D.W., Malik, R.A., Eds.; Diabetes and the Nervous System; Elsevier: Amsterdam, The Netherlands, 2014; Volume 126, pp. 195–209. [Google Scholar]
- Naranjo, C.; Del Reguero, L.; Moratalla, G.; Hercberg, M.; Valenzuela, M.; Failde, I. Anxiety, Depression and Sleep Disorders in Patients with Diabetic Neuropathic Pain: A Systematic Review. Expert Rev. Neurother. 2019, 19, 1201–1209. [Google Scholar] [CrossRef]
- Normahani, P.; Shalhoub, J. Diabetic Foot Disease. Surg. Oxf. 2022, 40, 53–61. [Google Scholar] [CrossRef]
- Kerr, M.; Barron, E.; Chadwick, P.; Evans, T.; Kong, W.M.; Rayman, G.; Sutton-Smith, M.; Todd, G.; Young, B.; Jeffcoate, W.J. The Cost of Diabetic Foot Ulcers and Amputations to the National Health Service in England. Diabet. Med. 2019, 36, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.B.; Desai, U.; Cummings, A.K.G.; Birnbaum, H.G.; Skornicki, M.; Parsons, N.B. Burden of Diabetic Foot Ulcers for Medicare and Private Insurers. Diabetes Care 2014, 37, 651–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrakis, I.; Kyriopoulos, I.J.; Ginis, A.; Athanasakis, K. Losing a Foot versus Losing a Dollar; a Systematic Review of Cost Studies in Diabetic Foot Complications. Expert Rev. Pharmacoecon. Outcomes Res. 2017, 17, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Chalk, C.; Benstead, T.J.; Moore, F. Aldose Reductase Inhibitors for the Treatment of Diabetic Polyneuropathy. Cochrane Database Syst. Rev. 2007, CD004572. [Google Scholar] [CrossRef]
- Braffett, B.H.; Gubitosi-Klug, R.A.; Albers, J.W.; Feldman, E.L.; Martin, C.L.; White, N.H.; Orchard, T.J.; Lopes-Virella, M.; Lachin, J.M.; Pop-Busui, R. Risk Factors for Diabetic Peripheral Neuropathy and Cardiovascular Autonomic Neuropathy in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study. Diabetes 2020, 69, 1000–1010. [Google Scholar] [CrossRef]
- England, J.D.; Asbury, A.K. Peripheral Neuropathy. Lancet 2004, 363, 2151–2161. [Google Scholar] [CrossRef]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic Neuropathy. Nat. Rev. Dis. Primers 2019, 5, 41. [Google Scholar] [CrossRef]
- Selvarajah, D.; Wilkinson, I.D.; Davies, J.; Gandhi, R.; Tesfaye, S. Central Nervous System Involvement in Diabetic Neuropathy. Curr. Diabetes Rep. 2011, 11, 310–322. [Google Scholar] [CrossRef]
- Shillo, P.; Sloan, G.; Greig, M.; Hunt, L.; Selvarajah, D.; Elliott, J.; Gandhi, R.; Wilkinson, I.D.; Tesfaye, S. Painful and Painless Diabetic Neuropathies: What Is the Difference? Curr. Diabetes Rep. 2019, 19, 32. [Google Scholar] [CrossRef] [Green Version]
- Malik, R.A.; Tesfaye, S.; Newrick, P.G.; Walker, D.; Rajbhandari, S.M.; Siddique, I.; Sharma, A.K.; Boulton, A.J.M.; King, R.H.M.; Thomas, P.K.; et al. Sural Nerve Pathology in Diabetic Patients with Minimal but Progressive Neuropathy. Diabetologia 2005, 48, 578–585. [Google Scholar] [CrossRef]
- Said, G.; Slama, G.; Selva, J. Progressive Centripetal Degeneration of Axons in Small Fibre Diabetic Polyneuropathy: A Clinical and Pathological Study. Brain 1983, 106, 791–807. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Shao, S.-J.; Guo, H.-D. Schwann Cells Apoptosis Is Induced by High Glucose in Diabetic Peripheral Neuropathy. Life Sci. 2020, 248, 117459. [Google Scholar] [CrossRef]
- Dey, I.; Midha, N.; Singh, G.; Forsyth, A.; Walsh, S.K.; Singh, B.; Kumar, R.; Toth, C.; Midha, R. Diabetic Schwann Cells Suffer from Nerve Growth Factor and Neurotrophin-3 Underproduction and Poor Associability with Axons. Glia 2013, 61, 1990–1999. [Google Scholar] [CrossRef]
- Padilla, A.; Descorbeth, M.; Almeyda, A.L.; Payne, K.; De Leon, M. Hyperglycemia Magnifies Schwann Cell Dysfunction and Cell Death Triggered by PA-Induced Lipotoxicity. Brain Res. 2011, 1370, 64–79. [Google Scholar] [CrossRef] [Green Version]
- Naruse, K. Schwann Cells as Crucial Players in Diabetic Neuropathy. In Myelin: Basic and Clinical Advances; Sango, K., Yamauchi, J., Ogata, T., Susuki, K., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; pp. 345–356. ISBN 978-981-329-636-7. [Google Scholar]
- Baptista, F.I.; Pinheiro, H.; Gomes, C.A.; Ambrósio, A.F. Impairment of Axonal Transport in Diabetes: Focus on the Putative Mechanisms Underlying Peripheral and Central Neuropathies. Mol. Neurobiol. 2019, 56, 2202–2210. [Google Scholar] [CrossRef]
- Ryle, C.; Donaghy, M. Non-Enzymatic Glycation of Peripheral Nerve Proteins in Human Diabetics. J. Neurol. Sci. 1995, 129, 62–68. [Google Scholar] [CrossRef]
- Fernyhough, P.; Diemel, L.T.; Tomlinson, D.R. Target Tissue Production and Axonal Transport of Neurotrophin-3 Are Reduced in Streptozotocin-Diabetic Rats. Diabetologia 1998, 41, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Sima, A.A.; Lattimer, S.A.; Yagihashi, S.; Greene, D.A. Axo-glial dysjunction. A novel structural lesion that accounts for poorly reversible slowing of nerve conduction in the spontaneously diabetic bio-breeding rat. J. Clin. Investig. 1986, 77, 474–484. [Google Scholar] [CrossRef] [Green Version]
- Zochodne, D.W. Diabetes Mellitus and the Peripheral Nervous System: Manifestations and Mechanisms. Muscle Nerve 2007, 36, 144–166. [Google Scholar] [CrossRef]
- Zochodne, D.W.; Ho, L.T. The Influence of Sulindac on Experimental Streptozotocin-Induced Diabetic Neuropathy. Can. J. Neurol. Sci. 1994, 21, 194–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zochodne, D.W.; Ho, L.T.; Allison, J.A. Dorsal Root Ganglia Microenvironment of Female BB Wistar Diabetic Rats with Mild Neuropathy. J. Neurol. Sci. 1994, 127, 36–42. [Google Scholar] [CrossRef]
- Zochodne, D.W. Chapter 26-Mechanisms of Diabetic Neuron Damage: Molecular Pathways. In Handbook of Clinical Neurology; Zochodne, D.W., Malik, R.A., Eds.; Diabetes and the Nervous System; Elsevier: Amsterdam, The Netherlands, 2014; Volume 126, pp. 379–399. [Google Scholar]
- Selvarajah, D.; Wilkinson, I.D.; Emery, C.J.; Harris, N.D.; Shaw, P.J.; Witte, D.R.; Griffiths, P.D.; Tesfaye, S. Early Involvement of the Spinal Cord in Diabetic Peripheral Neuropathy. Diabetes Care 2006, 29, 2664–2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, R.A.; Newrick, P.G.; Sharma, A.K.; Jennings, A.; Ah-See, A.K.; Mayhew, T.M.; Jakubowski, J.; Boulton, A.J.; Ward, J.D. Microangiopathy in Human Diabetic Neuropathy: Relationship between Capillary Abnormalities and the Severity of Neuropathy. Diabetologia 1989, 32, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Dyck, P.J. Abnormalities of Endoneurial Microvessels and Sural Nerve Pathology in Diabetic Neuropathy. Neurology 1987, 37, 20–28. [Google Scholar] [CrossRef]
- Tesfaye, S.; Harris, N.; Jakubowski, J.J.; Mody, C.; Wilson, R.M.; Rennie, I.G.; Ward, J.D. Impaired Blood Flow and Arterio-Venous Shunting in Human Diabetic Neuropathy: A Novel Technique of Nerve Photography and Fluorescein Angiography. Diabetologia 1993, 36, 1266–1274. [Google Scholar] [CrossRef] [Green Version]
- Dyck, P.J.; Lais, A.; Karnes, J.L.; O’Brien, P.; Rizza, R. Fiber Loss Is Primary and Multifocal in Sural Nerves in Diabetic Polyneuropathy. Ann. Neurol. 1986, 19, 425–439. [Google Scholar] [CrossRef]
- Quattrini, C.; Jeziorska, M.; Boulton, A.J.M.; Malik, R.A. Reduced Vascular Endothelial Growth Factor Expression and Intra-Epidermal Nerve Fiber Loss in Human Diabetic Neuropathy. Diabetes Care 2008, 31, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Britland, S.T.; Young, R.J.; Ashutosh, S.K.; Clarke, B.F. Relationship of Endoneurial Capillary Abnormalities to Type and Severity of Diabetic Polyneuropathy. Diabetes 1990, 39, 909–913. [Google Scholar] [CrossRef]
- Malik, R.A.; Veves, A.; Masson, E.A.; Sharma, A.K.; Ah-See, A.K.; Schady, W.; Lye, R.H.; Boulton, A.J. Endoneurial Capillary Abnormalities in Mild Human Diabetic Neuropathy. J. Neurol. Neurosurg. Psychiatry 1992, 55, 557–561. [Google Scholar] [CrossRef] [Green Version]
- Cameron, N.E.; Eaton, S.E.M.; Cotter, M.A.; Tesfaye, S. Vascular Factors and Metabolic Interactions in the Pathogenesis of Diabetic Neuropathy. Diabetologia 2001, 44, 1973–1988. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, N.; Barma, S.; Konwar, N.; Dewanjee, S.; Manna, P. Mechanistic Insight of Diabetic Nephropathy and Its Pharmacotherapeutic Targets: An Update. Eur. J. Pharmacol. 2016, 791, 8–24. [Google Scholar] [CrossRef]
- Chau, J.F.L.; Lee, M.K.; Law, J.W.S.; Chung, S.K.; Chung, S.S.M. Sodium/Myo-Inositol Cotransporter-1 Is Essential for the Development and Function of the Peripheral Nerves. FASEB J. 2005, 19, 1887–1889. [Google Scholar] [CrossRef] [Green Version]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; Timms, J.; Waterfield, M.D. Cellular Function of Phosphoinositide 3-Kinases: Implications for Development, Immunity, Homeostasis, and Cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 616–675. [Google Scholar] [CrossRef]
- Greene, D.A.; Lattimer-Greene, S.; Sima, A.A. Pathogenesis of Diabetic Neuropathy: Role of Altered Phosphoinositide Metabolism. Crit. Rev. Neurobiol. 1989, 5, 143–219. [Google Scholar]
- Uehara, K.; Yamagishi, S.-I.; Otsuki, S.; Chin, S.; Yagihashi, S. Effects of Polyol Pathway Hyperactivity on Protein Kinase C Activity, Nociceptive Peptide Expression, and Neuronal Structure in Dorsal Root Ganglia in Diabetic Mice. Diabetes 2004, 53, 3239–3247. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Eichberg, J. 1,2-Diacylglycerol Content and Its Arachidonyl-Containing Molecular Species Are Reduced in Sciatic Nerve from Streptozotocin-Induced Diabetic Rats. J. Neurochem. 1990, 55, 1087–1090. [Google Scholar] [CrossRef]
- Kamiya, H.; Nakamura, J.; Hamada, Y.; Nakashima, E.; Naruse, K.; Kato, K.; Yasuda, Y.; Hotta, N. Polyol Pathway and Protein Kinase C Activity of Rat Schwannoma Cells. Diabetes Metab. Res. Rev. 2003, 19, 131–139. [Google Scholar] [CrossRef]
- Vague, P.; Coste, T.C.; Jannot, M.F.; Raccah, D.; Tsimaratos, M. C-Peptide, Na+,K+-ATPase, and Diabetes. Exp. Diabesity Res. 2004, 5, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Sima, A.A. C-Peptide and Diabetic Neuropathy. Expert Opin. Investig. Drugs 2003, 12, 1471–1488. [Google Scholar] [CrossRef]
- Greene, D.A.; Chakrabarti, S.; Lattimer, S.A.; Sima, A.A. Role of Sorbitol Accumulation and Myo-Inositol Depletion in Paranodal Swelling of Large Myelinated Nerve Fibers in the Insulin-Deficient Spontaneously Diabetic Bio-Breeding Rat. Reversal by Insulin Replacement, an Aldose Reductase Inhibitor, and Myo-Inositol. J. Clin. Investig. 1987, 79, 1479–1485. [Google Scholar] [CrossRef]
- Callaghan, B.C.; Cheng, H.T.; Stables, C.L.; Smith, A.L.; Feldman, E.L. Diabetic Neuropathy: Clinical Manifestations and Current Treatments. Lancet Neurol. 2012, 11, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Dewanjee, S.; Das, S.; Das, A.K.; Bhattacharjee, N.; Dihingia, A.; Dua, T.K.; Kalita, J.; Manna, P. Molecular Mechanism of Diabetic Neuropathy and Its Pharmacotherapeutic Targets. Eur. J. Pharmacol. 2018, 833, 472–523. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Bierhaus, A.; Hofmann, M.A.; Ziegler, R.; Nawroth, P.P. AGEs and Their Interaction with AGE-Receptors in Vascular Disease and Diabetes Mellitus. I. The AGE Concept. Cardiovasc. Res. 1998, 37, 586–600. [Google Scholar] [CrossRef] [Green Version]
- Wada, R.; Yagihashi, S. Role of Advanced Glycation End Products and Their Receptors in Development of Diabetic Neuropathy. Ann. N. Y. Acad. Sci. 2005, 1043, 598–604. [Google Scholar] [CrossRef]
- Sugimoto, K.; Nishizawa, Y.; Horiuchi, S.; Yagihashi, S. Localization in Human Diabetic Peripheral Nerve of Nε-Carboxymethyllysine-Protein Adducts, an Advanced Glycation Endproduct. Diabetologia 1997, 40, 1380–1387. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.K.; Howarth, N.L.; Devenny, J.J.; Bitensky, M.W. Structural and Functional Consequences of Increased Tubulin Glycosylation in Diabetes Mellitus. Proc. Natl. Acad. Sci. USA 1982, 79, 6546–6550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucala, R.; Tracey, K.J.; Cerami, A. Advanced Glycosylation Products Quench Nitric Oxide and Mediate Defective Endothelium-Dependent Vasodilatation in Experimental Diabetes. J. Clin. Investig. 1991, 87, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinksy, D.; Stern, D. Enhanced Cellular Oxidant Stress by the Interaction of Advanced Glycation End Products with Their Receptors/Binding Proteins. J Biol. Chem. 1994, 13, 9889–9897. [Google Scholar] [CrossRef]
- Knott, H.M.; Brown, B.E.; Davies, M.J.; Dean, R.T. Glycation and Glycoxidation of Low-Density Lipoproteins by Glucose and Low-Molecular Mass Aldehydes. Formation of Modified and Oxidized Particles. Eur. J. Biochem. 2003, 270, 3572–3582. [Google Scholar] [CrossRef] [Green Version]
- Aronson, D.; Rayfield, E.J. How Hyperglycemia Promotes Atherosclerosis: Molecular Mechanisms. Cardiovasc. Diabetol. 2002, 1, 1. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced Glycation End-Products: A Review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [Green Version]
- Geraldes, P.; King, G.L. Activation of Protein Kinase C Isoforms & Its Impact on Diabetic Complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Yagihashi, S. Chapter Eight-Glucotoxic Mechanisms and Related Therapeutic Approaches. In International Review of Neurobiology; Calcutt, N.A., Fernyhough, P., Eds.; Controversies in Diabetic Neuropathy; Academic Press: Cambridge, MA, USA, 2016; Volume 127, pp. 121–149. [Google Scholar]
- Cassese, A.; Esposito, I.; Fiory, F.; Barbagallo, A.P.M.; Paturzo, F.; Mirra, P.; Ulianich, L.; Giacco, F.; Iadicicco, C.; Lombardi, A.; et al. In Skeletal Muscle Advanced Glycation End Products (AGEs) Inhibit Insulin Action and Induce the Formation of Multimolecular Complexes Including the Receptor for AGEs. J. Biol. Chem. 2008, 283, 36088–36099. [Google Scholar] [CrossRef] [Green Version]
- Scivittaro, V.; Ganz, M.B.; Weiss, M.F. AGEs Induce Oxidative Stress and Activate Protein Kinase C-ΒII in Neonatal Mesangial Cells. Am. J. Physiol.-Ren. Physiol. 2000, 278, F676–F683. [Google Scholar] [CrossRef] [Green Version]
- Lal, M.A.; Brismar, H.; Eklöf, A.-C.; Aperia, A. Role of Oxidative Stress in Advanced Glycation End Product-Induced Mesangial Cell Activation. Kidney Int. 2002, 61, 2006–2014. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.S.; Koppenol, W.H. Nitric Oxide, Superoxide, and Peroxynitrite: The Good, the Bad, and Ugly. Am. J. Physiol.-Cell Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [Green Version]
- Negi, G.; Kumar, A.; Sharma, S.S. Concurrent Targeting of Nitrosative Stress–PARP Pathway Corrects Functional, Behavioral and Biochemical Deficits in Experimental Diabetic Neuropathy. Biochem. Biophys. Res. Commun. 2010, 391, 102–106. [Google Scholar] [CrossRef]
- Obrosova, I.G.; Li, F.; Abatan, O.I.; Forsell, M.A.; Komjáti, K.; Pacher, P.; Szabó, C.; Stevens, M.J. Role of Poly(ADP-Ribose) Polymerase Activation in Diabetic Neuropathy. Diabetes 2004, 53, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Szabó, C. Role of Poly(ADP-Ribose) Polymerase 1 (PARP-1) in Cardiovascular Diseases: The Therapeutic Potential of PARP Inhibitors. Cardiovasc. Drug Rev. 2007, 25, 235–260. [Google Scholar] [CrossRef] [Green Version]
- Ha, H.C.; Hester, L.D.; Snyder, S.H. Poly(ADP-Ribose) Polymerase-1 Dependence of Stress-Induced Transcription Factors and Associated Gene Expression in Glia. Proc. Natl. Acad. Sci. USA 2002, 99, 3270. [Google Scholar] [CrossRef] [Green Version]
- Buse, M.G. Hexosamines, Insulin Resistance, and the Complications of Diabetes: Current Status. Am. J. Physiol. -Endocrinol. Metab. 2006, 290, E1–E8. [Google Scholar] [CrossRef]
- Kaneto, H.; Xu, G.; Song, K.-H.; Suzuma, K.; Bonner-Weir, S.; Sharma, A.; Weir, G.C. Activation of the Hexosamine Pathway Leads to Deterioration of Pancreatic β-Cell Function through the Induction of Oxidative Stress. J. Biol. Chem. 2001, 276, 31099–31104. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.M.; Calabek, B.; Roberts, L.; Feldman, E.L. Chapter 34-Biology of Diabetic Neuropathy. In Handbook of Clinical Neurology; Said, G., Krarup, C., Eds.; Peripheral Nerve Disorders; Elsevier: Amsterdam, The Netherlands, 2013; Volume 115, pp. 591–606. [Google Scholar]
- National Centre for Biotechnology Information. SP1 Sp1 Transcription Factor [Homo Sapiens (Human)]-Gene-NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/6667 (accessed on 16 November 2021).
- Du, X.-L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-Induced Mitochondrial Superoxide Overproduction Activates the Hexosamine Pathway and Induces Plasminogen Activator Inhibitor-1 Expression by Increasing Sp1 Glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, H.J.; Whiteside, C.I.; Fantus, I.G. The Hexosamine Pathway Regulates the Plasminogen Activator Inhibitor-1 Gene Promoter and Sp1 Transcriptional Activation through Protein Kinase C-beta I and -delta. J. Biol. Chem. 2002, 277, 33833–33841. [Google Scholar] [CrossRef] [Green Version]
- Viader, A.; Sasaki, Y.; Kim, S.; Strickland, A.; Workman, C.S.; Yang, K.; Gross, R.W.; Milbrandt, J. Aberrant Schwann Cell Lipid Metabolism Linked to Mitochondrial Deficits Leads to Axon Degeneration and Neuropathy. Neuron 2013, 77, 886–898. [Google Scholar] [CrossRef] [Green Version]
- Folli, F.; Bonfanti, L.; Renard, E.; Kahn, C.R.; Merighi, A. Insulin Receptor Substrate-1 (IRS-1) Distribution in the Rat Central Nervous System. J. Neurosci. 1994, 14, 6412–6422. [Google Scholar] [CrossRef] [PubMed]
- Walcher, D.; Marx, N. C-Peptide in the Vessel Wall. Rev. Diabet. Stud. 2009, 6, 180–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Feldman, E.L. Insulin Resistance in the Nervous System. Trends Endocrinol. Metab. 2012, 23, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerging Risk Factors Collaboration; Sarwar, N.; Gao, P.; Seshasai, S.R.K.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; et al. Diabetes Mellitus, Fasting Blood Glucose Concentration, and Risk of Vascular Disease: A Collaborative Meta-Analysis of 102 Prospective Studies. Lancet Lond. Engl. 2010, 375, 2215–2222. [Google Scholar] [CrossRef] [Green Version]
- Farooqi, A.; Gillies, C.; Sathanapally, H.; Abner, S.; Seidu, S.; Davies, M.J.; Polonsky, W.H.; Khunti, K. A Systematic Review and Meta-Analysis to Compare the Prevalence of Depression between People with and without Type 1 and Type 2 Diabetes. Prim. Care Diabetes 2022, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Huang, C.; Deng, H.; Wang, H. Diabetes as a Risk Factor for Dementia and Mild Cognitive Impairment: A Meta-Analysis of Longitudinal Studies. Intern. Med. J. 2012, 42, 484–491. [Google Scholar] [CrossRef]
- Eaton, S.E.; Harris, N.D.; Rajbhandari, S.M.; Greenwood, P.; Wilkinson, I.D.; Ward, J.D.; Griffiths, P.D.; Tesfaye, S. Spinal-Cord Involvement in Diabetic Peripheral Neuropathy. Lancet 2001, 358, 35–36. [Google Scholar] [CrossRef]
- Wilkinson, I.D.; Slevarajah, D.; Hutton, M.; Griffiths, P.D.; Gandhi, R.; Solomon, T. Imaging of the cortico-spinal tracts in diabetic neuropathy using Diffusion tensor magnetic resonance. In Proceedings of the American Diabetes Association Diabetes Professional 69th Scientific Sessions Congress, New Orleans, LA, USA, 5–9 June 2009. [Google Scholar]
- Segerdahl, A.R.; Themistocleous, A.C.; Fido, D.; Bennett, D.L.; Tracey, I. A Brain-Based Pain Facilitation Mechanism Contributes to Painful Diabetic Polyneuropathy. Brain 2018, 141, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, D.; Wilkinson, I.D.; Emery, C.J.; Shaw, P.J.; Griffiths, P.D.; Gandhi, R.; Tesfaye, S. Thalamic Neuronal Dysfunction and Chronic Sensorimotor Distal Symmetrical Polyneuropathy in Patients with Type 1 Diabetes Mellitus. Diabetologia 2008, 51, 2088. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, D.; Wilkinson, I.D.; Gandhi, R.; Griffiths, P.D.; Tesfaye, S. Microvascular Perfusion Abnormalities of the Thalamus in Painful but Not Painless Diabetic Polyneuropathy: A Clue to the Pathogenesis of Pain in Type 1 Diabetes. Diabetes Care 2011, 34, 718–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvarajah, D.; Wilkinson, I.D.; Maxwell, M.; Davies, J.; Sankar, A.; Boland, E.; Gandhi, R.; Tracey, I.; Tesfaye, S. Magnetic Resonance Neuroimaging Study of Brain Structural Differences in Diabetic Peripheral Neuropathy. Diabetes Care 2014, 37, 1681–1688. [Google Scholar] [CrossRef] [Green Version]
- Ghandi, R.; Slevarajah, D.; Emery, C.; Wilkinson, I.D.; Tesfaye, S. Sensory pathway neurochemical abnormalities within the brain in diabetic neuropathy. In Proceedings of the American Diabetes Association Diabetes Professional 68th Scientific Sessions Congress, San Francisco, CA, USA, 6–10 June 2008. [Google Scholar]
- Nordengen, K.; Heuser, C.; Rinholm, J.E.; Matalon, R.; Gundersen, V. Localisation of N-Acetylaspartate in Oligodendrocytes/Myelin. Brain Struct. Funct. 2015, 220, 899–917. [Google Scholar] [CrossRef]
- Selvarajah, D.; Wilkinson, I.D.; Fang, F.; Sankar, A.; Davies, J.; Boland, E.; Harding, J.; Rao, G.; Gandhi, R.; Tracey, I.; et al. Structural and Functional Abnormalities of the Primary Somatosensory Cortex in Diabetic Peripheral Neuropathy: A Multimodal MRI Study. Diabetes 2019, 68, 796–806. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Qu, M.; Yi, X.; Zhuo, P.; Tang, J.; Chen, X.; Zhou, G.; Hu, P.; Qiu, T.; Xing, W.; et al. Sensorimotor and Pain-Related Alterations of the Gray Matter and White Matter in Type 2 Diabetic Patients with Peripheral Neuropathy. Hum. Brain Mapp. 2020, 41, 710–725. [Google Scholar] [CrossRef] [Green Version]
- Tesfaye, S.; Sloan, G. Involvement of the Central Nervous System in Diabetic Distal Symmetrical Polyneuropathy. J. Xiangya Med. 2021, 6. [Google Scholar] [CrossRef]
- The Diabetes Control and Complications Trial Research Group. The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive Blood-Glucose Control with Sulphonylureas or Insulin Compared with Conventional Treatment and Risk of Complications in Patients with Type 2 Diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar] [CrossRef]
- Callaghan, B.C.; Little, A.A.; Feldman, E.L.; Hughes, R.A. Enhanced Glucose Control for Preventing and Treating Diabetic Neuropathy. Cochrane Database Syst. Rev. 2012, CD007543. [Google Scholar] [CrossRef]
- Ang, L.; Jaiswal, M.; Martin, C.; Pop-Busui, R. Glucose Control and Diabetic Neuropathy: Lessons from Recent Large Clinical Trials. Curr. Diabetes Rep. 2014, 14, 528. [Google Scholar] [CrossRef]
- Gæde, P.; Vedel, P.; Larsen, N.; Jensen, G.V.H.; Parving, H.-H.; Pedersen, O. Multifactorial Intervention and Cardiovascular Disease in Patients with Type 2 Diabetes. N. Engl. J. Med. 2009, 348, 580–591. [Google Scholar] [CrossRef] [Green Version]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for Neuropathic Pain in Adults: A Systematic Review and Meta-Analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Derry, S.; Bell, R.F.; Straube, S.; Wiffen, P.J.; Aldington, D.; Moore, R.A. Pregabalin for Neuropathic Pain in Adults. Cochrane Database Syst. Rev. 2019, CD007076. [Google Scholar] [CrossRef]
- Wiffen, P.J.; Derry, S.; Bell, R.F.; Rice, A.S.; Tölle, T.R.; Phillips, T.; Moore, R.A. Gabapentin for Chronic Neuropathic Pain in Adults. Cochrane Database Syst. Rev. 2017, CD007938. [Google Scholar] [CrossRef] [Green Version]
- Lunn, M.P.; Hughes, R.A.; Wiffen, P.J. Duloxetine for Treating Painful Neuropathy, Chronic Pain or Fibromyalgia. Cochrane Database Syst. Rev. 2014, CD007115. [Google Scholar] [CrossRef]
- National Institute for Health and Care Excellence (NICE). British National Formulary. Pregabalin. Available online: https://bnf.nice.org.uk/drug/pregabalin.html (accessed on 11 April 2022).
- National Institute for Health and Care Excellence (NICE). British National Formulary. Gabapentin. Available online: https://bnf.nice.org.uk/drug/gabapentin.html#indicationsAndDoses (accessed on 11 April 2022).
- National Institute for Health and Care Excellence (NICE). British National Formulary. Duloxetine. Available online: https://bnf.nice.org.uk/drug/duloxetine.html (accessed on 11 April 2022).
- Pregabalin (Lyrica): Reports of Severe Respiratory Depression. Available online: https://www.gov.uk/drug-safety-update/pregabalin-lyrica-reports-of-severe-respiratory-depression (accessed on 11 April 2022).
- Moore, R.A.; Derry, S.; Aldington, D.; Cole, P.; Wiffen, P.J. Amitriptyline for Neuropathic Pain in Adults. Cochrane Database Syst. Rev. 2015, 2015, CD008242. [Google Scholar] [CrossRef]
- Duehmke, R.M.; Derry, S.; Wiffen, P.J.; Bell, R.F.; Aldington, D.; Moore, R.A. Tramadol for Neuropathic Pain in Adults. Cochrane Database Syst. Rev. 2017, CD003726. [Google Scholar] [CrossRef]
- Staudt, M.D.; Prabhala, T.; Sheldon, B.L.; Quaranta, N.; Zakher, M.; Bhullar, R.; Pilitsis, J.G.; Argoff, C.E. Current Strategies for the Management of Painful Diabetic Neuropathy. J. Diabetes Sci. Technol. 2020, 16, 1932296820951829. [Google Scholar] [CrossRef]
- van Nooten, F.; Treur, M.; Pantiri, K.; Stoker, M.; Charokopou, M. Capsaicin 8% Patch Versus Oral Neuropathic Pain Medications for the Treatment of Painful Diabetic Peripheral Neuropathy: A Systematic Literature Review and Network Meta-Analysis. Clin. Ther. 2017, 39, 787–803.e18. [Google Scholar] [CrossRef] [Green Version]
- Polydefkis, M.; Hauer, P.; Sheth, S.; Sirdofsky, M.; Griffin, J.W.; McArthur, J.C. The Time Course of Epidermal Nerve Fibre Regeneration: Studies in Normal Controls and in People with Diabetes, with and without Neuropathy. Brain 2004, 127, 1606–1615. [Google Scholar] [CrossRef] [Green Version]
- Henson, J.V.; Varhabhatla, N.C.; Bebic, Z.; Kaye, A.D.; Yong, R.J.; Urman, R.D.; Merkow, J.S. Spinal Cord Stimulation for Painful Diabetic Peripheral Neuropathy: A Systematic Review. Pain Ther. 2021, 10, 895–908. [Google Scholar] [CrossRef]
- de Vos, C.C.; Meier, K.; Zaalberg, P.B.; Nijhuis, H.J.A.; Duyvendak, W.; Vesper, J.; Enggaard, T.P.; Lenders, M.W.P.M. Spinal Cord Stimulation in Patients with Painful Diabetic Neuropathy: A Multicentre Randomized Clinical Trial. Pain 2014, 155, 2426–2431. [Google Scholar] [CrossRef]
- Slangen, R.; Schaper, N.C.; Faber, C.G.; Joosten, E.A.; Dirksen, C.D.; van Dongen, R.T.; Kessels, A.G.; van Kleef, M. Spinal Cord Stimulation and Pain Relief in Painful Diabetic Peripheral Neuropathy: A Prospective Two-Center Randomized Controlled Trial. Diabetes Care 2014, 37, 3016–3024. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, N.E.; Ferraro, M.C.; Gibson, W.; Rice, A.S.; Vase, L.; Coyle, D.; Eccleston, C. Implanted Spinal Neuromodulation Interventions for Chronic Pain in Adults. Cochrane Database Syst. Rev. 2021, 12, CD013756. [Google Scholar] [CrossRef]
- Chapman, K.B.; Van Roosendaal, B.-K.W.; Van Helmond, N.; Yousef, T.A. Unilateral Dorsal Root Ganglion Stimulation Lead Placement with Resolution of Bilateral Lower Extremity Symptoms in Diabetic Peripheral Neuropathy. Cureus 2020, 12, e10735. [Google Scholar] [CrossRef]
- Eldabe, S.; Espinet, A.; Wahlstedt, A.; Kang, P.; Liem, L.; Patel, N.K.; Vesper, J.; Kimber, A.; Cusack, W.; Kramer, J. Retrospective Case Series on the Treatment of Painful Diabetic Peripheral Neuropathy with Dorsal Root Ganglion Stimulation. Neuromodul. J. Int. Neuromodul. Soc. 2018, 21, 787–792. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, S.; Normahani, P.; Lane, T.; Hohenschurz-Schmidt, D.; Oliver, N.; Davies, A.H. Pathogenesis of Distal Symmetrical Polyneuropathy in Diabetes. Life 2022, 12, 1074. https://doi.org/10.3390/life12071074
Smith S, Normahani P, Lane T, Hohenschurz-Schmidt D, Oliver N, Davies AH. Pathogenesis of Distal Symmetrical Polyneuropathy in Diabetes. Life. 2022; 12(7):1074. https://doi.org/10.3390/life12071074
Chicago/Turabian StyleSmith, Sasha, Pasha Normahani, Tristan Lane, David Hohenschurz-Schmidt, Nick Oliver, and Alun Huw Davies. 2022. "Pathogenesis of Distal Symmetrical Polyneuropathy in Diabetes" Life 12, no. 7: 1074. https://doi.org/10.3390/life12071074