
The Fate of Leydig Cells in Men with Spermatogenic Failure


Abstract
:1. Introduction
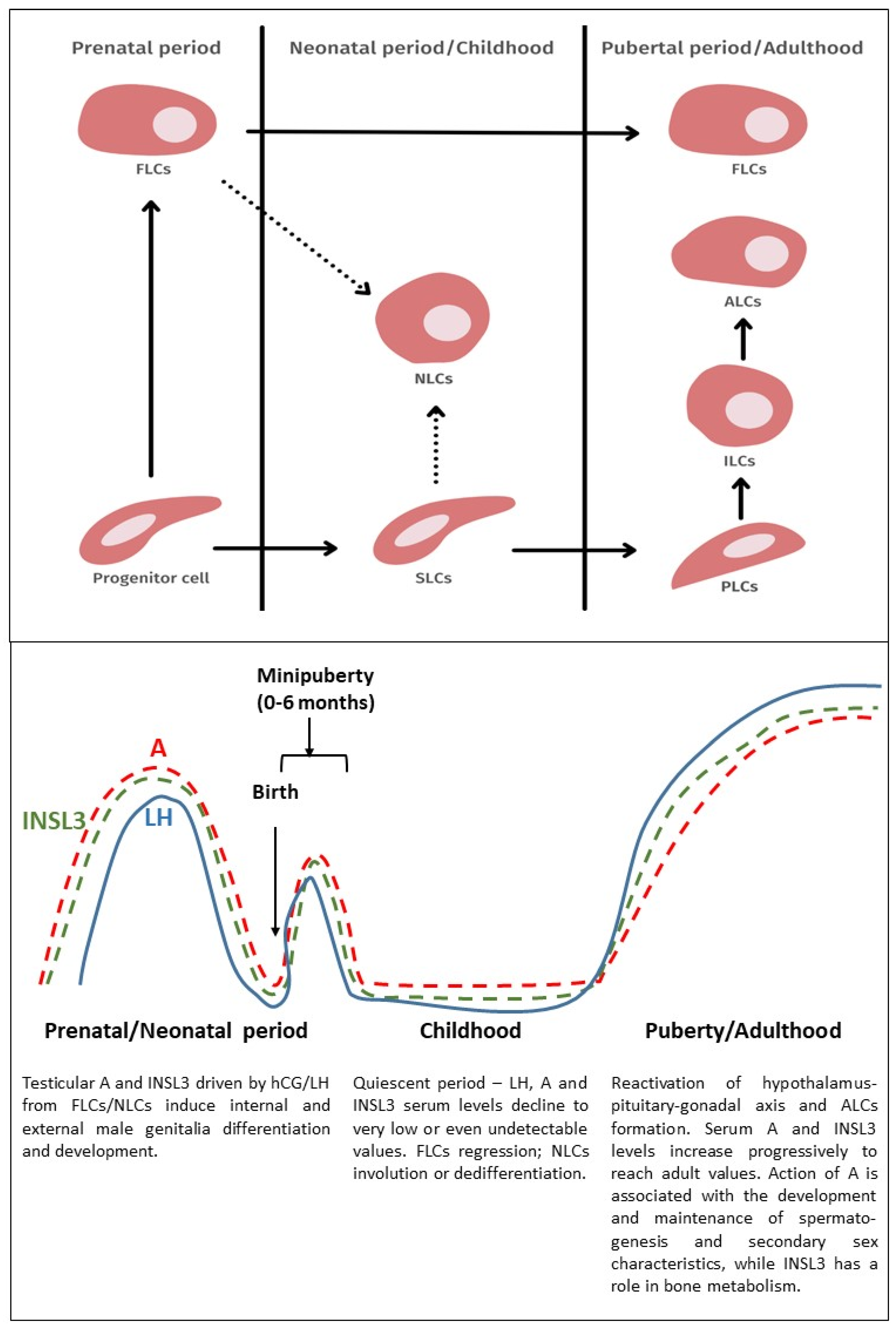
2. The Development and Function of Leydig Cells
2.1. Fetal Leydig Cells
2.2. Neonatal Leydig Cells
2.3. Adult Leydig Cells
2.4. The Relationship between Fetal and Adult Leydig Cells
3. The Contribution of Leydig Cells to Spermatogenesis
3.1. Steroidogenesis
3.2. Testosterone
3.3. Estradiol
3.4. Other Factors
4. Spermatogenic Failure and Leydig Cell Function
4.1. Non-Obstructive Azoospermia
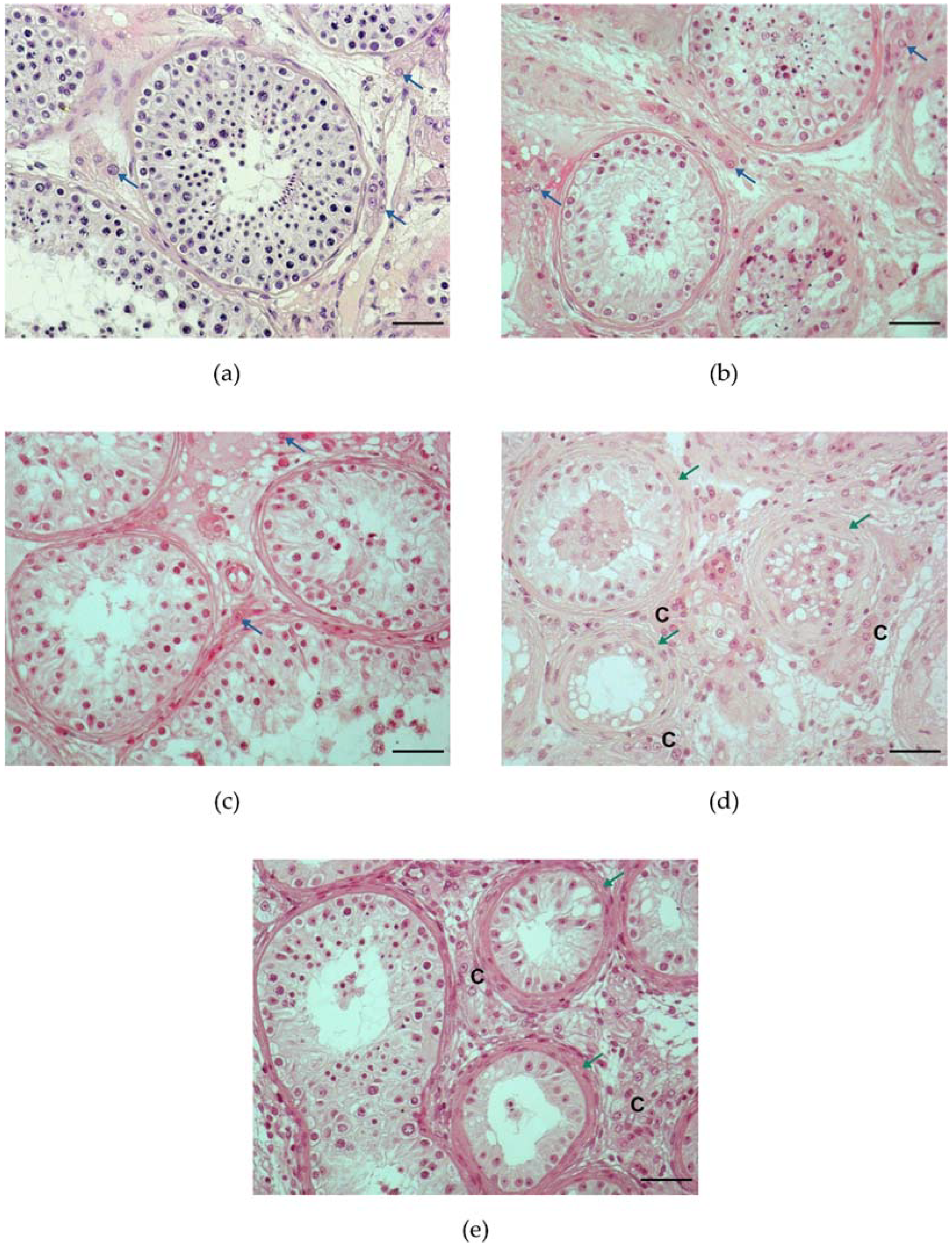
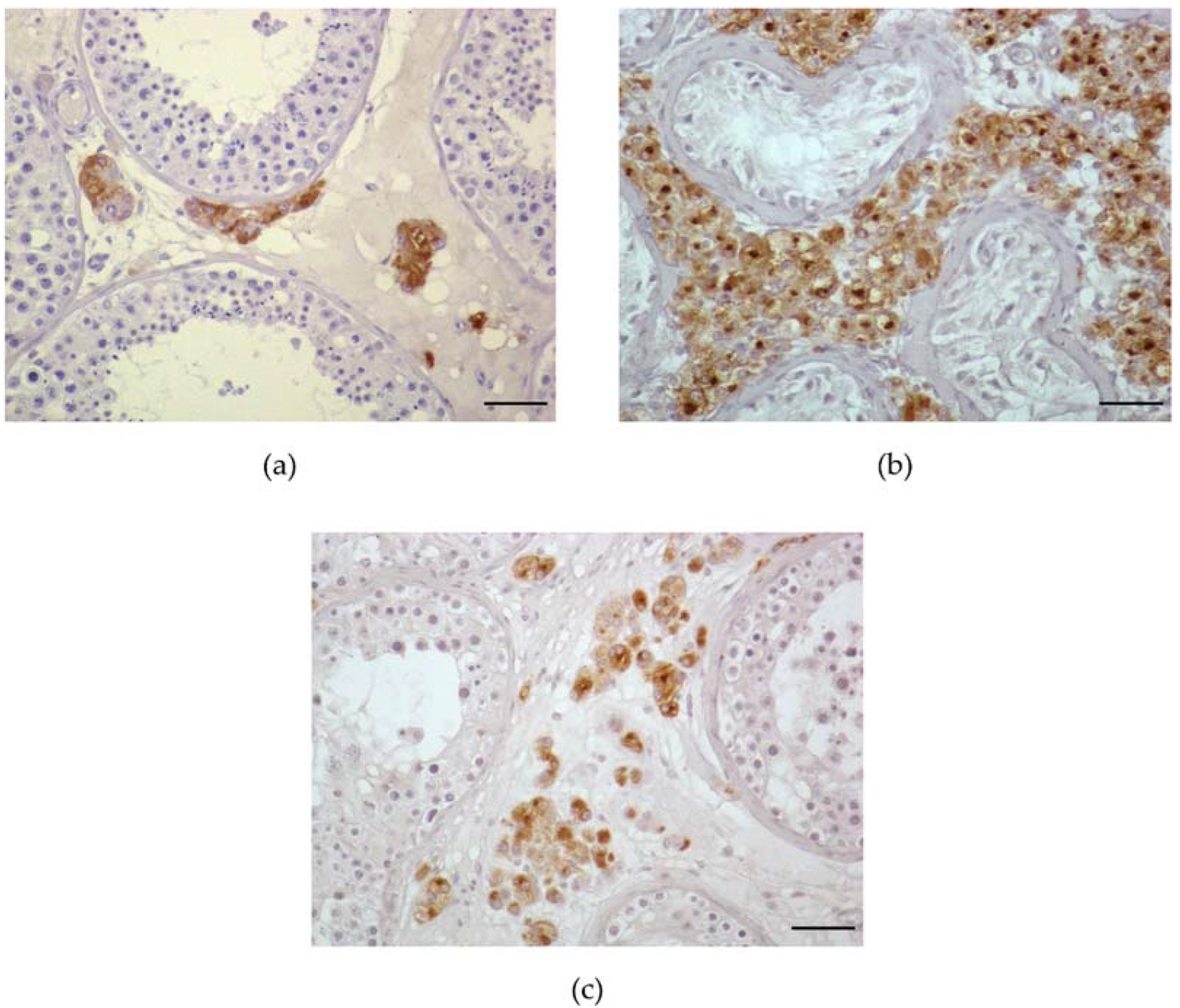
4.2. Histological Pattern
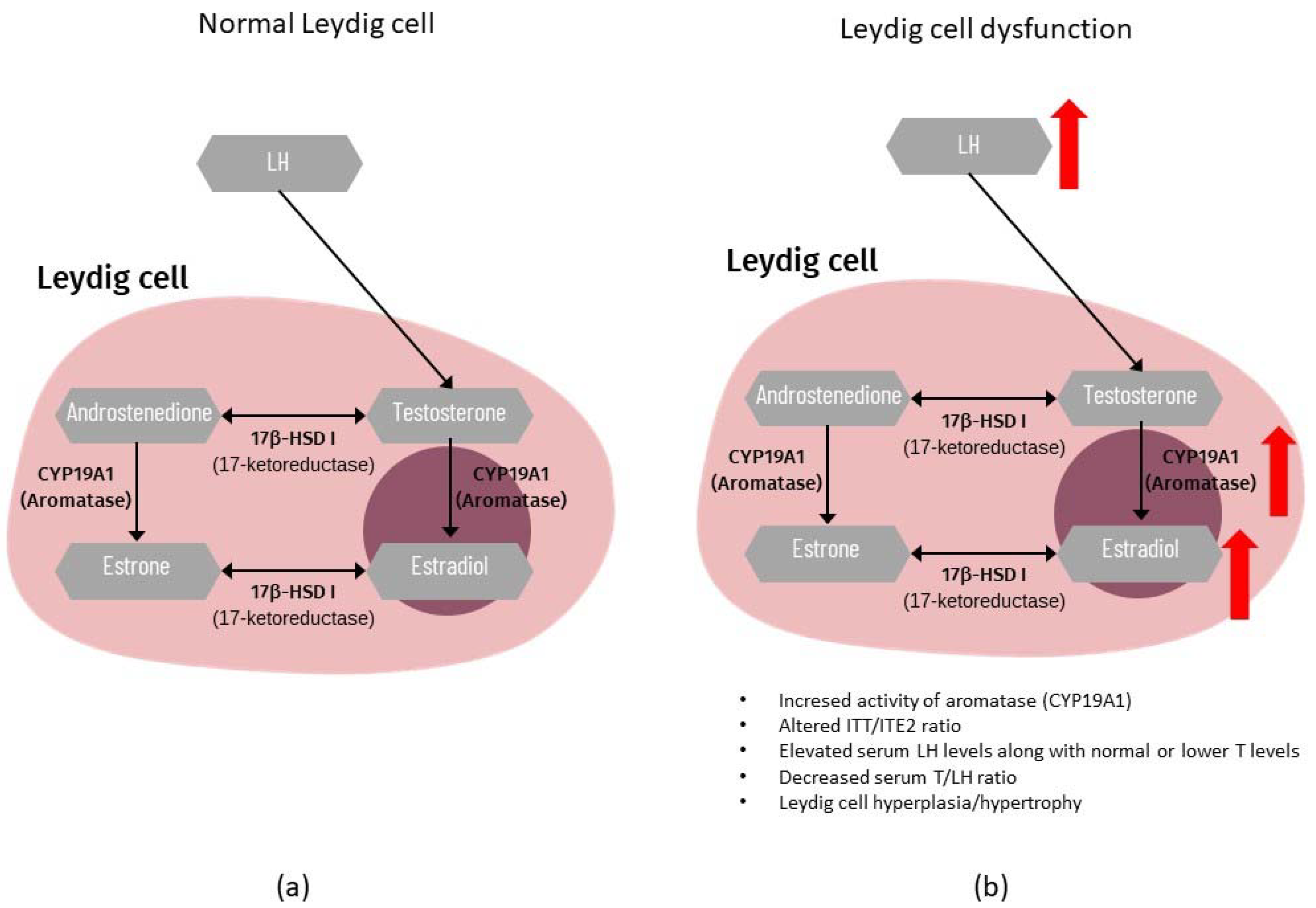
4.3. Endocrine Profile
4.4. Testicular Dysgenesis Syndrome
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lara, N.L.M.; Costa, G.M.J.; Avelar, G.F.; Lacerda, S.M.S.N.; Hess, R.A.; de França, L.R. Testis physiology—Overview and histology. In Encyclopedia of Reproduction, 2nd ed.; Skinner, M., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 105–116. [Google Scholar]
- Svingen, T.; Koopman, P. Building the mammalian testis: Origins, differentiation, and assembly of the component cell populations. Genes Dev. 2013, 27, 2409–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, M.H.; Pawlina, W. Male reproductive system. In Histology: A Text and Atlas, 7th ed.; LWW: Philadelphia, PA, USA, 2015; pp. 790–807. [Google Scholar]
- Fietz, D.; Bergmann, M. Functional Anatomy and Histology of the Testis. In Endocrinology of the Testis and Male Reproduction; Simoni, M., Huhtaniemi, I.T., Eds.; Springer International Publishing AG: Berlin, Germany, 2017; pp. 313–341. [Google Scholar]
- Jones, R.E.; Lopez, K.H. The male reproductive system. In Human Reproductive Biology, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 67–83. [Google Scholar]
- O’Shaughnessy, P.J. The human Leydig cell. In Male Hypogonadism, 2nd ed.; Winters, S., Huhtaniem, I.T., Eds.; Springer International Publishing AG: Berlin, Germany, 2017; pp. 25–47. [Google Scholar]
- Wallace, E.M.; Groome, N.P.; Riley, S.C.; Parker, A.C.; Wu, F.C.W. Effects of chemotherapy-induced testicular damage on inhibin, gonadotropin, and testosterone secretion: A prospective longitudinal study. J. Clin. Endocrinol. Metab. 1997, 82, 3111–3115. [Google Scholar] [CrossRef] [PubMed]
- Holm, M.; De Meyts, E.R.; Andersson, A.M.; Skakkebæk, N.E. Leydig cell micronodules are a common finding in testicular biopsies from men with impaired spermatogenesis and are associated with decreased testosterone/LH ratio. J. Pathol. 2003, 199, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.-M.; Jørgensen, N.; Frydelund-Larsen, L.; Meyts, E.R.-D.; Skakkebæk, N.E. Impaired Leydig Cell Function in Infertile Men: A Study of 357 Idiopathic Infertile Men and 318 Proven Fertile Controls. J. Clin. Endocrinol. Metab. 2004, 89, 3161–3167. [Google Scholar] [CrossRef] [Green Version]
- De Kretser, D.M. Is Spermatogenic Damage Associated with Leydig Cell Dysfunction? J. Clin. Endocrinol. Metab. 2004, 89, 3158–3160. [Google Scholar] [CrossRef] [Green Version]
- Wikström, A.M.; Dunkel, L. Klinefelter syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 239–250. [Google Scholar] [CrossRef]
- Adamczewska, D.; Slowikowska-Hilczer, J.; Marchlewska, K.; Walczak-Jedrzejowska, R. Features of gonadal dysgenesis and Leydig cell impairment in testes with Sertoli cell-only syndrome. Folia Histochem. Cytobiol. 2020, 58, 73–82. [Google Scholar] [CrossRef]
- Lardone, M.C.; Piottante, A.; Valdevenito, R.; Ebensperger, M.; Castro, A. Histological and hormonal testicular function in oligo/azoospermic infertile men. Andrologia 2012, 45, 379–385. [Google Scholar] [CrossRef]
- Joensen, U.N.; Jørgensen, N.; Meyts, E.R.; De Skakkebæk, N.E. Testicular dysgenesis syndrome and Leydig cell function. Basic Clin. Pharmacol. Toxicol. 2008, 102, 155–161. [Google Scholar] [CrossRef]
- Sharpe, R.M. Androgens and the masculinization programming window: Human–rodent differences. Biochem. Soc. Trans. 2020, 48, 1725–1735. [Google Scholar] [CrossRef]
- Skakkebæk, N.; Meyts, E.R.-D.; Main, K.M. Testicular dysgenesis syndrome: An increasingly common developmental disorder with environmental aspects: Opinion. Hum. Reprod. 2001, 16, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Zirkin, B.R.; Papadopoulos, V. Leydig cells: Formation, function, and regulation. Biol. Reprod. 2018, 99, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Griswold, S.; Behringer, R. Fetal Leydig cell origin and development. Sex. Dev. 2009, 3, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teerds, K.J.; Huhtaniemi, I.T. Morphological and functional maturation of Leydig cells: From rodent models to primates. Hum. Reprod. Updat. 2015, 21, 310–328. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wang, Y.; Ge, R.; Zirkin, B.R. Leydig cell stem cells: Identification, proliferation and differentiation. Mol. Cell. Endocrinol. 2017, 445, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nistal, M.; Paniagua, R.; Regadera, J.; Santamarìa, L.; Amat, P. A quantitative morphological study of human Leydig cells from birth to adulthood. Cell Tissue Res. 1986, 246, 229–236. [Google Scholar] [CrossRef]
- Prince, F.P. Ultrastructural evidence of mature Leydig cells and Leydig cell regression in the neonatal human testis. Anat. Rec. 1990, 228, 405–417. [Google Scholar] [CrossRef]
- Prince, F.P. Ultrastructural evidence of indirect and direct autonomic innervation of human Leydig cells: Comparison of neonatal, childhood and pubertal ages. Cell Tissue Res. 1992, 269, 383–390. [Google Scholar] [CrossRef]
- Prince, F.P. The triphasic nature of Leydig cell development in humans, and comments on nomenclature. J. Endocrinol. 2001, 168, 213–216. [Google Scholar] [CrossRef] [Green Version]
- Habert, R.; Lejeune, H.; Saez, J.M. Origin, differentiation and regulation of fetal and adult Leydig cells. Mol. Cell. Endocrinol. 2001, 179, 47–74. [Google Scholar] [CrossRef]
- Yao, H.H.-C.; Whoriskey, W.; Capel, B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev. 2002, 16, 1433–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyabayashi, K.; Katoh-Fukui, Y.; Ogawa, H.; Baba, T.; Shima, Y.; Sugiyama, N.; Kitamura, K.; Morohashi, K.-I. Aristaless Related Homeobox Gene, Arx, Is Implicated in Mouse Fetal Leydig Cell Differentiation Possibly through Expressing in the Progenitor Cells. PLoS ONE 2013, 8, e68050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codesal, J.; Regadera, J.; Nistal, M.; Regadera-Sejas, J.; Paniagua, R. Involution of human fetal Leydig cells. An immunohistochemical, ultrastructural and quantitative study. J. Anat. 1990, 172, 103–114. [Google Scholar]
- Gautier, C.; Levacher, C.; Saez, J.M.; Habert, R. Expression and regulation of transforming growth factor beta1 mRNA and protein in rat fetal testis in vitro. Biochem. Biophys. Res. Commun. 1997, 236, 135–139. [Google Scholar] [CrossRef]
- Rouiller-Fabre, V.; Carmona, S.; Merhi, R.A.; Cate, R.; Habert, R.; Vigier, B. Effect of Anti-Mullerian Hormone on Sertoli and Leydig Cell Functions in Fetal and Immature Rats. Endocrinology 1998, 139, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Svechnikov, K.; Landreh, L.; Weisser, J.; Izzo, G.; Colón, E.; Svechnikova, I.; Söder, O. Origin, Development and Regulation of Human Leydig Cells. Horm. Res. Paediatr. 2010, 73, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kuopio, T.; Paranko, J.; Pelliniemi, L.J. Basement membrane and epithelial features of fetal-type Leydig cells in rat and human testis. Differentiation 1989, 40, 198–206. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J.; Baker, P.J.; Heikkilä, M.; Vainio, S.; Mcmahon, A.P. Localization of 17beta-hydroxysteroid dehydrogenase/17-ketosteroid reductase isoform expression in the developing mouse testis--androstenedione is the major androgen secreted by fetal/neonatal leydig cells. Endocrinology 2000, 141, 2631–2637. [Google Scholar] [CrossRef]
- Shima, Y.; Miyabayashi, K.; Haraguchi, S.; Arakawa, T.; Otake, H.; Baba, T.; Matsuzaki, S.; Shishido, Y.; Akiyama, H.; Tachibana, T.; et al. Contribution of Leydig and Sertoli Cells to Testosterone Production in Mouse Fetal Testes. Mol. Endocrinol. 2013, 27, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Blaschko, S.D.; Cunha, G.R.; Baskin, L.S. Molecular mechanisms of external genitalia development. Differentiation 2012, 84, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Baskin, L.; Shen, J.; Sinclair, A.; Cao, M.; Liu, X.; Liu, G.; Isaacson, D.; Overland, M.; Li, Y.; Cunha, G.R. Development of the human penis and clitoris. Differentiation 2018, 103, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, P.K.; Burnett, A.L. Development of the Male Reproductive System. In Clinical Urologic Endocrinology; Kavoussi, P., Costabile, R., Salonia, A., Eds.; Springer: London, UK, 2013; pp. 11–24. [Google Scholar]
- Hutson, J.M. Embryology of the human genital tract. In Disorders of Sex Development; Hutson, J.M., Warne, G.L., Grover, S.R., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 11–21. [Google Scholar]
- Ge, R.; Chen, G.; Tanrikut, C.; Hardy, M. Phthalate ester toxicity in Leydig cells: Developmental timing and dosage considerations. Reprod. Toxicol. 2007, 23, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, J.; Hsu, S.Y.; Matsumi, H.; Roh, J.S.; Fu, P.; Wade, J.D.; Bathgate, R.D.A.; Hsueh, A.J.W. INSL3/Leydig insulin-like peptide activates the LGR8 receptor important in testis descent. J. Biol. Chem. 2002, 277, 31283–31286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogatcheva, N.V.; Truong, A.; Feng, S.; Engel, W.; Adham, I.M.; Agoulnik, A.I. GREAT/LGR8 is the only receptor for insulin-like 3 peptide. Mol. Endocrinol. 2003, 17, 2639–2646. [Google Scholar] [CrossRef] [Green Version]
- Ivell, R.; Hartung, S. The molecular basis of cryptorchidism. Mol. Hum. Reprod. 2003, 9, 175–181. [Google Scholar] [CrossRef]
- Bay, K.; Hartung, S.; Ivell, R.; Schumacher, M.; Jürgensen, D.; Jørgensen, N.; Holm, M.; Skakkebaek, N.E.; Andersson, A.-M. Insulin-Like Factor 3 Serum Levels in 135 Normal Men and 85 Men with Testicular Disorders: Relationship to the Luteinizing Hormone-Testosterone Axis. J. Clin. Endocrinol. Metab. 2005, 90, 3410–3418. [Google Scholar] [CrossRef] [Green Version]
- Bay, K.; Matthiesson, K.L.; McLachlan, R.I.; Andersson, A.-M. The Effects of Gonadotropin Suppression and Selective Replacement on Insulin-Like Factor 3 Secretion in Normal Adult Men. J. Clin. Endocrinol. Metab. 2006, 91, 1108–1111. [Google Scholar] [CrossRef] [Green Version]
- Ferlin, A.; Bogatcheva, N.; Gianesello, L.; Pepe, A.; Vinanzi, C.; Agoulnik, A.; Foresta, C. Insulin-like factor 3 gene mutations in testicular dysgenesis syndrome: Clinical and functional characterization. Mol. Hum. Reprod. 2006, 12, 401–406. [Google Scholar] [CrossRef]
- Foresta, C.; Bettella, A.; Vinanzi, C.; Dabrilli, P.; Meriggiola, M.C.; Garolla, A.; Ferlin, A. A Novel Circulating Hormone of Testis Origin in Humans. J. Clin. Endocrinol. Metab. 2004, 89, 5952–5958. [Google Scholar] [CrossRef] [Green Version]
- Berensztein, E.; Belgorosky, A.; De Dávila, M.T.G.; Rivarola, M.A. Basal Testosterone Secretion and Response to Human Luteinizing, Follicle-Stimulating, and Growth Hormones in Culture of Cells Isolated from Testes of Infants and Children. Pediatr. Res. 1995, 38, 592–597. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Li, X.; Li, L.; Chen, H.; Ge, R.-S. Insights into the Development of the Adult Leydig Cell Lineage from Stem Leydig Cells. Front. Physiol. 2017, 8, 430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, D.J.; Sharpe, R.M.; Welsh, M.; Fisken, M.; Scott, H.M.; Hutchison, G.R.; Drake, A.J.; van den Driesche, S. Androgen action in the masculinization programming window and development of male reproductive organs. Int. J. Androl. 2010, 33, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.-S.; Dong, Q.; Sottas, C.M.; Chen, H.; Zirkin, B.R.; Hardy, M.P. Gene Expression in Rat Leydig Cells During Development from the Progenitor to Adult Stage: A Cluster Analysis. Biol. Reprod. 2005, 72, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.-S.; Dong, Q.; Sottas, C.M.; Papadopoulos, V.; Zirkin, B.R.; Hardy, M.P. In search of rat stem Leydig cells: Identification, isolation, and lineage-specific development. Proc. Natl. Acad. Sci. USA 2006, 103, 2719–2724. [Google Scholar] [CrossRef] [Green Version]
- Stanley, E.L.; Johnston, D.S.; Fan, J.; Papadopoulos, V.; Chen, H.; Ge, R.-S.; Zirkin, B.R.; Jelinsky, S.A. Stem Leydig Cell Differentiation: Gene Expression During Development of the Adult Rat Population of Leydig Cells. Biol. Reprod. 2011, 85, 1161–1166. [Google Scholar] [CrossRef] [Green Version]
- Lottrup, G.; Nielsen, J.; Maroun, L.; Møller, L.; Yassin, M.; Leffers, H.; Skakkebæk, N.; Meyts, E.R.-D. Expression patterns of DLK1 and INSL3 identify stages of Leydig cell differentiation during normal development and in testicular pathologies, including testicular cancer and Klinefelter syndrome. Hum. Reprod. 2014, 29, 1637–1650. [Google Scholar] [CrossRef] [Green Version]
- Ariyaratne, S.; Kim, I.; Mills, N.; Mason, I.; Mendis-Handagama, C. Effects of ethane dimethane sulfonate on the functional structure of the adult rat testis. Arch. Androl. 2003, 49, 313–326. [Google Scholar] [CrossRef]
- Davidoff, M.S.; Middendorff, R.; Enikolopov, G.; Riethmacher, D.; Holstein, A.F.; Müller, D. Progenitor cells of the testosterone-producing Leydig cells revealed. J. Cell Biol. 2004, 167, 935–944. [Google Scholar] [CrossRef]
- Hardy, M.P.; Sharma, R.S.; Arambepola, N.K.; Sottas, C.M.; Russell, L.D.; Bunick, D.; Hess, R.; Cooke, P.S. Increased proliferation of Leydig cells induced by neonatal hypothyroidism in the rat. J. Androl. 1996, 17, 231–238. [Google Scholar]
- Sharpe, R.M.; Rivas, A.; Walker, M.; Mckinnell, C.; Fisher, J.S. Effect of neonatal treatment of rats with potent or weak (environmental) oestrogens, or with a GnRH antagonist, on Leydig cell development and function through puberty into adulthood. Int. J. Androl. 2003, 26, 26–36. [Google Scholar] [CrossRef]
- Rajpert-De Meyts, E.; Almstrup, K.; Nielsen, J.E.; Skakkebæk, N.E. The Testis in Childhood between Birth and Puberty. In Atlas on the Human Testis; Ježek, D., Ed.; Springer: London, UK, 2013; pp. 69–75. [Google Scholar]
- Haider, S.G. Cell Biology of Leydig Cells in the Testis. Int. Rev. Cytol. 2004, 233, 181–241. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, I.B.; Kaur, J.; Ge, R.S.; Cooke, P.S.; Yao, H.H. Dynamic changes in fetal Leydig cell populations influence adult Leydig cell populations in mice. FASEB J. 2013, 27, 2657–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaftanovskaya, E.M.; Lopez, C.; Ferguson, L.; Myhr, C.; Agoulnik, A.I. Genetic ablation of androgen receptor signaling in fetal Leydig cell lineage affects Leydig cell functions in adult testis. FASEB J. 2015, 29, 2327–2337. [Google Scholar] [CrossRef] [PubMed]
- Shima, Y.; Matsuzaki, S.; Miyabayashi, K.; Otake, H.; Baba, T.; Kato, S.; Huhtaniemi, I.; Morohashi, K.-I. Fetal Leydig Cells Persist as an Androgen-Independent Subpopulation in the Postnatal Testis. Mol. Endocrinol. 2015, 29, 1581–1593. [Google Scholar] [CrossRef] [Green Version]
- O’Shaughnessy, P.; Willerton, L.; Baker, P. Changes in Leydig Cell Gene Expression during Development in the Mouse. Biol. Reprod. 2002, 66, 966–975. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Ge, R.-S.; Zirkin, B.R. Leydig cells: From stem cells to aging. Mol. Cell. Endocrinol. 2009, 306, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Stévant, I.; Neirijnck, Y.; Borel, C.; Escoffier, J.; Smith, L.B.; Antonarakis, S.E.; Dermitzakis, E.T.; Nef, S. Deciphering Cell Lineage Specification during Male Sex Determination with Single-Cell RNA Sequencing. Cell Rep. 2018, 22, 1589–1599. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Shima, Y.; Miyabayashi, K.; Tokunaga, K.; Sato, T.; Baba, T.; Ohkawa, Y.; Akiyama, H.; Suyama, M.; Morohashi, K.-I. Isolation and Characterization of Fetal Leydig Progenitor Cells of Male Mice. Endocrinology 2016, 157, 1222–1233. [Google Scholar] [CrossRef] [Green Version]
- Pelosi, E.; Koopman, P. Development of the Testis. Ref. Modul. Biomed. Sci. 2017, 204, 112040. [Google Scholar] [CrossRef]
- Su, D.-M.; Feng, Y.; Wang, L.; Wu, Y.-L.; Ge, R.-S.; Ma, X. Influence of fetal Leydig cells on the development of adult Leydig cell population in rats. J. Reprod. Dev. 2018, 64, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Kilcoyne, K.R.; Smith, L.B.; Atanassova, N.; Macpherson, S.; McKinnell, C.; van den Driesche, S.; Jobling, M.S.; Chambers, T.J.G.; De Gendt, K.; Verhoeven, G.; et al. Fetal programming of adult Leydig cell function by androgenic effects on stem/progenitor cells. Proc. Natl. Acad. Sci. USA 2014, 111, E1924–E1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shima, Y.; Morohashi, K.-i. Leydig progenitor cells in fetal testis. Mol. Cell Endocrinol. 2017, 445, 55–64. [Google Scholar] [CrossRef]
- Heinrich, A.; DeFalco, T. Essential roles of interstitial cells in testicular development and function. Andrology 2020, 8, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Bott, R.C.; McFee, R.M.; Clopton, D.T.; Toombs, C.; Cupp, A.S. Vascular Endothelial Growth Factor and Kinase Domain Region Receptor Are Involved in Both Seminiferous Cord Formation and Vascular Development During Testis Morphogenesis in the Rat. Biol. Reprod. 2006, 75, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Combes, A.N.; Wilhelm, D.; Davidson, T.; Dejana, E.; Harley, V.; Sinclair, A.; Koopman, P. Endothelial cell migration directs testis cord formation. Dev. Biol. 2009, 326, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFalco, T.; Bhattacharya, I.; Williams, A.V.; Sams, D.M.; Capel, B. Yolk-sac–derived macrophages regulate fetal testis vascularization and morphogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, E2384–E2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, S.J.; DeFalco, T. Role of the testis interstitial compartment in spermatogonial stem cell function. Reproduction 2017, 154, R151–R162. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Wu, J.; Liu, B.; Jiang, Y.; Chen, W.; Li, J.; He, Q.; He, Z. The roles and mechanisms of Leydig cells and myoid cells in regulating spermatogenesis. Cell Mol. Life Scixp. 2019, 76, 2681–2695. [Google Scholar] [CrossRef]
- Miller, W.L.; Auchus, R.J. The Molecular Biology, Biochemistry, and Physiology of Human Steroidogenesis and Its Disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [Green Version]
- Stocco, D.M. StAR Protein and the Regulation of Steroid Hormone Biosynthesis. Annu. Rev. Physiol. 2001, 63, 193–213. [Google Scholar] [CrossRef]
- Hauet, T.; Liu, J.; Li, H.; Gazouli, M.; Culty, M.; Papadopoulos, V. PBR, Star, and PKA: Partners in cholesterol transport in steroidogenic cells. Endocr. Res. 2002, 28, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.H.; Hales, D.B. Overview of Steroidogenic Enzymes in the Pathway from Cholesterol to Active Steroid Hormones. Endocr. Rev. 2004, 25, 947–970. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.M.; Mason, J.I.; Sharpe, R.M. Steroidogenesis in the Fetal Testis and Its Susceptibility to Disruption by Exogenous Compounds. Endocr. Rev. 2009, 30, 883–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghazadeh, Y.; Zirkin, B.R.; Papadopoulos, V. Pharmacological Regulation of the Cholesterol Transport Machinery in Steroidogenic Cells of the Testis. Vitam. Horm. 2015, 98, 189–227. [Google Scholar] [CrossRef]
- Schiffer, L.; Arlt, W.; Storbeck, K.-H. Intracrine androgen biosynthesis, metabolism and action revisited. Mol. Cell. Endocrinol. 2018, 465, 4–26. [Google Scholar] [CrossRef]
- Walker, W.H. Molecular mechanisms of testosterone action in spermatogenesis. Steroids 2009, 74, 602–607. [Google Scholar] [CrossRef]
- Connan-Perrot, S.; Léger, T.; Lelandais, P.; Desdoits-Lethimonier, C.; David, A.; Fowler, P.; Mazaud-Guittot, S. Six Decades of Research on Human Fetal Gonadal Steroids. Int. J. Mol. Sci. 2021, 22, 6681. [Google Scholar] [CrossRef]
- Walker, W.H. Testosterone signaling and the regulation of spermatogenesis. Spermatogenesis 2011, 1, 116–120. [Google Scholar] [CrossRef] [Green Version]
- De Gent, K.; Swinnen, J.K.; Saunders, P.T.K.; Schoonjas, L.; Dewerchin, M.; Devoss, A.; Tan, K.; Atanassova, N.; Claessens, F.; Lecureuil, C.; et al. Faculty Opinions recommendation of A Sertoli cell-selective knockout of the androgen receptor causes spermatogenic arrest in meiosis. Proc. Natl. Acad. Sci. USA 2004, 101, 1327–1332. [Google Scholar] [CrossRef] [Green Version]
- Welsh, M.; Saunders, P.; Atanassova, N.; Sharpe, R.; Smith, L.B. Androgen action via testicular peritubular myoid cells is essential for male fertility. FASEB J. 2009, 23, 4218–4230. [Google Scholar] [CrossRef] [Green Version]
- O’Shaughnessy, P.J.; Verhoeven, G.; De Gendt, K.; Monteiro, A.; Abel, M.H. Direct Action through the Sertoli Cells Is Essential for Androgen Stimulation of Spermatogenesis. Endocrinology 2010, 151, 2343–2348. [Google Scholar] [CrossRef] [Green Version]
- Willems, A.; Roesl, C.; Mitchell, R.; Milne, L.; Jeffery, N.; Smith, S.; Verhoeven, G.; Brown, P.; Smith, L. Sertoli cell androgen receptor signalling in adulthood is essential for post-meiotic germ cell development. Mol. Reprod. Dev. 2015, 82, 626–627. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-Y.; Yeh, S.-D.; Wang, R.-S.; Yeh, S.; Zhang, C.; Lin, H.-Y.; Tzeng, C.-R.; Chang, C. Differential effects of spermatogenesis and fertility in mice lacking androgen receptor in individual testis cells. Proc. Natl. Acad. Sci. USA 2006, 103, 18975–18980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Nie, R.; Prins, G.S.; Saunders, P.; Katzenellenbogen, B.S.; Hess, R.A. Localization of androgen and estrogen receptors in adult male mouse reproductive tract. J. Androl. 2002, 23, 870–881. [Google Scholar] [PubMed]
- Smith, L.B.; Walker, W.H. The regulation of spermatogenesis by androgens. Semin. Cell Dev. Biol. 2014, 30, 2–13. [Google Scholar] [CrossRef] [Green Version]
- Willems, A.; Batlouni, S.R.; Esnal, A.; Swinnen, J.V.; Saunders, P.T.K.; Sharpe, R.M.; França, L.R.; De Gendt, K.; Verhoeven, G. Selective Ablation of the Androgen Receptor in Mouse Sertoli Cells Affects Sertoli Cell Maturation, Barrier Formation and Cytoskeletal Development. PLoS ONE 2010, 5, e14168. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Greenlee, A.R.; Taub, C.J.; Braun, R.E. Sertoli Cell-Specific Deletion of the Androgen Receptor Compromises Testicular Immune Privilege in Mice. Biol. Reprod. 2011, 85, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Chen, Y.-T.; Yeh, S.-D.; Xu, Q.; Wang, R.-S.; Guillou, F.; Lardy, H.; Yeh, S. Infertility with defective spermatogenesis and hypotestosteronemia in male mice lacking the androgen receptor in Sertoli cells. Proc. Natl. Acad. Sci. USA 2004, 101, 6876–6881. [Google Scholar] [CrossRef] [Green Version]
- Holdcraft, R.W.; Braun, R.E. Androgen receptor function is required in Sertoli cells for the terminal differentiation of haploid spermatids. Development 2004, 131, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.-S.; Yeh, S.; Chen, L.-M.; Lin, H.-Y.; Zhang, C.; Ni, J.; Wu, C.-C.; di Sant’Agnese, P.A.; Demesy-Bentley, K.L.; Tzeng, C.-R.; et al. Androgen Receptor in Sertoli Cell Is Essential for Germ Cell Nursery and Junctional Complex Formation in Mouse Testes. Endocrinology 2006, 147, 5624–5633. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C.; Ádám, M.; Walenta, L.; Schmid, N.; Heikela, H.; Schubert, K.; Flenkenthaler, F.; Dietrich, K.-G.; Gruschka, S.; Arnold, G.J.; et al. Insights into the role of androgen receptor in human testicular peritubular cells. Andrology 2018, 6, 756–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreau, S.; Wolczynski, S.; Galeraud-Denis, I. Aromatase, oestrogens and human male reproduction. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Fietz, D.; Ratzenböck, C.; Hartmann, K.; Raabe, O.; Kliesch, S.; Weidner, W.; Klug, J.; Bergmann, M. Expression pattern of estrogen receptors α and β and G-protein-coupled estrogen receptor 1 in the human testis. Histochem. Cell Biol. 2014, 142, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.F.; Alves, M.G.; Martins, A.D.; Correia, S.; Bernardino, R.L.; Silva, J.; Barros, A.; Sousa, M.; Cavaco, J.E.; Socorro, S. Expression pattern of G protein-coupled receptor 30 in human seminiferous tubular cells. Gen. Comp. Endocrinol. 2014, 201, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Bernardino, R.; Alves, M.G.; Silva, J.; Barros, A.; Ferraz, L.; Sousa, M.; Sa, R.; Oliveira, P.F. Expression of Estrogen Receptors Alpha (ER-α), Beta (ER-β), and G Protein-Coupled Receptor 30 (GPR30) in Testicular Tissue of Men with Klinefelter Syndrome. Horm. Metab. Res. 2016, 48, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Abney, T.O. The potential roles of estrogens in regulating Leydig cell development and function: A review. Steroids 1999, 64, 610–617. [Google Scholar] [CrossRef]
- Chen, B.; Chen, D.; Jiang, Z.; Li, J.; Liu, S.; Dong, Y.; Yao, W.; Akingbemi, B.; Ge, R.; Li, X. Effects of Estradiol and Methoxychlor on Leydig Cell Regeneration in the Adult Rat Testis. Int. J. Mol. Sci. 2014, 15, 7812–7826. [Google Scholar] [CrossRef] [Green Version]
- Vaucher, L.; Funaro, M.G.; Mehta, A.; Mielnik, A.; Bolyakov, A.; Prossnitz, E.R.; Schlegel, P.N.; Paduch, D. Activation of GPER-1 Estradiol Receptor Downregulates Production of Testosterone in Isolated Rat Leydig Cells and Adult Human Testis. PLoS ONE 2014, 9, e92425. [Google Scholar] [CrossRef]
- Pentikäinen, V.; Erkkilä, K.; Suomalainen, L.; Parvinen, M.; Dunkel, L. Estradiol Acts as a Germ Cell Survival Factor in the Human Testis in Vitro. J. Clin. Endocrinol. Metab. 2000, 85, 2057–2067. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.M.; O’Donnell, L.; Simpson, E.R.; Jones, M.E.E. The Phenotype of the Aromatase Knockout Mouse Reveals Dietary Phytoestrogens Impact Significantly on Testis Function. Endocrinology 2002, 143, 2913–2921. [Google Scholar] [CrossRef]
- Walczak-Jędrzejowska, R.; Slowikowska-Hilczer, J.; Marchlewska, K.; Kula, K. Maturation, proliferation and apoptosis of seminal tubule cells at puberty after administration of estradiol, follicle stimulating hormone or both. As. J. Androl. 2008, 10, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; Sirianni, R.; Delalande, C.; Silandre, D.; Bois, C.; Andò, S.; Maggiolini, M.; Carreau, S.; Pezzi, V. 17 beta-estradiol activates rapid signaling pathways involved in rat pachytene spermatocytes apoptosis through GPR30 and ER alpha. Mol. Cell Endocrinol. 2010, 320, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Royer, C.; Lucas, T.F.G.; Lazari, M.F.M.; Porto, C.S. 17Beta-Estradiol Signaling and Regulation of Proliferation and Apoptosis of Rat Sertoli Cells. Biol. Reprod. 2012, 86, 108. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; Sirianni, R.; Casaburi, I.; Pezzi, V. Role of estrogen receptors and G protein-coupled estrogen receptor in regulation of hypothalamus-pituitary-testis axis and spermatogenesis. Front Endocrinol. (Lausanne) 2014, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Guarducci, E.; Nuti, F.; Becherini, L.; Rotondi, M.; Balercia, G.; Forti, G.; Krausz, C. Estrogen receptor α promoter polymorphism: Stronger estrogen action is coupled with lower sperm count. Hum. Reprod. 2006, 21, 994–1001. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.W.; Nie, R.; Carnes, K.; Zhou, Q.; Sharief, N.A.Q.; Hess, R.A. The antiestrogen ICI 182,780 induces early effects on the adult male mouse reproductive tract and long-term decreased fertility without testicular atrophy. Reprod. Biol. Endocrinol. 2003, 1, 57. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.M.; O’Donnell, L.; Jones, M.E.E.; Meachem, S.J.; Boon, W.C.; Fisher, C.R.; Graves, K.H.; McLachlan, R.I.; Simpson, E.R. Impairment of spermatogenesis in mice lacking a functional aromatase (cyp 19) gene. Proc. Natl. Acad. Sci. USA 1999, 96, 7986–7991. [Google Scholar] [CrossRef] [Green Version]
- Griffeth, R.J.; Bianda, V.; Nef, S. The emerging role of insulin-like growth factors in testis development and function. Basic Clin. Androl. 2014, 24, 12. [Google Scholar] [CrossRef] [Green Version]
- Cannarella, R.; Condorelli, R.A.; La Vignera, S.; Calogero, A.E. Effects of the insulin-like growth factor system on testicular differentiation and function: A review of the literature. Andrology 2018, 6, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Cho, K.-S.; Li, Y.; Tchedre, K.; Antolik, C.; Ma, J.; Chew, J.; Utheim, T.P.; Huang, X.A.; Yu, H.; et al. IGFBPL1 Regulates Axon Growth through IGF-1-mediated Signaling Cascades. Sci. Rep. 2018, 8, 2054. [Google Scholar] [CrossRef] [Green Version]
- Hermann, B.P.; Cheng, K.; Singh, A.; Roa-De La Cruz, L.; Mutoji, K.N.; Chen, I.-C.; Gildersleeve, H.; Lehle, J.D.; Mayo, M.; Westernströer, B.; et al. The Mammalian Spermatogenesis Single-Cell Transcriptome, from Spermatogonial Stem Cells to Spermatids. Cell Rep. 2018, 25, 1650–1667.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neirijnck, Y.; Papaioannou, M.D.; Nef, S. The Insulin/IGF System in Mammalian Sexual Development and Reproduction. Int. J. Mol. Sci. 2019, 20, 4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neirijnck, Y.; Calvel, P.; Kilcoyne, K.R.; Kühne, F.; Stévant, I.; Griffeth, R.J.; Pitetti, J.-L.; Andric, S.A.; Hu, M.-C.; Pralong, F.; et al. Insulin and IGF1 receptors are essential for the development and steroidogenic function of adult Leydig cells. FASEB J. 2018, 32, 3321–3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thackare, H.; Nicholson, H.D.; Whittington, K. Oxytocin—Its role in male reproduction and new potential therapeutic uses. Hum. Reprod. Updat. 2006, 12, 437–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assinder, S.J.; Rezvani, A.; Nicholson, H.D. Oxytocin promotes spermiation and sperm transfer in the mouse. Int. J. Androl. 2002, 25, 19–26. [Google Scholar] [CrossRef]
- Kawamura, K.; Kumagai, J.; Sudo, S.; Chun, S.-Y.; Pisarska, M.; Morita, H.; Toppari, J.; Fu, P.; Wade, J.D.; Bathgate, R.A.D.; et al. Paracrine regulation of mammalian oocyte maturation and male germ cell survival. Proc. Natl. Acad. Sci. USA 2004, 101, 7323–7328. [Google Scholar] [CrossRef] [Green Version]
- Amory, J.K.; Page, S.T.; Anawalt, B.D.; Coviello, A.D.; Matsumoto, A.M.; Bremner, W.J. Elevated End-of-Treatment Serum INSL3 Is Associated with Failure to Completely Suppress Spermatogenesis in Men Receiving Male Hormonal Contraception. J. Androl. 2007, 28, 548–554. [Google Scholar] [CrossRef]
- Francomano, D.; Sanguigni, V.; Capogrosso, P.; Deho, F.; Antonini, G. New Insight into Molecular and Hormonal Connection in Andrology. Int. J. Mol. Sci. 2021, 22, 11908. [Google Scholar] [CrossRef]
- Meinhardt, A.; Bacher, M.; McFarlane, J.R.; Metz, C.N.; Seitz, J.; Hedger, M.P.; de Kretser, D.M.; Bucala, M. Macrophage migration inhibitory factor production by Leydig cells: Evidence for a role in the regulation of testicular function. Endocrinology 1996, 137, 5090–5095. [Google Scholar] [CrossRef]
- Wennemuth, G.; Aumüller, G.; Bacher, M.; Meinhardt, A. Macrophage Migration Inhibitory Factor-Induced Ca2+ Response in Rat Testicular Peritubular Cells. Biol. Reprod. 2000, 62, 1632–1639. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-Q.; He, X.-Z.; Zhang, J.-S.; Wang, R.-A.; Zhou, J.; Xu, R.-J. Stage-specific localization of transforming growth factor beta1 and beta3 and their receptors during spermatogenesis in men. As. J. Androl. 2004, 6, 105–109. [Google Scholar]
- Gonzalez, C.R.; Gonzalez, B.; Rulli, S.B.; Huhtaniemi, I.; Calandra, R.S.; Gonzalez-Calvar, S.I. TGF-.BETA.1 System in Leydig Cells. Part I: Effect of hCG and Progesterone. J. Reprod. Dev. 2010, 56, 389–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, C.R.; Calandra, R.S.; Gonzalez-Calvar, I.S. Influence of the photoperiod on TGF-β1 and p15 expression in hamster Leydig cells. Reprod. Biol. 2012, 12, 201–218. [Google Scholar] [CrossRef]
- Hart, P.J.; Deep, S.; Taylor, A.B.; Shu, Z.; Hinck, C.S.; Hinck, A.P. Crystal structure of the human TβR2 ectodomain–TGF-β3 complex. Nat. Struct. Biol. 2002, 9, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Ebner, R.; Chen, R.-H.; Lawler, S.; Zioncheck, T.; Derynck, R. Determination of Type I Receptor Specificity by the Type II Receptors for TGF-β or Activin. Science 1993, 262, 900–902. [Google Scholar] [CrossRef] [PubMed]
- Loveland, K.L.; Klein, B.; Pueschl, D.; Indumathy, S.; Bergmann, M.; Loveland, B.E.; Hedger, M.P.; Schuppe, H.-C. Cytokines in Male Fertility and Reproductive Pathologies: Immunoregulation and Beyond. Front. Endocrinol. 2017, 8, 307. [Google Scholar] [CrossRef]
- Eldamnhoury, E.M.; Elatrash, G.A.; Rashwan, H.M.; El-Sakka, A.I. Association between leukocytospermia and semen interleukin-6 and tumor necrosis factor-alpha in infertile men. Andrology 2018, 6, 775–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boivin, J.; Bunting, L.; Collins, J.A.; Nygren, K.G. International estimates of infertility prevalence and treatment-seeking: Potential need and demand for infertility medical care. Hum. Reprod. 2007, 22, 1506–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungwirth, A.; Giwercman, A.; Tournaye, H.; Diemer, T.; Kopa, Z.; Dohle, G.; Krausz, C.; EAU Working Group on Male Infertility. European Association of Urology Guidelines on Male Infertility: The 2012 Update. Eur. Urol. 2012, 62, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Massart, A.; Lissens, W.; Tournaye, H.; Stouffs, K. Genetic causes of spermatogenic failure. As. J. Androl. 2012, 14, 40–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, K.; Enatsu, N.; Fujisawa, M. Management of non-obstructive azoospermia. Reprod. Med. Biol. 2016, 15, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Buck Louis, G.M.; Toppari, J.; Andersson, A.-M.; Eisenberg, M.L.; Jensen, T.K.; Jørgensen, N.; Swan, S.H.; Sapra, K.J.; et al. Male Reproductive Disorders and Fertility Trends: Influences of Environment and Genetic Susceptibility. Physiol. Rev. 2016, 96, 55–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteves, S.C.; Agarwal, A. The azoospermic male: Current knowledge and future perspectives. Clinics 2013, 68, 1. [Google Scholar] [CrossRef]
- Tüttelmann, F.; Werny, F.; Cooper, T.G.; Kliesch, S.; Simoni, M.; Nieschlag, E. Clinical experience with azoospermia: Aetiology and chances for spermatozoa detection upon biopsy. Int. J. Androl. 2011, 34, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Olesen, I.A.; Andersson, A.-M.; Aksglaede, L.; Skakkebaek, N.E.; Meyts, E.R.; Joergensen, N.; Juul, A. Clinical, genetic, biochemical, and testicular biopsy findings among 1,213 men evaluated for infertility. Fertil. Steril. 2017, 107, 74–82.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, P.N. Causes of azoospermia and their management. Reprod. Fertil. Dev. 2004, 16, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R. Medical management of non-obstructive azoospermia. Clinics 2013, 68, 75–79. [Google Scholar] [CrossRef]
- Jarvi, K.; Lo, K.; Fischer, A.; Grantmyre, J.; Zini, A.; Chow, V.; Mak, V. CUA Guideline: The workup of azoospermic males. Can. Urol. Assoc. J. 2010, 4, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Berookhim, B.M.; Schlegel, P.N. Azoospermia due to Spermatogenic Failure. Urol. Clin. N. Am. 2014, 41, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Punjani, N.; Schlegel, P. Reproductive Chances of Men with Azoospermia Due to Spermatogenic Dysfunction. J. Clin. Med. 2021, 10, 1400. [Google Scholar] [CrossRef] [PubMed]
- Fakhro, K.A.; Elbardisi, H.; Arafa, M.; Robay, A.; Rodriguez-Flores, J.L.; Mezey, J.G.; Crystal, R.G.; Al-Shakaki, A.; Syed, N.; Khalil, C.A.; et al. Point-of-care whole-exome sequencing of idiopathic male infertility. Genet. Med. 2018, 20, 1365–1373. [Google Scholar] [CrossRef]
- Tang, Z.-R.; Xu, X.-L.; Deng, S.-L.; Lian, Z.-X.; Yu, K. Oestrogenic Endocrine Disruptors in the Placenta and the Fetus. Int. J. Mol. Sci. 2020, 21, 1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamischke, A.; Baumgardt, A.; Ju, J.; Horst, J.; Nieschlag, E. Clinical and Diagnostic Features of Patients with Suspected Klinefelter Syndrome. J. Androl. 2003, 24, 41–48. [Google Scholar] [PubMed]
- Blagosklonova, O.; Fellmann, F.; Clavequin, M.-C.; Roux, C.; Bresson, J.-L. AZFa deletions in Sertoli cell-only syndrome: A retrospective study. Mol. Hum. Reprod. 2000, 6, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Hopps, C.V.; Mielnik, A.; Goldstein, M.; Palermo, G.D.; Rosenwaks, Z.; Schlegel, P.N. Detection of sperm in men with Y chromosome microdeletions of the AZFa, AZFb and AZFc regions. Hum. Reprod. 2003, 18, 1660–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Ma, M.Y.; Xiao, C.Y.; Li, L.; Li, S.W.; Zhang, S.Z. Massive deletion in AZFb/b+c and azoospermia with Sertoli cell only and/or maturation arrest. Int J. Androl. 2008, 31, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.J.; Masson, P.; Mielnik, A.; Marean, M.B.; Schlegel, P.N.; Paduch, D.A. A decade of experience emphasizes that testing for Y microdeletions is essential in American men with azoospermia and severe oligozoospermia. Fertil. Steril. 2010, 94, 1753–1756. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Hasuike, S.; Yogev, L.; Maduro, M.R.; Ishikawa, M.; Westphal, H.; Lamb, D.J. Azoospermia in patients heterozygous for a mutation in SYCP. Lancet 2003, 362, 1714–1719. [Google Scholar] [CrossRef]
- Bashamboo, A.; Ferraz-De-Souza, B.; Loureno, D.; Lin, L.; Sebire, N.J.; Montjean, D.; Bignon-Topalovic, J.; Mandelbaum, J.; Siffroi, J.-P.; Christin-Maitre, S.; et al. Human male infertility associated with mutations in NR5A1 encoding steroidogenic factor. Am. J. Hum. Genet. 2010, 87, 505–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Silber, S.J.; Leu, N.A.; Oates, R.D.; Marszalek, J.D.; Skaletsky, H.; Brown, L.G.; Rozen, S.G.; Page, D.C.; Wang, P.J. TEX 11 is mutated in infertile men with azoospermia and regulates genome-wide recombination rates in mouse. EMBO Mol. Med. 2015, 7, 1198–1210. [Google Scholar] [CrossRef] [PubMed]
- Yatsenko, A.N.; Georgiadis, A.P.; Röpke, A.; Berman, A.J.; Jaffe, T.; Olszewska, M.; Westernströer, B.; Sanfilippo, J.; Kurpisz, M.; Rajkovic, A.; et al. X-Linked TEX11 Mutations, Meiotic Arrest, and Azoospermia in Infertile Men. N. Engl. J. Med. 2015, 372, 2097–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Khelifa, M.; Ghieh, F.; Boudjenah, R.; Hue, C.; Fauvert, D.; Dard, R.; Garchon, H.J.; Vialard, F. A MEI1 homozygous missense mutation associated with meiotic arrest in a consanguineous family. Hum. Reprod. 2018, 33, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, J.; Dai, L.; Zhu, Y.; Hu, H.; Tan, L.; Chen, T.; Liang, D.; He, J.; Tu, M.; et al. Original article: XRCC2 mutation causes meiotic arrest, azoospermia and infertility. J. Med. Genet. 2018, 55, 628. [Google Scholar] [CrossRef] [PubMed]
- Ghieh, F.; Mitchell, V.; Mandon-Pepin, B.; Vialard, F. Genetic defects in human azoospermia. Basic Clin. Androl. 2019, 29, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koc, G.; Ozdemir, A.A.; Girgin, G.; Akbal, C.; Kirac, D.; Avcilar, T.; Guney, A.I. Male infertility in Sertoli cell-only syndrome: An investigation of autosomal gene defects. Int. J. Urol. 2018, 26, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babakhanzadeh, E.; Khodadadian, A.; Rostami, S.; Alipourfard, I.; Aghaei, M.; Nazari, M.; Hosseinnia, M.; Mehrjardi, M.Y.V.; Jamshidi, Y.; Ghasemi, N. Testicular expression of TDRD1, TDRD5, TDRD9 and TDRD12 in azoospermia. BMC Med. Genet. 2020, 21, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Martín, M.C.; Castilla, J.A.; Palomino-Morales, R.J.; Carmona, F.D. Genetic Landscape of Nonobstructive Azoospermia and New Perspectives for the Clinic. J. Clin. Med. 2020, 9, 300. [Google Scholar] [CrossRef] [Green Version]
- Thakker, S.; Persily, J.; Najari, B.B. Kallman syndrome and central non-obstructive azoospermia. Best Pr. Res. Clin. Endocrinol. Metab. 2020, 34, 101475. [Google Scholar] [CrossRef]
- Akarsu, C.; Caglar, G.; Vicdan, K.; Isik, A.; Tuncay, G. Pregnancies achieved by testicular sperm recovery in male hypogonadotrophic hypogonadism with persistent azoospermia. Reprod. Biomed. Online 2009, 18, 455–459. [Google Scholar] [CrossRef]
- Esteves, S.; Papanikolaou, V. Clinical efficacy, safety and tolerability of recombinant human chorionic gonadotropin to restore spermatogenesis and androgen production of hypogonadotropic hypogonadal men. Fertil. Steril. 2011, 96, S230. [Google Scholar] [CrossRef]
- Fraietta, R.; Zylberstejn, D.S.; Esteves, S.C. Hypogonadotropic Hypogonadism Revisited. Clinics 2013, 68, 81–88. [Google Scholar] [CrossRef]
- Singh, P.; Cugati, G.; Singh, M.; Singh, A.K. Hyperprolactinemia: An often missed cause of male infertility. J. Hum. Reprod. Sci. 2011, 4, 102–103. [Google Scholar] [CrossRef] [PubMed]
- Dabbous, Z.; Atkin, S.L. Hyperprolactinaemia in male infertility: Clinical case scenarios. Arab J. Urol. 2017, 16, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiman, J.; Griffin, J.E. The Frequency of Androgen Receptor Deficiency in Infertile Men. J. Clin. Endocrinol. Metab. 1982, 54, 725–732. [Google Scholar] [CrossRef]
- Akin, J.W.; Behzadian, A.; Tho, S.P.; McDonough, P.G. Evidence for a partial deletion in the androgen receptor gene in a phenotypic male with azoospermia. Am. J. Obstet. Gynecol. 1991, 165, 1891–1894. [Google Scholar] [CrossRef]
- Huff, D.S.; Fenig, D.M.; Canning, D.A.; Carr, M.G.; Zderic, S.A.; Snyder, H.M. Abnormal germ cell development in cryptorchidism. Horm. Res. 2001, 55, 11–17. [Google Scholar] [CrossRef]
- Sijstermans, K.; Hack, W.W.M.; Meijer, R.W.; Van Der Voort-Doedens, L.M. The frequency of undescended testis from birth to adulthood: A review. Int. J. Androl. 2008, 31, 1–11. [Google Scholar] [CrossRef]
- Fedder, J. History of cryptorchidism and ejaculate volume as simple predictors for the presence of testicular sperm. Syst. Biol. Reprod. Med. 2011, 57, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Mostafa, T.; Anis, M.T.; El-Nashar, A.; Imam, H.; Othman, I.A. Varicocelectomy reduces reactive oxygen species levels and increases antioxidant activity of seminal plasma from infertile men with varicocele. Int. J. Androl. 2001, 24, 261–265. [Google Scholar] [CrossRef]
- Poulakis, V.; Ferakis, N.; De Vries, R.; Witzsch, U.; Becht, E. Induction of spermatogenesis in men with azoospermia or severe oligoteratoasthenospermia after antegrade internal spermatic vein sclerotherapy for the treatment of varicocele. As. J. Androl. 2006, 8, 613–619. [Google Scholar] [CrossRef]
- Gat, Y.; Gornish, M.; Perlow, A.; Chakraborty, J.; Levinger, U.; Ben-Shlomo, I.; Pasqualotto, F. Azoospermia and Sertoli-cell-only syndrome: Hypoxia in the sperm production site due to impairment in venous drainage of male reproductive system. Andrologia 2010, 42, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Inci, K.; Gunay, L.M. The role of varicocele treatment in the management of non-obstructive azoospermia. Clinics 2013, 68, 89. [Google Scholar] [CrossRef]
- Zampieri, N.; Bosaro, L.; Costantini, C.; Zaffagnini, S.; Zampieri, G. Relationship between Testicular Sperm Extraction and Varicocelectomy in Patients with Varicocele and Nonobstructive Azoospermia. Urology 2013, 82, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Kavoussi, P.K.; Hunn, C.; Gilkey, M.S.; Chen, S.-H.; Kavoussi, K.M.; Wininger, J.D.; Kavoussi, S.K. Sertoli cell only syndrome induced by a varicocele. Transl. Androl. Urol. 2019, 8, 405–408. [Google Scholar] [CrossRef]
- Huyghe, E.; Matsuda, T.; Daudin, M.; Chevreau, C.; Bachaud, J.M.; Plante, P.; Bujan, L.; Thonneau, P. Fertility after testicular cancer treatments: Results of a large multicenter study. Cancer 2004, 100, 732–737. [Google Scholar] [CrossRef]
- Gandini, L.; Sgrò, P.; Lombardo, F.; Paoli, D.; Culasso, F.; Toselli, L.; Tsamatropoulos, P.; Lenzi, A. Effect of chemo- or radiotherapy on sperm parameters of testicular cancer patients. Hum. Reprod. 2006, 21, 2882–2889. [Google Scholar] [CrossRef]
- Green, D.M.; Kawashima, T.; Stovall, M.; Leisenring, W.; Sklar, C.A.; Mertens, A.C.; Donaldson, S.S.; Byrne, J.; Robison, L.L. Fertility of Male Survivors of Childhood Cancer: A Report from the Childhood Cancer Survivor Study. J. Clin. Oncol. 2010, 28, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Greenfield, D.M.; Walters, S.J.; Coleman, R.E.; Hancock, B.W.; Eastell, R.; Davies, H.A.; Snowden, J.A.; Derogatis, L.; Shalet, S.M.; Ross, R.J.M. Prevalence and Consequences of Androgen Deficiency in Young Male Cancer Survivors in a Controlled Cross-Sectional Study. J. Clin. Endocrinol. Metab. 2007, 92, 3476–3482. [Google Scholar] [CrossRef] [Green Version]
- Benedict, C.; Shuk, E.; Ford, J.S. Fertility Issues in Adolescent and Young Adult Cancer Survivors. J. Adolesc. Young Adult Oncol. 2016, 5, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Chiba, K.; Yamaguchi, K.; Li, F.; Ando, M.; Fujisawa, M. Finasteride-associated male infertility. Fertil. Steril. 2011, 95, 1786.e9–1786.e11. [Google Scholar] [CrossRef]
- Samplaski, M.K.; Nangia, A.K. Adverse effects of common medications on male fertility. Nat. Rev. Urol. 2015, 12, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Shang, X.; Zhang, Z.; Jing, H.; Shao, J.; Fei, Q.; Rayburn, E.R.; Li, H. FDA-approved medications that impair human spermatogenesis. Oncotarget 2017, 8, 10714–10725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakhem, G.A.; Motosko, C.C.; Mu, E.W.; Ho, R.S. Infertility and teratogenicity after paternal exposure to systemic dermatologic medications: A systematic review. J. Am. Acad. Dermatol. 2019, 80, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Bermas, B.L. Paternal safety of anti-rheumatic medications. Best Pr. Res. Clin. Obstet. Gynaecol. 2020, 64, 77–84. [Google Scholar] [CrossRef]
- Mylchreest, E.; Sar, M.; Wallace, D.G.; Foster, P.M. Fetal testosterone insufficiency and abnormal proliferation of Leydig cells and gonocytes in rats exposed to di(n-butyl) phthalate. Reprod. Toxicol. 2002, 16, 19–28. [Google Scholar] [CrossRef]
- Barlow, N.J.; Foster, P.M.D. Pathogenesis of male reproductive tract lesions from gestation through adulthood following in utero exposure to Di(n-butyl) phthalate. Toxicol. Pathol. 2003, 31, 397–410. [Google Scholar]
- Fisher, J.S.; MacPherson, S.; Marchetti, N.; Sharpe, R.M. Human ‘testicular dysgenesis syndrome’: A possible model using in-utero exposure of the rat to dibutyl phthalate. Hum. Reprod. 2003, 18, 1383–1394. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.-X.; Lian, Q.-Q.; Ge, R.-S.; Hardy, D.O.; Li, X.-K. Phthalate-induced testicular dysgenesis syndrome: Leydig cell influence. Trends Endocrinol. Metab. 2009, 20, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Ren, X.; Zhang, T.; Zhou, X.; Chen, X.; Lu, H.; Zhou, X.; Zhang, X.; Wang, S.; Qin, C.; et al. Comprehensive Analysis of the Association Between Human Non-obstructive Azoospermia and Plasticisers via Single-Cell and Traditional RNA Sequencing Methods. Expo. Health 2022, 1–14. [Google Scholar] [CrossRef]
- Liu, C.; Duan, W.; Li, R.; Xu, S.; Zhang, L.; Chen, C.; He, M.; Lu, Y.; Wu, H.; Pi, H.; et al. Exposure to bisphenol A disrupts meiotic progression during spermatogenesis in adult rats through estrogen-like activity. Cell Death Dis. 2013, 4, e676. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.S.; Kwon, W.-S.; Lee, J.-S.; Yoon, S.-J.; Ryu, B.-Y.; Pang, M.-G. Bisphenol-A Affects Male Fertility via Fertility-related Proteins in Spermatozoa. Sci. Rep. 2015, 5, 9169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wen, Z.; Wang, Y.; Mo, J.; Zhong, Y.; Ge, R.-S. Bisphenols and Leydig Cell Development and Function. Front. Endocrinol. 2020, 11, 447. [Google Scholar] [CrossRef] [PubMed]
- Pajarinen, J.T.; Karhunen, P.J. Spermatogenic Arrest and “Sertoli Cell-Only” Syndrome--Common Alcohol-Induced Disorders of the Human Testis. Obstet. Gynecol. Surv. 1994, 17, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Sermondade, N.; Elloumi, H.; Berthaut, I.; Mathieu, E.; Delarouzière, V.; Ravel, C.; Mandelbaum, J. Progressive alcohol-induced sperm alterations leading to spermatogenic arrest, which was reversed after alcohol withdrawal. Reprod. Biomed. Online 2010, 20, 324–327. [Google Scholar] [CrossRef] [Green Version]
- Masuda, H.; Inamoto, T.; Azuma, H.; Katsuoka, Y.; Tawara, F. Successful testicular sperm extraction in an azoospermic man with postpubertal mumps orchitis. Hinyokika Kiyo. Acta Urol. J. 2011, 57, 529–530. [Google Scholar]
- Zhang, S.; An, Y.; Li, J.; Guo, J.; Zhou, G.; Li, J.; Xu, Y. Relation between the testicular sperm assay and sex hormone level in patients with azoospermia induced by mumps. Int. J. Clin. Exp. Med. 2015, 8, 21669–21673. [Google Scholar]
- Schuppe, H.-C.; Pilatz, A.; Hossain, H.; Diemer, T.; Wagenlehner, F.; Weidner, W. Urogenital Infection as a Risk Factor for Male Infertility. Dtsch Arztebl Int. 2017, 114, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Fijak, M.; Pilatz, A.; Hedger, M.P.; Nicolas, N.; Bhushan, S.; Michel, V.; Tung, K.S.K.; Schuppe, H.-C.; Meinhastd, A. Infectious, inflammatory and ‘autoimmune’ male factor infertility: How do rodent models inform clinical practice? Hum. Reprod. 2018, 24, 416. [Google Scholar] [CrossRef] [Green Version]
- Gacci, M.; Coppi, M.; Baldi, E.; Sebastianelli, A.; Zaccaro, C.; Morselli, S.; Pecorano, S.; Manera, A.; Nicoletti, S.; Liaci, A.; et al. Semen impairment and occurrence of SARS-CoV-2 virus in semen after recovery from COVID-19. Hum. Reprod. 2021, 36, 1520–1529. [Google Scholar] [CrossRef]
- Hagiuda, J.; Ishikawa, H.; Hanawa, Y.; Marumo, K. Recovery from azoospermia caused by a testicular injury: A case report. Andrologia 2014, 46, 447–448. [Google Scholar] [CrossRef]
- Alawamlh, O.A.H.; Flannigan, R.; Hayden, R.; Goldstein, M.; Li, P.S.; Lee, R.K. Testicular Torsion and Spermatogenesis. Adv. Exp. Med. Biol. 2021, 1288, 287–306. [Google Scholar] [CrossRef]
- Li, Z.; Tian, J.; Cui, G.; Wang, M.; Yu, D. Effects of local testicular heat treatment on Leydig cell hyperplasia and testosterone biosynthesis in rat testes. Reprod. Fertil. Dev. 2015, 28, 1424–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziaeipour, S.; Piryaei, A.; Aliaghaei, A.; Nazarian, H.; Naserzadeh, P.; Ebrahimi, V.; Abdi, S.; Shahi, F.; Ahmadi, H.; Fathabadi, F.F.; et al. Chronic scrotal hyperthermia induces azoospermia and severe damage to testicular tissue in mice. Acta Histochem. 2021, 123, 151712. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhang, J.; Xiong, J.; Ma, C.; Yang, B.; Li, H. New insights into the potential mechanisms of spermatogenic failure in patients with idiopathic azoospermia. Mol. Hum. Reprod. 2020, 26, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Brannigan, R.E.; Das, A.; Halpern, J.A.; Darves-Bornoz, A.L.; Patel, M.; Wren, J.; Keeter, M.K. Sperm retrieval success and testicular histopathology in idiopathic nonobstructive azoospermia. As. J. Androl. 2020, 22, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, J.S.; Petit, C.; Rose, R.S.; Frapsauce, C.; Dijols, L.; Rigot, J.M.; Guérif, F. Non-obstructive idiopathic azoospermia vs azoospermia with antecedents of cryptorchidism: Ways and probabilities of becoming parents. Basic Clin. Androl. 2021, 31, 3. [Google Scholar] [CrossRef] [PubMed]
- Baksi, A.; Vasan, S.S.; Dighe, R.R. DNA Flow cytometric analysis of the human testicular tissues to investigate the status of spermatogenesis in azoospermic patients. Sci. Rep. 2018, 8, 11117. [Google Scholar] [CrossRef]
- Goluža, T.; Boscanin, A.; Cvetko, J.; Kozina, V.; Kosovic, M.; Bernat, M.M.; Kasum, M.; Kastelan, Z.; Jezek, D. Macrophages and Leydig Cells in Testicular Biopsies of Azoospermic Men. BioMed Res. Int. 2014, 2014, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Madsen, A.; Oehme, N.B.; Roelants, M.; Bruserud, I.S.; Eide, G.E.; Viste, K.; Bjerknes, R.; Almås, B.; Rosendahl, K.; Sagen, J.V.; et al. Testicular Ultrasound to Stratify Hormone References in a Cross-Sectional Norwegian Study of Male Puberty. J. Clin. Endocrinol. Metab. 2020, 105, 1888–1898. [Google Scholar] [CrossRef]
- Ruiz-Olvera, S.F.; Rajmil, O.; Sanchez-Curbelo, J.R.; Vinay, J.; Rodriguez-Espinosa, J.; Ruiz-Castañé, E. Association of serum testosterone levels and testicular volume in adult patients. Andrologia 2018, 50, 3. [Google Scholar] [CrossRef]
- Bergmann, M.; Ježek, D. Damage of Spermatogenesis. In Atlas on the Human Testis; Ježek, D., Ed.; Springer: London, UK, 2013; pp. 99–112. [Google Scholar]
- Esteves, S.C.; Prudencio, C.; Seol, B.; Verza, S.; Knoedler, C.; Agarwal, A. Comparison of sperm retrieval and reproductive outcome in azoospermic men with testicular failure and obstructive azoospermia treated for infertility. As. J. Androl. 2014, 16, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Cito, G.; Coccia, M.E.; Picone, R.; Nesi, G.; Cocci, A.; Dabizzi, S.; Garaffa, G.; Fucci, R.; Falcone, P.; Bertocci, F.; et al. Novel method of histopathological analysis after testicular sperm extraction in patients with nonobstructive and obstructive azoospermia. Clin. Exp. Reprod. Med. 2018, 45, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Toksöz, S.; Kizilkan, Y. Comparison of the Histopathological Findings of Testis Tissues of Non-Obstructive Azoospermia with the Findings after Microscopic Testicular Sperm Extraction. Urol. J. 2019, 16, 212–215. [Google Scholar] [PubMed]
- Nistal, M.; Paniagua, R.; Riestra, M.L.; Reyes-Múgica, M.; Cajaiba, M.M. Bilateral prepubertal testicular biopsies predict significance of cryptorchidism-associated mixed testicular atrophy, and allow assessment of fertility. Am. J. Surg. Pathol. 2007, 31, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Van Casteren, N.J.; Looijenga, L.H.J.; Dohle, G.R. Testicular microlithiasis and carcinoma in situ overview and proposed clinical guideline. Int. J. Androl. 2009, 32, 279–287. [Google Scholar] [CrossRef]
- Barbonetti, A.; Martorella, A.; Minaldi, E.; D’Andrea, S.; Bardhi, D.; Castellini, C.; Francavilla, F.; Francavilla, S. Testicular Cancer in Infertile Men with and Without Testicular Microlithiasis: A Systematic Review and Meta-Analysis of Case-Control Studies. Front. Endocrinol. 2019, 10, 164. [Google Scholar] [CrossRef]
- Eisenberg, M.L.; Betts, P.; Herder, D.; Lamb, D.J.; Lipshultz, L.I. Increased risk of cancer among azoospermic men. Fertil. Steril. 2013, 100, 681–685.e1. [Google Scholar] [CrossRef] [Green Version]
- Bay, K.; Asklund, C.; Skakkebaek, N.E.; Andersson, A.-M. Testicular dysgenesis syndrome: Possible role of endocrine disrupters. Best Pr. Res. Clin. Endocrinol. Metab. 2006, 20, 77–90. [Google Scholar] [CrossRef]
- Slowikowska-Hilczer, J.; Szarras-Czapnik, M.; Wolski, J.K.; Oszukowska, E.; Hilczer, M.; Jakubowski, L.; Walczak-Jedrzejowska, R.; Marchlewska, K.; Filipiak, E.; Kaluzewski, B.; et al. The risk of neoplasm associated with dysgenetic testes in prepubertal and pubertal/adult patients. Folia Histochem. Cytobiol. 2015, 53, 218–226. [Google Scholar] [CrossRef]
- Niedzielski, J.K.; Oszukowska, E.; Słowikowska-Hilczer, J. Undescended testis—Current trends and guidelines: A review of the literature. Arch. Med. Sci. 2016, 12, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Nozawa, S.; Iwamoto, T. Study of spermatogenesis and thickening of lamina propria in the human seminiferous tubules. Fertil. Steril. 2008, 90, 1310–1312. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, J.; Muller, D.; Feuerstacke, C.; Kliesch, S.; Bergmann, M.; Mühlfeld, C.; Middendorff, R. Disturbed spermatogenesis associated with thickened lamina propria of seminiferous tubules is not caused by dedifferentiation of myofibroblasts. Hum. Reprod. 2011, 26, 1450–1461. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Nozawa, S.; Yoshiike, M.; Otoi, T.; Iwamoto, T. Glycoconjugates recognized by peanut agglutinin lectin in the inner acellular layer of the lamina propria of seminiferous tubules in human testes showing impaired spermatogenesis. Hum. Reprod. 2012, 27, 659–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooba, T.; Ishikawa, T.; Yamaguchi, K.; Kondo, Y.; Sakamoto, Y.; Fujisawa, M. Expression and Distribution of Laminin Chains in the Testis for Patients with Azoospermia. J. Androl. 2008, 29, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Shiraishi, K.; Matsuyama, H. Effects of human chorionic gonadotropin on testicular interstitial tissues in men with non-obstructive azoospermia. Andrology 2017, 5, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Mahran, A.M.; Elgamal, D.A.; Ghafeer, H.H.; Abdel-Maksoud, S.A.; Farrag, A.A. Histological alterations in Leydig cells and macrophages in azoospermic men. Andrologia 2016, 49, e12714. [Google Scholar] [CrossRef]
- Ježek, D.; Knežević, N.; Banek, L.; Krhen, I.; Muzic, V.; Kalanj-Bognar, S.; Sincic, S.; Zimak, Z.; Kastelan, Z.; Jezej, V. Fine Structure of Leydig Cells in Patients with Non-Obstructive Azoospermia. Acta Clin. Croat. 2002, 41, 1266. [Google Scholar]
- Carucci, L.R.; Tirkes, A.T.; Pretorius, E.S.; Genega, E.M.; Weinstein, S.P. Testicular Leydig’s Cell Hyperplasia: MR Imaging and Sonographic Findings. AJR Am. J. Roentgenol. 2003, 180, 501–503. [Google Scholar] [CrossRef]
- Li, X.; Strauss, L.; Kaatrasalo, A.; Mayerhofer, A.; Huhtaniemi, I.; Santti, R.; Mäkelä, S.; Poutanen, M. Transgenic Mice Expressing P450 Aromatase as a Model for Male Infertility Associated with Chronic Inflammation in the Testis. Endocrinology 2006, 147, 1271–1277. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, H.; Jia, L.; Li, X.; Rahman, N. Oestrogen action and male fertility: Experimental and clinical findings. Cell Mol. Life Scixp. 2015, 72, 3915–3930. [Google Scholar] [CrossRef]
- Aksglæde, L.; Skakkebæk, N.E.; Almstrup, K.; Juul, A. Clinical and biological parameters in 166 boys, adolescents and adults with nonmosaic Klinefelter syndrome: A Copenhagen experience. Acta Paediatr. 2011, 100, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zheng, H.; Lin, W.; Tajima, A.; Zhang, Y.; Zhang, X.; Zhang, H.; Wu, J.; Han, D.; Rahman, N.A.; et al. Estrogen promotes Leydig cell engulfment by macrophages in male infertility. J. Clin. Investig. 2014, 124, 2709–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, C.; Connolly, S.; MacEneaney, O.; O’Keane, C.; McQuaid, S.E. Leydig Cell Hyperplasia Mimicking a Testicular Tumour in a Patient with Klinefelter Syndrome. Eur. J. Case Rep. Intern. Med. 2019, 6, 001129. [Google Scholar] [CrossRef] [PubMed]
- Tash, J.A.; McCallum, S.; Hardy, M.P.; Knudsen, B.; Schlegel, P.N. Men with nonobstructive azoospermia have Leydig cell hypertrophy but not hyperplasia. J. Urol. 2002, 168, 1068–1070. [Google Scholar] [CrossRef]
- McKinnell, C.; Sharpe, R.M.; Mahood, K.; Hallmark, N.; Scott, H.; Ivell, R.; Staub, C.; Jegou, B.; Haag, F.; Koch-Nolte, F.; et al. Expression of Insulin-Like Factor 3 Protein in the Rat Testis during Fetal and Postnatal Development and in Relation to Cryptorchidism Induced by in Utero Exposure to Di (n-Butyl) Phthalate. Endocrinology 2005, 146, 4536–4544. [Google Scholar] [CrossRef]
- Lin, H.; Ge, R.-S.; Chen, G.-R.; Hu, G.-X.; Dong, L.; Lian, Q.-Q.; Hardy, D.O.; Sottas, C.M.; Li, X.-K.; Hardy, M.P. Involvement of testicular growth factors in fetal Leydig cell aggregation after exposure to phthalate in utero. Proc. Natl. Acad. Sci. USA 2008, 105, 7218–7222. [Google Scholar] [CrossRef] [Green Version]
- Hauptman, D.; Perić, M.H.; Marić, T.; Bojanac, A.K.; Sinčić, N.; Zimak, Z.; Kastelan, Z.; Jezevk, D. Leydig Cells in Patients with Non-Obstructive Azoospermia: Do They Really Proliferate? Life 2021, 11, 1266. [Google Scholar] [CrossRef]
- Shono, T.; Suita, S. Reasonable explanation for both the antiandrogenic mechanism of DBP and DBP-induced Leydig cell hyperplasia in prenatally DBP-treated rats. Toxicol. Appl. Pharmacol. 2009, 164, 336. [Google Scholar] [CrossRef]
- Mahood, I.K.; Hallmark, N.; McKinnell, C.; Walker, M.; Fisher, J.S.; Sharpe, R.M. Abnormal Leydig Cell Aggregation in the Fetal Testis of Rats Exposed to Di (n-Butyl) Phthalate and Its Possible Role in Testicular Dysgenesis. Endocrinology 2005, 146, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Winge, S.B.; Dalgaard, M.D.; Belling, K.G.; Jensen, J.M.; Nielsen, J.E.; Aksglaede, L.; Schierup, M.H.; Brunak, S.; Skakkebæk, N.E.; Juul, A.; et al. Transcriptome analysis of the adult human Klinefelter testis and cellularity-matched controls reveals disturbed differentiation of Sertoli- and Leydig cells. Cell Death Dis. 2018, 9, 586. [Google Scholar] [CrossRef]
- Welsh, M.; Moffat, L.; Belling, K.; de França, L.R.; Segatelli, T.M.; Saunders, P.T.K.; Sharpe, R.M.; Smith, L.B. Androgen receptor signalling in peritubular myoid cells is essential for normal differentiation and function of adult Leydig cells. Int. J. Androl. 2012, 35, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Tanrikut, C.; Goldstein, M.; Rosoff, J.S.; Lee, R.K.; Nelson, C.J.; Mulhall, J.P. Varicocele as a risk factor for androgen deficiency and effect of repair. Br. J. Urol. 2011, 108, 1480–1484. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Razic, M.M.; Abdel-Hamid, I.A.; Elsobky, E.; El-Dahtory, F. Further Evidence of the Clinical, Hormonal, and Genetic Heterogeneity of Klinefelter Syndrome: A Study of 216 Infertile Egyptian Patients. J. Androl. 2012, 33, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobjer, J.; Naumovska, M.; Giwercman, Y.; Giwercman, A. High prevalence of androgen deficiency and abnormal lipid profile in infertile men with non-obstructive azoospermia. Int. J. Androl. 2012, 35, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Rohayem, J.; Luberto, A.; Nieschlag, E.; Zitzmann, M.; Kliesch, S. Delayed treatment of undescended testes may promote hypogonadism and infertility. Endocrine 2017, 55, 914–924. [Google Scholar] [CrossRef]
- Isaksson, S.; Bogefors, K.; Ståhl, O.; Eberhard, J.; Giwercman, Y.; Leijonhufvud, I.; Link, K.; Øra, I.; Romerius, P.; Bobjer, J.; et al. High risk of hypogonadism in young male cancer survivors. Clin. Endocrinol. 2018, 88, 432–441. [Google Scholar] [CrossRef]
- Ma, D.; Luo, N.; Xue, G. Trimethyltin (TMT) Reduces Testosterone Production in Adult Leydig Cells in Rats. Int. J. Toxicol. 2019, 38, 493–500. [Google Scholar] [CrossRef]
- Akingbemi, B.T.; Sottas, C.M.; Koulova, A.I.; Klinefelter, G.R.; Hardy, M.P. Inhibition of Testicular Steroidogenesis by the Xenoestrogen Bisphenol A Is Associated with Reduced Pituitary Luteinizing Hormone Secretion and Decreased Steroidogenic Enzyme Gene Expression in Rat Leydig Cells. Endocrinology 2004, 145, 592–603. [Google Scholar] [CrossRef]
- Welsh, M.; Saunders, P.T.; Fisken, M.; Scott, H.M.; Hutchison, G.R.; Smith, L.B.; Sharpe, R.M. Identification in rats of a programming window for reproductive tract masculinization, disruption of which leads to hypospadias and cryptorchidism. J. Clin. Investig. 2008, 118, 1479–1490. [Google Scholar] [CrossRef] [Green Version]
- van den Driesche, S.; Scott, H.M.; MacLeod, D.J.; Fisken, M.; Walker, M.; Sharpe, R.M. Relative importance of prenatal and postnatal androgen action in determining growth of the penis and anogenital distance in the rat before, during and after puberty. Int. J. Androl. 2011, 34, e578–e586. [Google Scholar] [CrossRef]
- Sato, Y.; Asahina, K.; Yoshiike, M.; Nozawa, S.; Otoi, T.; Iwamoto, T. A change in the steroid metabolic pathway in human testes showing deteriorated spermatogenesis. Reprod. Biol. 2020, 20, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Abid, S.; Maitra, A.; Meherji, P.; Patel, Z.; Kadam, S.; Shah, J.; Shah, R.; Kulkarni, V.; Baburao, V.; Gokral, J. Clinical and laboratory evaluation of idiopathic male infertility in a secondary referral center in India. J. Clin. Lab. Anal. 2008, 22, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Koşar, P.A.; Özçelik, N.; Koşar, A. Cytogenetic abnormalities detected in patients with non-obstructive azoospermia and severe oligozoospermia. J. Assist. Reprod. Genet. 2010, 27, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Ekman, B.; Fitts, D.; Marelli, C.; Murray, R.D.; Quinkler, M.; Zelissen, P.M. European Adrenal Insufficiency Registry (EU-AIR): A comparative observational study of glucocorticoid replacement therapy. BMC Endocr. Disord. 2014, 14, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, I.; Hizuka, N.; Muraoka, T.; Ichihara, A. Adult Growth Hormone Deficiency: Current Concepts. Neurol. Med.-Chirurg. 2014, 54, 599–605. [Google Scholar] [CrossRef] [Green Version]
- Gámez, J.M.; Penalba, R.; Cardoso, N.; Ponzo, O.; Carbone, S.; Pandolfi, M.; Scacchi, P.; Reynoso, R. Low dose of bisphenol A impairs the reproductive axis of prepuberal male rats. J. Physiol. Biochem. 2013, 70, 239–246. [Google Scholar] [CrossRef]
- Bahmanimehr, A.; Zeighami, S.; Jahromi, B.N.; Parsanezhad, M.E.; Davari, M.; Montazeri, S.; Vaziri, N.M.; Zarei, A. Detection of Y Chromosome Microdeletions and Hormonal Profile Analysis of Infertile Men undergoing Assisted Reproductive Technologies. Int. J. Fertil. Steril. 2018, 12, 173–177. [Google Scholar] [CrossRef]
- Johnson, M.; Raheem, A.; De Luca, F.; Hallerstrom, M.; Zainal, Y.; Poselay, S.; Mohammadi, B.; Moubasher, A.; Johnson, T.F.; Muneer, A.; et al. An analysis of the frequency of Y-chromosome microdeletions and the determination of a threshold sperm concentration for genetic testing in infertile men. Br. J. Urol. 2019, 123, 367–372. [Google Scholar] [CrossRef]
- Jørgensen, N.; Joensen, U.N.; Toppari, J.; Punab, M.; Erenpreiss, J.; Zilaitiene, B.; Paasch, U.; Salzbrunn, A.; Fernandez, M.F.; Virtanen, H.; et al. Compensated reduction in Leydig cell function is associated with lower semen quality variables: A study of 8182 European young men. Hum. Reprod. 2016, 31, 947–957. [Google Scholar] [CrossRef] [Green Version]
- Bandak, M.; Jørgensen, N.; Juul, A.; Lauritsen, J.; Oturai, P.; Mortensen, J.; Hojman, P.; Helge, J.; Daugaard, G. Leydig cell dysfunction, systemic inflammation and metabolic syndrome in long-term testicular cancer survivors. Eur. J. Cancer 2017, 84, 9–17. [Google Scholar] [CrossRef]
- Johnston, H.; Baker, P.J.; Abel, M.; Charlton, H.M.; Jackson, G.; Fleming, L.; Kumar, T.R.; O’Shaughnessy, P.J. Regulation of Sertoli Cell Number and Activity by Follicle-Stimulating Hormone and Androgen during Postnatal Development in the Mouse. Endocrinology 2004, 145, 318–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suomi, A.-M.; Main, K.M.; Kaleva, M.; Schmidt, I.M.; Chellakooty, M.; Virtanen, H.E.; Boisen, K.A.; Damgaard, I.N.; Kai, C.M.; Skakkebæk, N.E.; et al. Hormonal Changes in 3-Month-Old Cryptorchid Boys. J. Clin. Endocrinol. Metab. 2006, 91, 953–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skøtt, J.W.; Lauritsen, J.; Kreiberg, M.; Daugaard, G.; Bandak, M. Quality of Life in Long-Term Testicular Cancer Survivors with Compensated Leydig Cell Dysfunction. Clin. Genitourin. Cancer 2019, 17, e65–e71. [Google Scholar] [CrossRef] [PubMed]
- Lardone, M.C.; Castillo, P.; Valdevenito, R.; Ebensperger, M.; Ronco, A.M.; Pommer, R.; Piottante, A.; Castro, A. P450-aromatase activity and expression in human testicular tissues with severe spermatogenic failure. Int. J. Androl. 2010, 33, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Tarsitano, M.G.; Bandak, M.; Jørgensen, N.; Skakkebæk, N.E.; Juul, A.; Lenzi, A.; Daugaard, D.; Rajpert-De Meyts, E. Quantification of the Leydig cell compartment in testicular biopsies and association with biochemical Leydig cell dysfunction in testicular cancer survivors. Andrology 2018, 6, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Akingbemi, B.T.; Ge, R.; Klinefelter, G.R.; Zirkin, B.R.; Hardy, M.P. Phthalate-induced Leydig cell hyperplasia is associated with multiple endocrine disturbances. Proc. Natl. Acad. Sci. USA 2004, 101, 775–780. [Google Scholar] [CrossRef] [Green Version]
- Akingbemi, B.T.; Braden, T.D.; Kemppainen, B.W.; Hancock, K.D.; Sherrill, J.D.; Cook, S.J.; He, X.; Supko, J.G. Exposure to Phytoestrogens in the Perinatal Period Affects Androgen Secretion by Testicular Leydig Cells in the Adult Rat. Endocrinology 2007, 148, 4475–4488. [Google Scholar] [CrossRef]
- Shiraishi, K.; Oka, S.; Matsuyama, H. Testicular Testosterone and Estradiol Concentrations and Aromatase Expression in Men with Nonobstructive Azoospermia. J. Clin. Endocrinol. Metab. 2020, 106, 1803–1815. [Google Scholar] [CrossRef]
- Strauss, L.; Kallio, J.; Desai, N.; Pakarinen, P.; Miettinen, T.; Gylling, H.; Albercht, M.; Makela, S.; Mayerhofer, A.; Poutanen, M. Increased Exposure to Estrogens Disturbs Maturation, Steroidogenesis, and Cholesterol Homeostasis via Estrogen Receptor α in Adult Mouse Leydig Cells. Endocrinology 2009, 150, 2865–2872. [Google Scholar] [CrossRef] [Green Version]
- Lardone, M.C.; Argandoña, F.; Flórez, M.; Parada-Bustamante, A.; Ebensperger, M.; Palma, C.; Piottante, A.; Castro, A. Overexpression of CYP19A1 aromatase in Leydig cells is associated with steroidogenic dysfunction in subjects with Sertoli cell-only syndrome. Andrology 2017, 5, 41–48. [Google Scholar] [CrossRef]
- Lardone, M.C.; Reyes, I.N.; Ortiz, E.; Piottante, A.; Palma, C.; Ebensperger, M.; Castro, A. Testicular steroid sulfatase overexpression is associated with Leydig cell dysfunction in primary spermatogenic failure. Andrology 2021, 9, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Lardone, M.C.; Argandoña, F.; Lorca, M.; Piottante, A.; Flórez, M.; Palma, C.; Ebensperger, E.; Castro, A. Leydig cell dysfunction is associated with post-transcriptional deregulation of CYP17A1 in men with Sertoli cell-only syndrome. Mol. Hum. Reprod. 2018, 24, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Marsh, C.A.; Auchus, R.J. Fertility in patients with genetic deficiencies of cytochrome P450c17 (CYP17A1): Combined 17-hydroxylase/17,20-lyase deficiency and isolated 17,20-lyase deficiency. Fertil. Steril. 2014, 101, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Luboshitzky, R.; Kaplan-Zverling, M.; Shen-Orr, Z.; Nave, R.; Herer, P. Seminal plasma androgen/oestrogen balance in infertile men. Int. J. Androl. 2002, 25, 345–351. [Google Scholar] [CrossRef] [PubMed]
- McKinnell, C.; Atanassova, N.; Williams, K.; Fisher, J.S.; Walker, M.; Turner, K.J.; Saunders, T.K.; Sharpe, R.M. Suppression of androgen action and the induction of gross abnormalities of the reproductive tract in male rats treated neonatally with diethylstilbestrol. J. Androl. 2001, 22, 323–338. [Google Scholar] [PubMed]
- Rivas, A.; Fisher, J.S.; McKinnell, C.; Atanassova, N.; Sharpe, R.M. Induction of Reproductive Tract Developmental Abnormalities in the Male Rat by Lowering Androgen Production or Action in Combination with a Low Dose of Diethylstilbestrol: Evidence for Importance of the Androgen-Estrogen Balance. Endocrinology 2002, 143, 4797–4808. [Google Scholar] [CrossRef]
- Han, Y.; Feng, H.L.; Sandlow, J.I.; Haines, C.J. Comparing Expression of Progesterone and Estrogen Receptors in Testicular Tissue from Men With Obstructive and Nonobstructive Azoospermia. J. Androl. 2009, 30, 127–133. [Google Scholar] [CrossRef]
- Mizuno, K.; Kojima, Y.; Kurokawa, S.; Kamisawa, H.; Kohri, K.; Hayashi, Y. Altered Expression and Localization of Estrogen Receptors Alpha and Beta in the Testes of a Cryptorchid Rat Model. Urology 2011, 77, 251.e1–251.e6. [Google Scholar] [CrossRef]
- Sansone, A.; Kliesch, S.; Isidori, A.M.; Schlatt, S. AMH and INSL3 in testicular and extragonadal pathophysiology: What do we know? Andrology 2019, 7, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Overvad, S.; Bay, K.; Bojesen, A.; Gravholt, C.H. Low INSL3 in Klinefelter syndrome is related to osteocalcin, testosterone treatment and body composition, as well as measures of the hypothalamic-pituitary-gonadal axis. Andrology 2014, 2, 421–427. [Google Scholar] [CrossRef]
- Di Nisio, A.; De Toni, L.; Rocca, M.S.; Ghezzi, M.; Selice, R.; Taglialavoro, G.; Ferlin, A.; Forestra, C. Negative Association Between Sclerostin and INSL3 in Isolated Human Osteocytes and in Klinefelter Syndrome: New Hints for Testis–Bone Crosstalk. J. Clin. Endocrinol. Metab. 2018, 103, 2033–2041. [Google Scholar] [CrossRef] [PubMed]
- Trabado, S.; Maione, L.; Bry-Gauillard, H.; Affres, H.; Salenave, S.; Sarfati, J.; Bouvattier, C.; Delemer, B.; Chanson, P.; Le Bouc, Y.; et al. Insulin-like Peptide 3 (INSL3) in Men with Congenital Hypogonadotropic Hypogonadism/Kallmann Syndrome and Effects of Different Modalities of Hormonal Treatment: A Single-Center Study of 281 Patients. J. Clin. Endocrinol. Metab. 2014, 99, E268–E275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Cortessis, V.K.; Hwang, A.; Hardy, B.; Koh, C.J.; Bogatcheva, N.; Agoulnik, A.I. Mutation analysis of INSL3 and GREAT/LGR8 genes in familial cryptorchidism. Urology 2004, 64, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Foresta, C.; Ferlin, A. Role of INSL3 and LGR8 in cryptorchidism and testicular functions. Reprod. Biomed. Online 2004, 9, 294–298. [Google Scholar] [CrossRef]
- Ayers, K.; Kumar, R.; Robevska, G.; Bruell, S.; Bell, K.; Malik, M.A.; Bathgate, R.; Sinclair, A. Familial bilateral cryptorchidism is caused by recessive variants in RXFP. J. Med. Genet. 2019, 56, 727–733. [Google Scholar] [CrossRef] [Green Version]
- Bay, K.; Andersson, A.-M. Human testicular insulin-like factor 3: In relation to development, reproductive hormones and andrological disorders. Int. J. Androl. 2010, 34, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Park, E.; Kim, S.-C.; Ahn, R.-S.; Ko, C.; Lee, K. ERα/E2 signaling suppresses the expression of steroidogenic enzyme genes via cross-talk with orphan nuclear receptor Nur77 in the testes. Mol. Cell. Endocrinol. 2012, 362, 91–103. [Google Scholar] [CrossRef]
- Laguë, E.; Tremblay, J.J. Antagonistic Effects of Testosterone and the Endocrine Disruptor Mono-(2-Ethylhexyl) Phthalate on INSL3 Transcription in Leydig Cells. Endocrinology 2008, 149, 4688–4694. [Google Scholar] [CrossRef] [Green Version]
- Laguë, A.; Tremblay, J.J. Estradiol represses Insulin-like 3 expression and promoter activity in MA-10 Leydig cells. Toxicology 2009, 258, 101–105. [Google Scholar] [CrossRef]
- Soerensen, R.R.; Johannsen, T.H.; Skakkebaek, N.E.; Meyts, E.R.-D. Leydig cell clustering and Reinke crystal distribution in relation to hormonal function in adult patients with testicular dysgenesis syndrome (TDS) including cryptorchidism. Hormones 2016, 15, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Wohlfahrt-Veje, C.; Main, K.M.; Skakkebæk, N.E. Testicular dysgenesis syndrome: Foetal origin of adult reproductive problems. Clin. Endocrinol. 2009, 71, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Olesen, I.A.; Joensen, U.N.; Petersen, J.H.; Almstrup, K.; Rajpert-De Meyts, E.; Carlsen, E.; McLachlan, R.; Juul, A.; Jørgensen, N. Decrease in semen quality and Leydig cell function in infertile men: A longitudinal study. Hum. Reprod. 2018, 33, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, R.M.; Skakkebaek, N.E. Male reproductive disorders and the role of endocrine disruption: Advances in understanding and identification of areas for future research. Pure Appl. Chem. 2003, 75, 2023–2038. [Google Scholar] [CrossRef]
- O’Donnell, L.; Robertson, K.M.; Jones, M.E.; Simpson, E.R. Estrogen and Spermatogenesis. Endocr. Rev. 2001, 22, 289–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Etiology | Example | References |
| Chromosomal | Klinefelter syndrome | [11,149,151] |
| Y-chromosome microdeletions | [152,153,154,155] | |
| Genetic | Autosomal monogenic factors (TEX11, NR5A1, SYCP3, MEI1, and others) | [156,157,158,159,160,161,162,163,164,165] |
| Hormonal | Kallmann syndrome | [166] |
| Acquired hypogonadotropic hypogonadism | [167,168,169] | |
| Hyperprolactinemia | [170,171] | |
| Androgen resistance | [172,173] | |
| Developmental/Structural | Cryptorchidism | [174,175,176] |
| Varicocele | [177,178,179,180,181,182] | |
| Radiation and toxins | Radiotherapy | [183,184,185] |
| Chemotherapy | [184,186,187] | |
| Drugs | [188,189,190,191,192] | |
| EDCs (phthalates, bisphenol A) | [193,194,195,196,197,198,199,200] | |
| Alcohol abuse | [201,202] | |
| Infections | Mumps orchitis | [203,204] |
| Others | [205,206,207] | |
| Testicular trauma | Torsion | [208,209] |
| Other exogenous factors | Heat | [210,211] |
| Idiopathic | - | [212,213,214] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamczewska, D.; Słowikowska-Hilczer, J.; Walczak-Jędrzejowska, R. The Fate of Leydig Cells in Men with Spermatogenic Failure. Life 2022, 12, 570. https://doi.org/10.3390/life12040570
Adamczewska D, Słowikowska-Hilczer J, Walczak-Jędrzejowska R. The Fate of Leydig Cells in Men with Spermatogenic Failure. Life. 2022; 12(4):570. https://doi.org/10.3390/life12040570
Chicago/Turabian StyleAdamczewska, Daria, Jolanta Słowikowska-Hilczer, and Renata Walczak-Jędrzejowska. 2022. "The Fate of Leydig Cells in Men with Spermatogenic Failure" Life 12, no. 4: 570. https://doi.org/10.3390/life12040570