
The Innovative Informatics Approaches of High-Throughput Technologies in Livestock: Spearheading the Sustainability and Resiliency of Agrigenomics Research


Abstract
:1. Introduction
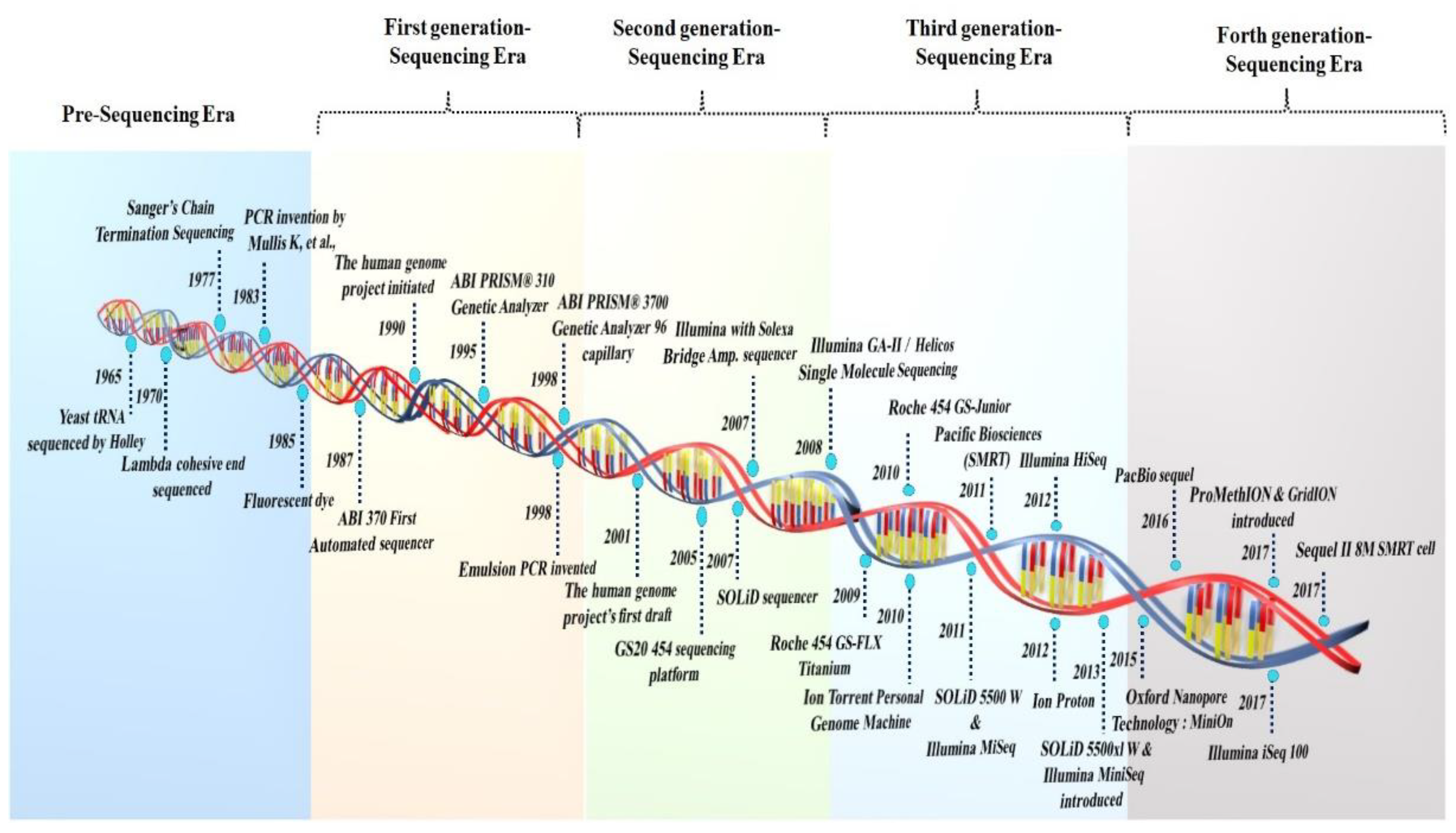
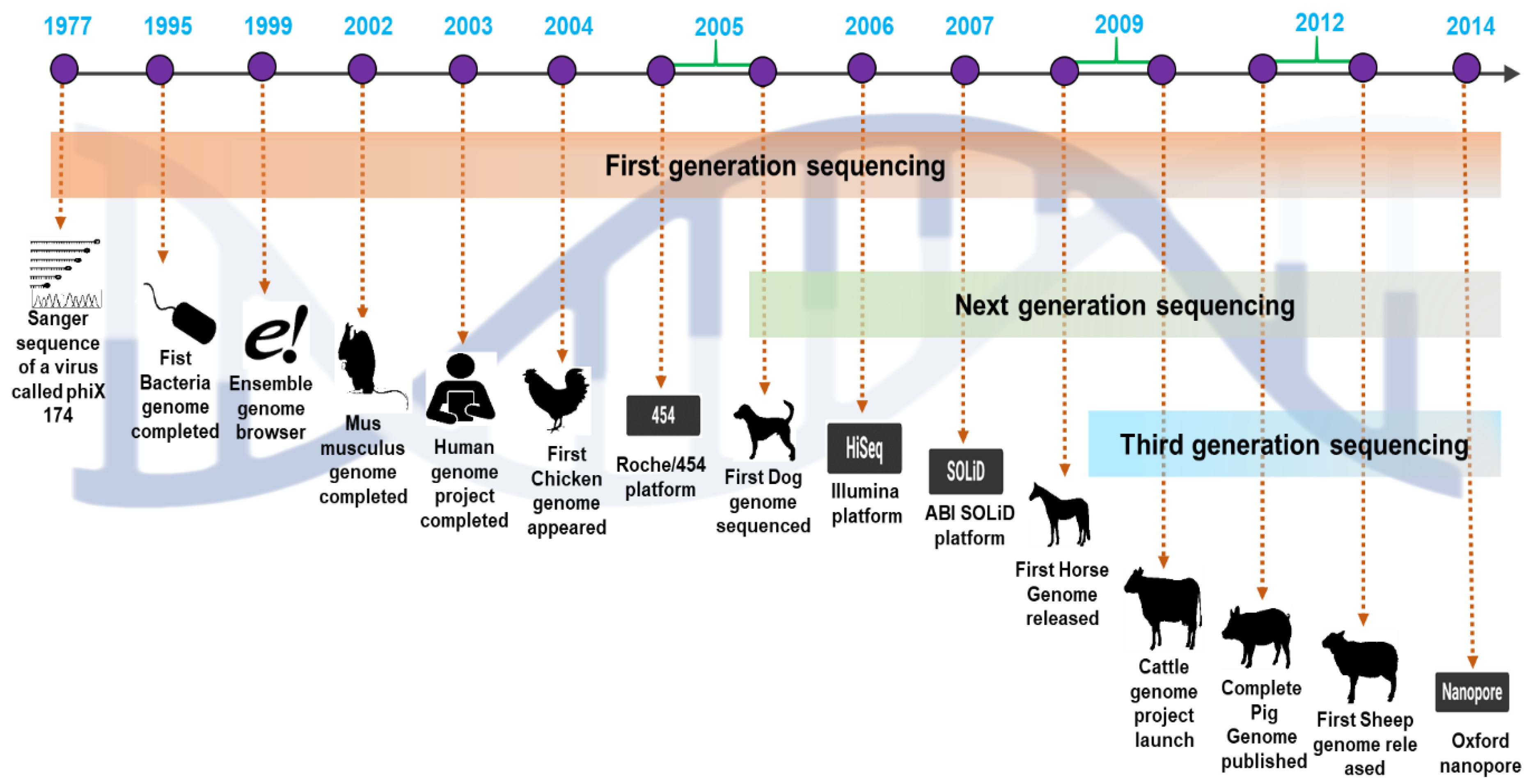
2. The Era of the Development of Sequencing Technologies
2.1. The First Generation of Sequencing Technologies
2.2. The Second Generation of Sequencing Technologies
2.3. Third-Generation Sequencing Platforms
2.4. Fourth-Generation Sequencing Platforms
3. The Perspective of Domestic Animal Reference Sequences
3.1. Insights into Cattle (Bos taurus) Genome Research
3.2. The Decade of Swine (Sus scrofa) Genomic Research
3.3. Genetic Assembly Research in Chickens (Gallus gallus) and Ducks (Anatidae)
3.4. Genome Architecture in Sheep (Ovis aries)
3.5. Inslight in the Horse (Equus caballus) Genomics
4. Databases and Online Resources
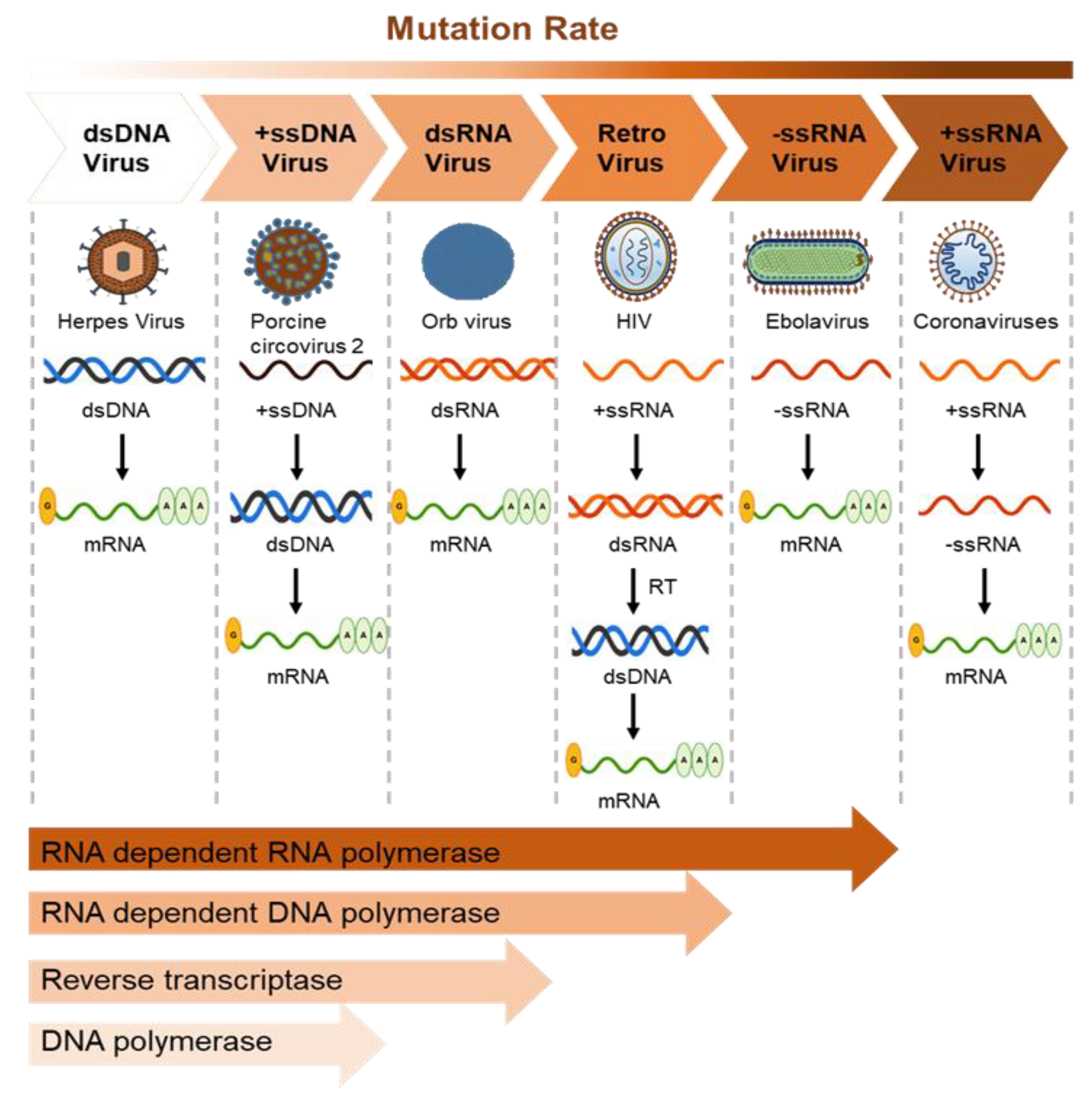
5. Outline of Zoonosis Infections

{kind=link}
{kind=link}
{kind=link}
| Agents | Category | Resource | Description | URL | References |
|---|---|---|---|---|---|
| Virus | Genome database | National Center for Biotechnology Information’s (NCBI’s) virus | The National Center for Biotechnology Information hosts the Virus Variation Resource, a valuation viral sequence data resource that contains modules for seven viral groups, including the influenza virus, Dengue virus, West Nile virus, Ebolavirus, MERS coronavirus, Rotavirus A, and Zika virus. Pipelines that scan recently made GenBank records, annotate genes and proteins, parse sample descriptors, and map them to controlled vocabulary support each module. | https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/ (accessed on 3 November 2022) | [99] |
| Genome database researching tool | Hmmer database | HMMER searches sequence databases for sequence homologs and performs sequence alignments. It is intended to detect distant homologs as sensitively as possible, relying on the robustness of its underlying probability models. | http://hmmer.org/ (accessed on 3 November 2022) | [100,101,102] | |
| Virus discovery and annotation tool | Cenote-Taker 2 | Cenote-Taker 2 was written in Bash, Perl, and Python. All scripts can be found on GitHub. This tool is a virus discovery and annotation tool available via the command line and graphical user interface with free computation access, employs highly sensitive models of hallmark virus genes to discover familiar or divergent viral sequences from user-input contigs. Furthermore, Cenote-Taker2 employs a versatile set of modules to automatically annotate the sequence features of contigs, providing more gene information than comparable tools. The BLAST and Hmmer databases created for this tool can be found on Zenodo. | https://github.com/mtisza1/Cenote-Taker2 (accessed on 3 November 2022) https://zenodo.org/record/4031657 (accessed on 3 November 2022) | [103] | |
| Viral genomes identification database | IMG/VR | The IMG/VR database contains the most comprehensive collection of viral sequences obtained from (meta)genomes. The IMG/VR V3 contains 18 373 cultivated and 2 314 329 uncultivated viral genomes (UViGs), nearly tripling the total number of sequences compared to the previous version. These were divided into 935 362 viral Operational Taxonomic Units (vOTUs), with 188 930 having two or more members. | https://img.jgi.doe.gov/cgi-bin/vr/main.cgi (accessed on 3 November 2022) | [104] | |
| Microbiome analysis resource | MGnify | It offers a free platform for assembling, analyzing, and archiving microbiome data derived from sequencing microbial populations found in specific environments. MGnify’s increased focus on metagenomic data assembly has resulted in a six-fold increase in the number of datasets assembled and analyzed. MGnify’s Notebook Server provides a no-installation Jupyter Lab environment for users to explore programmatic access to MGnify datasets using Python or R via the MGnifyR package. | https://www.ebi.ac.uk/metagenomics/ (accessed on 3 November 2022) https://shiny-portal.embl.de/shinyapps/app/06_mgnify-notebook-lab?jlpath=mgnify-examples/home.ipynb (accessed on 3 November 2022) | [105] | |
| Bacteria | Microbial Genome and Microbiomes database | IMG/M | The system serves as a public resource for genome and metagenome dataset analysis and annotation in a comprehensive comparative context. The IMG web user interface includes a number of analytical and visualization tools for comparing isolate genomes and metagenomes in IMG. | https://img.jgi.doe.gov/cgi-bin/m/main.cgi (accessed on 3 November 2022) https://img.jgi.doe.gov/ (accessed on 3 November 2022) | [106] |
| MetaGenome Gene Finding | MetaGeneMark/2 | MetaGeneMark’s developers, GENE PROBE Inc., have created and refined algorithms for gene prediction in metagenomic sequences for over fifteen years. This website provides access to gene prediction in metagenomes by utilizing metagenome parameters and gene prediction. This same MetaGeneMark-2 plugin has been further optimized for gene discovery in anonymous metagenomic sequences. In comparison to MetaGeneMark, estimated to be 2.7%, MetaGeneMark-2 reduces nearly twice the rate of false negative predictions and missed genes. MetaGeneMark-2 is a C++ program, and all experiments and results are run and analyzed in Python. All scripts can be found on GitHub. | http://opal.biology.gatech.edu/GeneMark/ (accessed on 3 November 2022) http://exon.gatech.edu/meta_gmhmmp.cgi (accessed on 3 November 2022) https://github.com/gatech-genemark/MetaGeneMark-2 (accessed on 3 November 2022) | [107,108,109] | |
| Genome database | Ensembl Bacteria | Ensembl Bacteria is a genome browser for bacteria and archaea. These are from the International Nucleotide Sequence Database Collaboration, the European Nucleotide Archive at the EBI, GenBank at the NCBI, and the Japanese DNA Database. The Ensembl Genomes project, launched in 2009, enhanced the Ensembl project by utilizing the same visualization, interactive, and programming tools to provide users with access to genome data from a further five domains: protists, bacteria, metazoa, plants, and fungi. | https://bacteria.ensembl.org/index.html (accessed on 3 November 2022) | [110,111,112] | |
| Bacterial Isolate Genome Sequence Database | BIGSdb | EBIGSdb is software that collects and evaluates sequencing data for bacterial isolates. BIGSdb extends the MLST concept to genomic data, allowing for the creation of many loci and assigning alleles based on sequence definition databases. The program is distributed under the GNU General Public License, version 3. The most recent version of this document may be obtained at https://bigsdb.readthedocs.org/ (accessed on 3 November 2022) | https://bigsdb.readthedocs.io/en/latest/ (accessed on 3 November 2022) | [113] | |
| Parasite | Malaria Genome database | UCSC Malaria | The UCSC Genome Browser is an online and downloadable genome browser created by the University of California, Santa Cruz’s Hughes Undergraduate Research Group, in collaboration with Prof. Manuel Ares Jr.’s laboratory. It combines the entire DNA sequences of multiple malaria parasite species (Plasmodium sp.) on a single screen, together with experimental data and found genes from the literature. Users may browse through the malaria parasite’s genome’s 14 chromosomes, insert their sequencing data and annotations, and compare results across species. | https://plasmodb.org/plasmo/app (accessed on 3 November 2022) | [114] |
| Eukaryotic Pathogen, Vector and Host Informatics Resource | PlasmoDB/ VEuPathDB | The database includes more than 500 organisms, including invertebrate vectors, eukaryotic pathogens (protists and fungus), and relevant free-living or non-pathogenic species or hosts. VEuPathDB projects integrate >1700 pre-analyzed datasets (and related metadata) with extensive search capabilities, visualizations, and analysis tools in a graphical interface to provide researchers with access to Omics data and bioinformatic studies. | https://plasmodb.org/plasmo/app (accessed on 3 November 2022) | [115] | |
| Model Organism Database for Caenorhabditis elegans | WormBase Parasite | It was established in 2000 and offered each species at WormBase a dependable and recognizable user interface. Furthermore, the WormBase Parasite V WBPS17 assembles the reliable, current information about the genetics, genomes, and biology of nematode Haemonchus contortus an animal endoparasite infecting wild and domesticated ruminants (including sheep and goats) worldwide. | http://www.wormbase.org (accessed on 3 November 2022) https://parasite.wormbase.org/Haemonchus_contortus_prjeb506/Info/Index/ (accessed on 3 November 2022) | [116,117,118] | |
| Global Mammal Parasite Database version 2.0 | GMPD | GMPD, a database of parasites of wild ungulates (artiodactyls and perissodactyls), carnivores, and primates, and is provided for download as complete flat files. The updated database contains over 24,000 entries from over 2700 literature sources. It included data on sampling method and sample size when obtainable, as well as “reported” and “corrected” binomials for each host and parasite species. Current higher taxonomies and data on transmission modes used by the majority of the parasite species in the database are also included. | parasites.nunn-lab.org (accessed on 3 November 2022) | [119] | |
| Fungi | Saccharomyces Genome Database | SGD | The SGD project delivers the highest-quality manually curated information from peer-reviewed literature and algorithms like sequence similarity searches, which leads to extensive details on genome characteristics and gene relationships. Researchers have public access to these data through online sites that are built for ease of use. | http://www.yeastgenome.org (accessed on 3 November 2022) | [120,121,122] |
| Common database microbial agents | Genome database for Archaea, Bacteria, Eukarya, Viruses | GOLD v.8 | It is a data management system that manually catalogs sequencing efforts from around the world and the supporting metadata. In GOLD, there were 387,480 different creatures divided throughout 305 different phyla and candidate phyla. The bulk of these organisms (88%) are bacteria, followed by eukaryotes (8.5%), viruses (2.5%), and archaea (1%). | https://gold.jgi.doe.gov/ (accessed on 3 November 2022) | [123] |
| Metagenomics RAST server | MG-RAST | The MG-RAST server is an open-source comparative genomics system based on the SEED platform. Users can upload raw fasta sequence data; the sequences will be normalized and analyzed, and summaries will be generated automatically. The service offers multiple methods for accessing the various data kinds, such as phylogenetic and metabolic reconstructions, as well as the ability to compare the metabolism and annotations of one or more metagenomes and genomes. | https://www.mg-rast.org/ (accessed on 3 November 2022) | [124,125] | |
| Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups | EggNOG Database | EggNOG is a publicly available database that analyzes thousands of genomes at once to determine orthology links between all of their genes. It included a significant upgrade to the underlying genome sets, which were enlarged to include 4445 representative bacteria and 168 archaea generated from 25 038 genomes, 477 eukaryotic species, and 2502 viral proteomes. | http://eggnog5.embl.de/#/app/home (accessed on 3 November 2022) | [126] |
6. The Mechanism of Zoonoses
7. HT-NGS and Bioinformatics Simulations for Pathogens Detection
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bai, Y.; Sartor, M.; Cavalcoli, J. Current status and future perspectives for sequencing livestock genomes. J. Anim. Sci. Biotechnol. 2012, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Sanchez, S.; Hanning, I.; Pendleton, S.; D’Souza, D. Next-generation sequencing: The future of molecular genetics in poultry production and food safety1 1Presented as part of the Next Generation Sequencing: Applications for Food Safety and Poultry Production Symposium at the Poultry Science Association’s annual meeting in Athens, Georgia, 10 July 2012. Poult. Sci. 2013, 92, 562–572. [Google Scholar] [CrossRef] [PubMed]
- McAdam, P.R.; Richardson, E.J.; Fitzgerald, J.R. High-throughput sequencing for the study of bacterial pathogen biology. Curr. Opin. Microbiol. 2014, 19, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasopoulou, K.; Boti, M.A.; Adamopoulos, P.G.; Skourou, P.C.; Scorilas, A. Third-Generation Sequencing: The Spearhead towards the Radical Transformation of Modern Genomics. Life 2021, 12, 30. [Google Scholar] [CrossRef]
- Next-Generation Sequencing Services Market Size, Share & Trends Analysis Report By Service Type (Human Genome Sequencing, Gene Regulation Services), By Workflow, By End-Use, And Segment Forecasts, 2022–2030; May 2022. Available online: https://www.grandviewresearch.com/industry-analysis/next-generation-sequencing-ngs-services-market (accessed on 2 November 2022).
- Wray-Cahen, D.; Bodnar, A.; Rexroad, C.; Siewerdt, F.; Kovich, D. Advancing genome editing to improve the sustainability and resiliency of animal agriculture. CABI Agric. Biosci. 2022, 3, 21. [Google Scholar] [CrossRef]
- Narrod, C.; Zinsstag, J.; Tiongco, M. A one health framework for estimating the economic costs of zoonotic diseases on society. EcoHealth 2012, 9, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Behravesh, C.B.; Brinson, D.; Hopkins, B.A.; Gomez, T.M. Backyard poultry flocks and salmonellosis: A recurring, yet preventable public health challenge. Clin. Infect. Dis. 2014, 58, 1432–1438. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, C.W.; Campagnolo, E.R.; Boktor, S.W.; Butler, C.L. Zoonotic disease awareness survey of backyard poultry and swine owners in southcentral Pennsylvania. Zoonoses Public Health 2020, 67, 280–290. [Google Scholar] [CrossRef]
- Benton, B.; King, S.; Greenfield, S.R.; Puthuveetil, N.; Reese, A.L.; Duncan, J.; Marlow, R.; Tabron, C.; Pierola, A.E.; Yarmosh, D.A., Jr. The ATCC Genome Portal: Microbial Genome Reference Standards with Data Provenance. Microbiol. Resour. Announc. 2021, 10, e00818–e00821. [Google Scholar] [CrossRef]
- Sinsheimer, R.L. A single-stranded DNA from bacteriophage phi X174. Brookhaven Symp. Biol. 1959, 12, 27–34. [Google Scholar]
- Ghosh, M.; Sharma, N.; Singh, A.K.; Gera, M.; Pulicherla, K.K.; Jeong, D.K. Transformation of animal genomics by next-generation sequencing technologies: A decade of challenges and their impact on genetic architecture. Crit. Rev. Biotechnol. 2018, 38, 1157–1175. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, C.A., 3rd. DNA sequencing: Bench to bedside and beyond. Nucleic Acids Res. 2007, 35, 6227–6237. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, S.; Liu, T.; Ren, X.; Li, W.; Chen, C.; Xie, Z. Benchmarking variant callers in next-generation and third-generation sequencing analysis. Brief. Bioinform. 2021, 22, bbaa148. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Diaz, M.H.; Winchell, J.M. The evolution of advanced molecular diagnostics for the detection and characterization of Mycoplasma pneumoniae. Front. Microbiol. 2016, 7, 232. [Google Scholar] [CrossRef]
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-generation sequencing: From basic research to diagnostics. Clin. Chem. 2009, 55, 641–658. [Google Scholar] [CrossRef] [Green Version]
- Applications of Clinical Microbial Next-Generation Sequencing: Report on an American Academy of Microbiology Colloquium held in Washington, DC, in April 2015; American Society for Microbiology: Washington, DC, USA, 2016.
- Golan, D.; Medvedev, P. Using state machines to model the Ion Torrent sequencing process and to improve read error rates. Bioinformatics 2013, 29, i344–i351. [Google Scholar] [CrossRef] [Green Version]
- Shendure, J.; Porreca, G.J.; Reppas, N.B.; Lin, X.; McCutcheon, J.P.; Rosenbaum, A.M.; Wang, M.D.; Zhang, K.; Mitra, R.D.; Church, G.M. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 2005, 309, 1728–1732. [Google Scholar] [CrossRef] [Green Version]
- Barba, E.; Tsermpini, E.-E.; Patrinos, G.P.; Koromina, M. Genome Informatics Pipelines and Genome Browsers. In Applied Genomics and Public Health; Elsevier: Amsterdam, The Netherlands, 2020; pp. 149–169. [Google Scholar]
- Gourle, H.; Karlsson-Lindsjo, O.; Hayer, J.; Bongcam-Rudloff, E. Simulating Illumina metagenomic data with InSilicoSeq. Bioinformatics 2019, 35, 521–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Enciso, M.; Ferretti, L. Massive parallel sequencing in animal genetics: Wherefroms and wheretos. Anim. Genet. 2010, 41, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhang, Y.; Ying, C.; Wang, D.; Du, C. Nanopore-based fourth-generation DNA sequencing technology. Genom. Proteom. Bioinform. 2015, 13, 4–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.K.; Gupta, U. Next generation sequencing and its applications. In Animal Biotechnology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 395–421. [Google Scholar]
- Kortenhoeven, C.; Joubert, F.; Bastos, A.D.; Abolnik, C. Virus genome dynamics under different propagation pressures: Reconstruction of whole genome haplotypes of West Nile viruses from NGS data. BMC Genom. 2015, 16, 118. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.R.; Koren, S.; Sutton, G. Assembly algorithms for next-generation sequencing data. Genomics 2010, 95, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, P.L. An overview of beef production from pasture and feedlot globally, as demand for beef and the need for sustainable practices increase. Animal 2021, 15, 100295. [Google Scholar] [CrossRef]
- Elsik, C.G.; Unni, D.R.; Diesh, C.M.; Tayal, A.; Emery, M.L.; Nguyen, H.N.; Hagen, D.E. Bovine Genome Database: New tools for gleaning function from the Bos taurus genome. Nucleic Acids Res. 2016, 44, D834–D839. [Google Scholar] [CrossRef] [Green Version]
- Arias, J.A.; Keehan, M.; Fisher, P.; Coppieters, W.; Spelman, R. A high density linkage map of the bovine genome. BMC Genet. 2009, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Snelling, W.M.; Chiu, R.; Schein, J.E.; Hobbs, M.; Abbey, C.A.; Adelson, D.L.; Aerts, J.; Bennett, G.L.; Bosdet, I.E.; Boussaha, M. A physical map of the bovine genome. Genome Biol. 2007, 8, R165. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Delcher, A.L.; Florea, L.; Kelley, D.R.; Schatz, M.C.; Puiu, D.; Hanrahan, F.; Pertea, G.; Van Tassell, C.P.; Sonstegard, T.S.; et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009, 10, R42. [Google Scholar] [CrossRef] [PubMed]
- McKay, S.D.; Schnabel, R.D.; Murdoch, B.M.; Aerts, J.; Gill, C.A.; Gao, C.; Li, C.; Matukumalli, L.K.; Stothard, P.; Wang, Z.; et al. Construction of bovine whole-genome radiation hybrid and linkage maps using high-throughput genotyping. Anim. Genet. 2007, 38, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Tellam, R.L.; Lemay, D.G.; Van Tassell, C.P.; Lewin, H.A.; Worley, K.C.; Elsik, C.G. Unlocking the bovine genome. BMC Genom. 2009, 10, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.P.; Wong, L.L.; Razali, S.A.; Afiqah-Aleng, N.; Mohd Nor, S.A.; Sung, Y.Y.; Van de Peer, Y.; Sorgeloos, P.; Danish-Daniel, M. Applications of Next-Generation Sequencing Technologies and Computational Tools in Molecular Evolution and Aquatic Animals Conservation Studies: A Short Review. Evol. Bioinform. Online 2019, 15, 1176934319892284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groenen, M.A. A decade of pig genome sequencing: A window on pig domestication and evolution. Genet. Sel. Evol. 2016, 48, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Outlook, F. Biannual Report on Global Food Markets; FAO: Rome, Italy, 2017. [Google Scholar]
- Groenen, M.A.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.J.; et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.; Xu, J.; Chen-Tsai, R.Y.; Li, K. Genome editing in livestock: Are we ready for a revolution in animal breeding industry? Transgenic Res. 2017, 26, 715–726. [Google Scholar] [CrossRef]
- Chen, K.; Baxter, T.; Muir, W.M.; Groenen, M.A.; Schook, L.B. Genetic resources, genome mapping and evolutionary genomics of the pig (Sus scrofa). Int. J. Biol. Sci. 2007, 3, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Prather, R.S. Pig genomics for biomedicine. Nat. Biotechnol. 2013, 31, 122–124. [Google Scholar] [CrossRef]
- Padhi, M.K. Importance of indigenous breeds of chicken for rural economy and their improvements for higher production performance. Scientifica 2016, 2016, 2604685. [Google Scholar] [CrossRef] [Green Version]
- Burt, D.W. Chicken genome: Current status and future opportunities. Genome Res. 2005, 15, 1692–1698. [Google Scholar] [CrossRef] [PubMed]
- Consortium, I.C.G.S. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar]
- Hackett, S.J.; Kimball, R.T.; Reddy, S.; Bowie, R.C.; Braun, E.L.; Braun, M.J.; Chojnowski, J.L.; Cox, W.A.; Han, K.L.; Harshman, J.; et al. A phylogenomic study of birds reveals their evolutionary history. Science 2008, 320, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Lee, J.; Heo, K.N.; Kwon, K.; Moon, Y.; Lim, D.; Lee, K.T.; Kim, J. Population analysis of the Korean native duck using whole-genome sequencing data. BMC Genom. 2020, 21, 554. [Google Scholar] [CrossRef]
- Zhang, Z.; Jia, Y.; Almeida, P.; Mank, J.E.; van Tuinen, M.; Wang, Q.; Jiang, Z.; Chen, Y.; Zhan, K.; Hou, S.; et al. Whole-genome resequencing reveals signatures of selection and timing of duck domestication. Gigascience 2018, 7, giy027. [Google Scholar] [CrossRef] [Green Version]
- Lewis, G.S. Present and future role of small ruminants in animal agriculture. Lat. Am. Arch. Anim. Prod. 2015, 23. Available online: https://ojs.alpa.uy/index.php/ojs_files/article/view/2669 (accessed on 2 November 2022).
- Morris, S.T. Overview of sheep production systems. In Advances in Sheep Welfare; Elsevier: Amsterdam, The Netherlands, 2017; pp. 19–35. [Google Scholar]
- Manach, C.; Donovan, J.L. Pharmacokinetics and metabolism of dietary flavonoids in humans. Free Radic. Res. 2004, 38, 771–785. [Google Scholar] [CrossRef]
- Upadhyay, M.; Hauser, A.; Kunz, E.; Krebs, S.; Blum, H.; Dotsev, A.; Okhlopkov, I.; Bagirov, V.; Brem, G.; Zinovieva, N.; et al. The First Draft Genome Assembly of Snow Sheep (Ovis nivicola). Genome Biol. Evol. 2020, 12, 1330–1336. [Google Scholar] [CrossRef]
- Sabir, J.; Mutwakil, M.; El-Hanafy, A.; Al-Hejin, A.; Sadek, M.A.; Abou-Alsoud, M.; Qureshi, M.; Saini, K.; Ahmed, M. Applying molecular tools for improving livestock performance: From DNA markers to next generation sequencing technologies. J. Food Agric. Environ. 2014, 12, 541–553. [Google Scholar]
- Smith, P.; Juengel, J.; Maclean, P.; Rand, C.; Stanton, J.L. Gestational nutrition 2: Gene expression in sheep fetal ovaries exposed to gestational under nutrition. Reproduction 2019, 157, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Reese, J.T.; Childers, C.P.; Sundaram, J.P.; Dickens, C.M.; Childs, K.L.; Vile, D.C.; Elsik, C.G. Bovine Genome Database: Supporting community annotation and analysis of the Bos taurus genome. BMC Genom. 2010, 11, 645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tozaki, T.; Ohnuma, A.; Kikuchi, M.; Ishige, T.; Kakoi, H.; Hirota, K.-i.; Kusano, K.; Nagata, S.-i. Rare and common variant discovery by whole-genome sequencing of 101 Thoroughbred racehorses. Sci. Rep. 2021, 11, 16057. [Google Scholar] [CrossRef] [PubMed]
- Felkel, S.; Vogl, C.; Rigler, D.; Dobretsberger, V.; Chowdhary, B.P.; Distl, O.; Fries, R.; Jagannathan, V.; Janecka, J.E.; Leeb, T.; et al. The horse Y chromosome as an informative marker for tracing sire lines. Sci. Rep. 2019, 9, 6095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowling, A.T.; Ruvinsky, A. The Genetics of the Horse; CABI: Wallingford, UK, 2000. [Google Scholar]
- Jagannathan, V.; Gerber, V.; Rieder, S.; Tetens, J.; Thaller, G.; Drogemuller, C.; Leeb, T. Comprehensive characterization of horse genome variation by whole-genome sequencing of 88 horses. Anim. Genet. 2019, 50, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Orlando, L.; Ginolhac, A.; Zhang, G.; Froese, D.; Albrechtsen, A.; Stiller, M.; Schubert, M.; Cappellini, E.; Petersen, B.; Moltke, I.; et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 2013, 499, 74–78. [Google Scholar] [CrossRef]
- Raudsepp, T.; Gustafson-Seabury, A.; Durkin, K.; Wagner, M.L.; Goh, G.; Seabury, C.M.; Brinkmeyer-Langford, C.; Lee, E.J.; Agarwala, R.; Stallknecht-Rice, E.; et al. A 4103 marker integrated physical and comparative map of the horse genome. Cytogenet. Genome Res. 2008, 122, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Zhao, Y.; Shiraigol, W.; Li, B.; Bai, D.; Ye, W.; Daidiikhuu, D.; Yang, L.; Jin, B.; Zhao, Q.; et al. Analysis of horse genomes provides insight into the diversification and adaptive evolution of karyotype. Sci. Rep. 2014, 4, 4958. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Qu, Z.; Das, P.J.; Fang, E.; Juras, R.; Cothran, E.G.; McDonell, S.; Kenney, D.G.; Lear, T.L.; Adelson, D.L.; et al. Copy number variation in the horse genome. PLoS Genet. 2014, 10, e1004712. [Google Scholar] [CrossRef] [Green Version]
- Bhati, M.; Kadri, N.K.; Crysnanto, D.; Pausch, H. Assessing genomic diversity and signatures of selection in Original Braunvieh cattle using whole-genome sequencing data. BMC Genom. 2020, 21, 27. [Google Scholar] [CrossRef] [Green Version]
- Rexroad, C.; Vallet, J.; Matukumalli, L.K.; Reecy, J.; Bickhart, D.; Blackburn, H.; Boggess, M.; Cheng, H.; Clutter, A.; Cockett, N.; et al. Genome to Phenome: Improving Animal Health, Production, and Well-Being—A New USDA Blueprint for Animal Genome Research 2018–2027. Front. Genet. 2019, 10, 327. [Google Scholar] [CrossRef]
- Hu, R.; Yao, R.; Li, L.; Xu, Y.; Lei, B.; Tang, G.; Liang, H.; Lei, Y.; Li, C.; Li, X.; et al. A database of animal metagenomes. Sci. Data 2022, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Ko, G.; Kim, P.G.; Cho, Y.; Jeong, S.; Kim, J.Y.; Kim, K.H.; Lee, H.Y.; Han, J.; Yu, N.; Ham, S.; et al. Bioinformatics services for analyzing massive genomic datasets. Genom. Inform. 2020, 18, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Developmental progress and current status of the Animal QTLdb. Nucleic Acids Res. 2015, 44, D827–D833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childers, C.P.; Reese, J.T.; Sundaram, J.P.; Vile, D.C.; Dickens, C.M.; Childs, K.L.; Salih, H.; Bennett, A.K.; Hagen, D.E.; Adelson, D.L.; et al. Bovine Genome Database: Integrated tools for genome annotation and discovery. Nucleic Acids Res. 2011, 39, D830–D834. [Google Scholar] [CrossRef]
- Chen, N.; Fu, W.; Zhao, J.; Shen, J.; Chen, Q.; Zheng, Z.; Chen, H.; Sonstegard, T.S.; Lei, C.; Jiang, Y. BGVD: An Integrated Database for Bovine Sequencing Variations and Selective Signatures. Genom. Proteom. Bioinform. 2020, 18, 186–193. [Google Scholar] [CrossRef]
- Nicolazzi, E.L.; Caprera, A.; Nazzicari, N.; Cozzi, P.; Strozzi, F.; Lawley, C.; Pirani, A.; Soans, C.; Brew, F.; Jorjani, H.; et al. SNPchiMp v.3: Integrating and standardizing single nucleotide polymorphism data for livestock species. BMC Genom. 2015, 16, 283. [Google Scholar] [CrossRef]
- Foroutan, A.; Fitzsimmons, C.; Mandal, R.; Piri-Moghadam, H.; Zheng, J.; Guo, A.; Li, C.; Guan, L.L.; Wishart, D.S. The Bovine Metabolome. Metabolites 2020, 10, 233. [Google Scholar] [CrossRef]
- Maity, S.; Bhat, A.H.; Giri, K.; Ambatipudi, K. BoMiProt: A database of bovine milk proteins. J. Proteom. 2020, 215, 103648. [Google Scholar] [CrossRef]
- Hu, Z.-L.; Fritz, E.R.; Reecy, J.M. AnimalQTLdb: A livestock QTL database tool set for positional QTL information mining and beyond. Nucleic Acids Res. 2006, 35, D604–D609. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.H.; Zhu, Q.H.; Li, X.; Zhu, J.W.; Tian, D.M.; Zhang, S.S.; Kang, H.L.; Li, C.P.; Dong, L.L.; Zhao, W.M.; et al. iSheep: An Integrated Resource for Sheep Genome, Variant and Phenotype. Front. Genet. 2021, 12, 714852. [Google Scholar] [CrossRef]
- Tian, X.; Li, R.; Fu, W.; Li, Y.; Wang, X.; Li, M.; Du, D.; Tang, Q.; Cai, Y.; Long, Y.; et al. Building a sequence map of the pig pan-genome from multiple de novo assemblies and Hi-C data. Sci. China Life Sci. 2020, 63, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Schook, L.B.; Beever, J.E.; Rogers, J.; Humphray, S.; Archibald, A.; Chardon, P.; Milan, D.; Rohrer, G.; Eversole, K. Swine Genome Sequencing Consortium (SGSC): A strategic roadmap for sequencing the pig genome. Comp. Funct. Genom. 2005, 6, 251–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uenishi, H.; Morozumi, T.; Toki, D.; Eguchi-Ogawa, T.; Rund, L.A.; Schook, L.B. Large-scale sequencing based on full-length-enriched cDNA libraries in pigs: Contribution to annotation of the pig genome draft sequence. BMC Genom. 2012, 13, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uenishi, H.; Eguchi, T.; Suzuki, K.; Sawazaki, T.; Toki, D.; Shinkai, H.; Okumura, N.; Hamasima, N.; Awata, T. PEDE (Pig EST Data Explorer): Construction of a database for ESTs derived from porcine full-length cDNA libraries. Nucleic Acids Res. 2004, 32, D484–D488. [Google Scholar] [CrossRef]
- Wang, M.-S.; Thakur, M.; Peng, M.-S.; Jiang, Y.; Frantz, L.A.F.; Li, M.; Zhang, J.-J.; Wang, S.; Peters, J.; Otecko, N.O.; et al. 863 genomes reveal the origin and domestication of chicken. Cell Res. 2020, 30, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-S.; Zhang, J.-J.; Guo, X.; Li, M.; Meyer, R.; Ashari, H.; Zheng, Z.-Q.; Wang, S.; Peng, M.-S.; Jiang, Y.; et al. Large-scale genomic analysis reveals the genetic cost of chicken domestication. BMC Biol. 2021, 19, 118. [Google Scholar] [CrossRef]
- Antin, P.B.; Yatskievych, T.A.; Davey, S.; Darnell, D.K. GEISHA: An evolving gene expression resource for the chicken embryo. Nucleic Acids Res. 2014, 42, D933–D937. [Google Scholar] [CrossRef] [Green Version]
- Wade, C.; Giulotto, E.; Sigurdsson, S.; Zoli, M.; Gnerre, S.; Imsland, F.; Lear, T.; Adelson, D.; Bailey, E.; Bellone, R. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science 2009, 326, 865–867. [Google Scholar] [CrossRef] [Green Version]
- Kalbfleisch, T.S.; Rice, E.S.; DePriest, M.S.; Walenz, B.P.; Hestand, M.S.; Vermeesch, J.R.; O’Connell, B.L.; Fiddes, I.T.; Vershinina, A.O.; Petersen, J.L. EquCab3, an updated reference genome for the domestic horse. BioRxiv 2018, 306928. [Google Scholar] [CrossRef]
- Gim, J.A.; Lee, S.; Kim, D.S.; Jeong, K.S.; Hong, C.P.; Bae, J.H.; Moon, J.W.; Choi, Y.S.; Cho, B.W.; Cho, H.G.; et al. HEpD: A database describing epigenetic differences between Thoroughbred and Jeju horses. Gene 2015, 560, 83–88. [Google Scholar] [CrossRef]
- Lee, J.-R.; Hong, C.P.; Moon, J.-W.; Jung, Y.-D.; Kim, D.-S.; Kim, T.-H.; Gim, J.-A.; Bae, J.-H.; Choi, Y.; Eo, J. Genome-wide analysis of DNA methylation patterns in horse. BMC Genom. 2014, 15, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Procop, G.W. Molecular diagnostics for the detection and characterization of microbial pathogens. Clin. Infect. Dis. 2007, 45 (Suppl. 2), S99–S111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammari, M.G.; Gresham, C.R.; McCarthy, F.M.; Nanduri, B. HPIDB 2.0: A curated database for host-pathogen interactions. Database 2016, 2016, baw103. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.T.; Sobur, M.A.; Islam, M.S.; Ievy, S.; Hossain, M.J.; El Zowalaty, M.E.; Rahman, A.T.; Ashour, H.M. Zoonotic Diseases: Etiology, Impact, and Control. Microorganisms 2020, 8, 1405. [Google Scholar] [CrossRef]
- Lange, S.; Ramirez, M.I. Tissue Remodeling in Health and Disease Caused by Bacteria, Parasites, Fungi, and Viruses. Front. Cell. Infect. Microbiol. 2021, 11, 642311. [Google Scholar] [CrossRef]
- Tenorio, J. Emerging zoonotic infectious diseases: A folly of human development. J. Livest. Sci. 2022, 13, 76–79. [Google Scholar] [CrossRef]
- van Doorn, H.R. Emerging infectious diseases. Medicine 2014, 42, 60–63. [Google Scholar] [CrossRef]
- Pal, S. Incidence of foodborne illness. US Pharm. 2017, 42, 14. [Google Scholar]
- Suminda, G.G.D.; Bhandari, S.; Won, Y.; Goutam, U.; Kanth Pulicherla, K.; Son, Y.-O.; Ghosh, M. High-throughput sequencing technologies in the detection of livestock pathogens, diagnosis, and zoonotic surveillance. Comput. Struct. Biotechnol. J. 2022, 20, 5378–5392. [Google Scholar] [CrossRef]
- Chen, J.; Yang, C.-C. The Impact of COVID-19 on the Revenue of the Livestock Industry: A Case Study of China. Animals 2021, 11, 3586. [Google Scholar] [CrossRef]
- Leifels, M.; Khalilur Rahman, O.; Sam, I.; Cheng, D.; Chua, F.J.D.; Nainani, D.; Kim, S.Y.; Ng, W.J.; Kwok, W.C.; Sirikanchana, K. The one health perspective to improve environmental surveillance of zoonotic viruses: Lessons from COVID-19 and outlook beyond. Isme Commun. 2022, 2, 107. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource—Improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Belford, A.K.; Dominguez-Huerta, G.; Bolduc, B.; Buck, C.B. Cenote-Taker 2 democratizes virus discovery and sequence annotation. Virus Evol. 2021, 7, veaa100. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Páez-Espino, D.; Chen, I.-M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Reddy, T.; Nayfach, S.; Schulz, F.; Call, L. IMG/VR v3: An integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 2021, 49, D764–D775. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Almeida, A.; Beracochea, M.; Boland, M.; Burgin, J.; Cochrane, G.; Crusoe, M.R.; Kale, V.; Potter, S.C.; Richardson, L.J. MGnify: The microbiome analysis resource in 2020. Nucleic Acids Res. 2020, 48, D570–D578. [Google Scholar] [CrossRef]
- Markowitz, V.M.; Korzeniewski, F.; Palaniappan, K.; Szeto, E.; Werner, G.; Padki, A.; Zhao, X.; Dubchak, I.; Hugenholtz, P.; Anderson, I. The integrated microbial genomes (IMG) system. Nucleic Acids Res. 2006, 34, D344–D348. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef] [Green Version]
- Besemer, J.; Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef] [Green Version]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.P.; Birney, E. Genome information resources—Developments at Ensembl. Trends Genet. 2004, 20, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Andrews, T.D.; Bevan, P.; Caccamo, M.; Chen, Y.; Clarke, L.; Coates, G.; Cuff, J.; Curwen, V.; Cutts, T.; et al. An overview of Ensembl. Genome Res. 2004, 14, 925–928. [Google Scholar] [CrossRef]
- Jolley, K.A.; Maiden, M.C.J. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 2010, 11, 595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhead, B.; Karolchik, D.; Kuhn, R.M.; Hinrichs, A.S.; Zweig, A.S.; Fujita, P.A.; Diekhans, M.; Smith, K.E.; Rosenbloom, K.R.; Raney, B.J. The UCSC genome browser database: Update 2010. Nucleic Acids Res. 2010, 38, D613–D619. [Google Scholar] [CrossRef]
- Amos, B.; Aurrecoechea, C.; Barba, M.; Barreto, A.; Basenko, E.Y.; Bażant, W.; Belnap, R.; Blevins, A.S.; Böhme, U.; Brestelli, J.; et al. VEuPathDB: The eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 2021, 50, D898–D911. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.W.; Antoshechkin, I.; Bieri, T.; Blasiar, D.; Chan, J.; Chen, W.J.; De La Cruz, N.; Davis, P.; Duesbury, M.; Fang, R. WormBase: A comprehensive resource for nematode research. Nucleic Acids Res. 2010, 38, D463–D467. [Google Scholar] [CrossRef] [Green Version]
- Laing, R.; Kikuchi, T.; Martinelli, A.; Tsai, I.J.; Beech, R.N.; Redman, E.; Holroyd, N.; Bartley, D.J.; Beasley, H.; Britton, C.; et al. The genome and transcriptome of Haemonchus contortus, a key model parasite for drug and vaccine discovery. Genome Biol. 2013, 14, R88. [Google Scholar] [CrossRef] [Green Version]
- Doyle, S.R.; Tracey, A.; Laing, R.; Holroyd, N.; Bartley, D.; Bazant, W.; Beasley, H.; Beech, R.; Britton, C.; Brooks, K.; et al. Genomic and transcriptomic variation defines the chromosome-scale assembly of Haemonchus contortus, a model gastrointestinal worm. Commun. Biol. 2020, 3, 656. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.R.; Pappalardo, P.; Huang, S.; Byers, J.E.; Farrell, M.J.; Gehman, A.; Ghai, R.R.; Haas, S.E.; Han, B.; Park, A.W.; et al. Global Mammal Parasite Database version 2.0. Ecology 2017, 98, 1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherry, J.M.; Ball, C.; Weng, S.; Juvik, G.; Schmidt, R.; Adler, C.; Dunn, B.; Dwight, S.; Riles, L.; Mortimer, R.K. Genetic and physical maps of Saccharomyces cerevisiae. Nature 1997, 387, 67. [Google Scholar] [CrossRef] [PubMed]
- Engel, S.R.; Dietrich, F.S.; Fisk, D.G.; Binkley, G.; Balakrishnan, R.; Costanzo, M.C.; Dwight, S.S.; Hitz, B.C.; Karra, K.; Nash, R.S.; et al. The reference genome sequence of Saccharomyces cerevisiae: Then and now. G3 Genes Genomes Genet. 2014, 4, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces Genome Database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40, D700–D705. [Google Scholar] [CrossRef]
- Mukherjee, S.; Stamatis, D.; Bertsch, J.; Ovchinnikova, G.; Sundaramurthi, J.C.; Lee, J.; Kandimalla, M.; Chen, I.-M.A.; Kyrpides, N.C.; Reddy, T. Genomes OnLine Database (GOLD) v. 8: Overview and updates. Nucleic Acids Res. 2021, 49, D723–D733. [Google Scholar] [CrossRef]
- Keegan, K.P.; Glass, E.M.; Meyer, F. MG-RAST, a metagenomics service for analysis of microbial community structure and function. In Microbial Environmental Genomics (MEG); Springer: Berlin/Heidelberg, Germany, 2016; pp. 207–233. [Google Scholar]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Shew, A.M.; Snell, H.A.; Nayga, R.M., Jr.; Lacity, M.C. Consumer valuation of blockchain traceability for beef in the U nited S tates. Appl. Econ. Perspect. Policy 2022, 44, 299–323. [Google Scholar] [CrossRef]
- Pelzer, K.D.; Currin, N. Zoonotic Diseases of Cattle; Virginia Cooperative Extension: Blacksburg, VA, USA, 2005. [Google Scholar]
- Masuda, T.; Nagai, M.; Yamasato, H.; Tsuchiaka, S.; Okazaki, S.; Katayama, Y.; Oba, M.; Nishiura, N.; Sassa, Y.; Omatsu, T.; et al. Identification of novel bovine group A rotavirus G15P[14] strain from epizootic diarrhea of adult cows by de novo sequencing using a next-generation sequencer. Vet. Microbiol. 2014, 171, 66–73. [Google Scholar] [CrossRef]
- Beato, M.S.; Marcacci, M.; Schiavon, E.; Bertocchi, L.; Di Domenico, M.; Peserico, A.; Mion, M.; Zaccaria, G.; Cavicchio, L.; Mangone, I.; et al. Identification and genetic characterization of bovine enterovirus by combination of two next generation sequencing platforms. J. Virol. Methods 2018, 260, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Jose, T.; Joseph, A. Domestic animals and zoonosis: A review. Pharma Innov. J. 2020, 9, 27–29. [Google Scholar]
- Olson, M.E.; Guselle, N. Are pig parasites a human health risk? Adv. Pork Prod. 2000, 11, 153–162. [Google Scholar]
- FAOSTAT. FAOSTAT Statistical Database; FAO (Food and Agriculture Organization of the United Nations): Rome, Italy, 2016. [Google Scholar]
- FAOSTAT. Agriculture Organization Corporate Statistical Database. Available online: https://www.fao.org/faostat/en/#home (accessed on 6 December 2018).
- Dodgson, J.; Delany, M.; Cheng, H. Poultry genome sequences: Progress and outstanding challenges. Cytogenet. Genome Res. 2011, 134, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Deeg, C.A.; Hauck, S.M.; Amann, B.; Pompetzki, D.; Altmann, F.; Raith, A.; Schmalzl, T.; Stangassinger, M.; Ueffing, M. Equine recurrent uveitis--a spontaneous horse model of uveitis. Ophthalmic Res. 2008, 40, 151–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenter, A.M.; Heckeroth, A.R.; Weiss, L.M. Toxoplasma gondii: From animals to humans. Int. J. Parasitol. 2000, 30, 1217–1258. [Google Scholar] [CrossRef] [Green Version]
- Skinner, L.J.; Timperley, A.C.; Wightman, D.; Chatterton, J.M.; Ho-Yen, D.O. Simultaneous diagnosis of toxoplasmosis in goats and goatowner’s family. Scand. J. Infect. Dis. 1990, 22, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Sacks, J.J.; Roberto, R.R.; Brooks, N.F. Toxoplasmosis infection associated with raw goat’s milk. JAMA 1982, 248, 1728–1732. [Google Scholar] [CrossRef] [PubMed]
- Dubey, J.P.; Lindsay, D.S.; Speer, C.A. Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin. Microbiol. Rev. 1998, 11, 267–299. [Google Scholar] [CrossRef] [Green Version]
- Gilchrist, C.A.; Turner, S.D.; Riley, M.F.; Petri, W.A., Jr.; Hewlett, E.L. Whole-genome sequencing in outbreak analysis. Clin. Microbiol. Rev. 2015, 28, 541–563. [Google Scholar] [CrossRef] [Green Version]
- Sobel Leonard, A.; McClain, M.T.; Smith, G.J.; Wentworth, D.E.; Halpin, R.A.; Lin, X.; Ransier, A.; Stockwell, T.B.; Das, S.R.; Gilbert, A.S.; et al. Deep Sequencing of Influenza A Virus from a Human Challenge Study Reveals a Selective Bottleneck and Only Limited Intrahost Genetic Diversification. J. Virol. 2016, 90, 11247–11258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imanian, B.; Donaghy, J.; Jackson, T.; Gummalla, S.; Ganesan, B.; Baker, R.C.; Henderson, M.; Butler, E.K.; Hong, Y.; Ring, B.; et al. The power, potential, benefits, and challenges of implementing high-throughput sequencing in food safety systems. NPJ Sci. Food 2022, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.J.; Attia, Y.; Ballah, F.M.; Islam, M.S.; Sobur, M.A.; Islam, M.A.; Ievy, S.; Rahman, A.; Nishiyama, A.; Islam, M.S. Zoonotic significance and antimicrobial resistance in Salmonella in poultry in Bangladesh for the period of 2011–2021. Zoonoticdis 2021, 1, 3–24. [Google Scholar] [CrossRef]
- Lynch, M.; Ackerman, M.S.; Gout, J.F.; Long, H.; Sung, W.; Thomas, W.K.; Foster, P.L. Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 2016, 17, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Aswad, A.; Katzourakis, A. Cell-Derived Viral Genes Evolve under Stronger Purifying Selection in Rhadinoviruses. J. Virol. 2018, 92, e00359-18. [Google Scholar] [CrossRef] [Green Version]
- Lauring, A.S. Within-host viral diversity: A window into viral evolution. Annu. Rev. Virol. 2020, 7, 63–81. [Google Scholar] [CrossRef]
- Hughes, A.L.; Hughes, M.A.K. More effective purifying selection on RNA viruses than in DNA viruses. Gene 2007, 404, 117–125. [Google Scholar] [CrossRef]
- Arcangeli, C.; Torricelli, M.; Sebastiani, C.; Lucarelli, D.; Ciullo, M.; Passamonti, F.; Giammarioli, M.; Biagetti, M. Genetic Characterization of Small Ruminant Lentiviruses (SRLVs) Circulating in Naturally Infected Sheep in Central Italy. Viruses 2022, 14, 686. [Google Scholar] [CrossRef]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Combe, M.; Sanjuan, R. Variability in the mutation rates of RNA viruses. Future Virol. 2014, 9, 605–615. [Google Scholar] [CrossRef]
- Pareek, C.S.; Smoczynski, R.; Tretyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011, 52, 413–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resources, N.; Council, N.R. Global considerations for animal agriculture research. In Critical Role of Animal Science Research in Food Security and Sustainability; National Academies Press: Washington, DC, USA, 2015. [Google Scholar]
- Rothschild, M.F.; Plastow, G.S. Applications of genomics to improve livestock in the developing world. Livest. Sci. 2014, 166, 76–83. [Google Scholar] [CrossRef]
- Jensen, K.; de Miranda Santos, I.K.; Glass, E.J. Using genomic approaches to unravel livestock (host)-tick-pathogen interactions. Trends Parasitol. 2007, 23, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Koehler, A.V.; Jabbar, A.; Hall, R.S.; Gasser, R.B. A Targeted “Next-Generation” Sequencing-Informatic Approach to Define Genetic Diversity in Theileria orientalis Populations within Individual Cattle: Proof-of-Principle. Pathogens 2020, 9, 448. [Google Scholar] [CrossRef]
- Pinnapureddy, A.R.; Stayner, C.; McEwan, J.; Baddeley, O.; Forman, J.; Eccles, M.R. Large animal models of rare genetic disorders: Sheep as phenotypically relevant models of human genetic disease. Orphanet J. Rare Dis. 2015, 10, 107. [Google Scholar] [CrossRef]
- Colitti, B.; Elisabetta, C.; Giantonella, P.; Capucchio, M.T.; Reina, R.; Bertolotti, L.; Rosati, S. A new NGS approach for SRLV full genome characterisation. In Proceedings of the XXXVIII Annual Meeting ECCO 2019, Torino, Italy, 12–14 June 2019; p. 20. [Google Scholar]



| Species | SRA Experiments (Direct Link) |
|---|---|
| Bos taurus (cattle) | 65,068 |
| Sus scrofa domesticus (domestic swine) | 4449 |
| Ovis aries (sheep) | 13,227 |
| Equus caballus (horse) | 13,398 |
| Gallus gallus domesticus (domestic chicken) | 38,902 |
| Anas platyrhynchos (mallard) | 6649 |
| Animal | Category | Resource | URL | Description | References |
|---|---|---|---|---|---|
| Bovine | Cattle Quantitative Trait Locus (QTL) Database | Cattle QTLdb | https://www.animalgenome.org/cgi-bin/QTLdb/BT/index (accessed on 2 November 2022) | The cattle QTL (contains 193,216 QTLs/associations) association data curated from published data (1111 publications). Those QTLs/associations represent 684 different traits. | [70] |
| Genome sequence | Bovine Genome Database (BGD) | http://bovinegenome.org/ (accessed on 2 November 2022) | Sequencing of the cattle genome first began in December 2003. The most recent version of BovineMine (BovineMine v1.6) now includes both the ARS-UCD1.2 and UMD3.1 genome assembly, whereas the previous version (BovineMine v1.4) only had UMD3.1.1. JBrowse is compatible with both ARS-UCD1.2 and UMD3.1. | [71] | |
| Variation database | BGVD | https://animal.nwsuaf.edu.cn/code/index.php/BosVar/ (accessed on 2 November 2022) http://222.90.83.22:88/code/index.php/BosVar (accessed on 2 November 2022) | The BGVD includes information on genomic variants of 432 samples from contemporary cattle worldwide, including ~60.44 million single-nucleotide polymorphisms (SNPs), ~6.86 million indels, and ~76,634 copy number variations with signs of selective sweeps. It can provide information about the selection scores for eight groups of European taurine, Eurasian taurine, East Asian taurine, Chinese indicine, Indian indicine, Africa taurine, Bos indicus, and Bos taurus by using six statistical terms. | [72] | |
| Bovine SNP database | SNPchiMp | https://bioinformaticshome.com/tools/descriptions/SNPchiMp_v.3.html (accessed on 2 November 2022) | SNPchiMp is a public MySQL database with a web-based interface officially attributed as an Ensembl web-based server. SNPchiMp v.3 analyzed six livestock species, ranging from one species for goats to more than ten for cattle, with a total of 23 SNP arrays. The interface includes SNP mapping information from the most recent genome assembly, information extraction from dbSNP for SNPs detected in all commercially available bovine chips, and identification of SNPs shared by two or more bovine chips. | [73] | |
| Metabolome database | The Bovine Metabolome | https://bovinedb.ca/ (accessed on 2 November 2022) | It is a free online resource that contains thorough information about small molecule metabolites identified in bovines. It is meant to be used to learn more about bovine biology and the micronutrients contained in bovine tissues and biofluids, as well as to improve beef and dairy cow veterinarian treatment. Serum, ruminal fluid, liver, longissimus thoracis (LT) muscle, semimembranosus (SM) muscle, and testis tissues are all characterized quantitatively in BMDB. Many data fields are connected to various databases (HMDB, PubChem, MetaCyc, ChEBI, UniProt, and GenBank) and applets for visualizing structure and pathways. | [74] | |
| Proteome database | BoMiProt | http://bomiprot.org/ (accessed on 2 November 2022) | BoMiProt, an online library of bovine milk proteome, contains approximately 3100 proteins from whey, fat globule membranes, and exosomes. Each entry in the database is thoroughly cross-referenced, comprising 397 proteins from various publications with well-defined information on protein function, biochemical characteristics, post-translational modifications, and relevance in milk. | [75] | |
| Sheep | Variantion database | SheepVar | http://222.90.83.22:88/code/index.php/SheepVar (accessed on 2 November 2022) | The database is an online resource led by Yu Jiang (Northwest A&F University, Yangling, Shaanxi, China). This comprehensive SheepVar database includes ~83 M SNPs and ~7 M Indels derived from 1116 samples of seven wild sheep relatives and 135 domestic sheep breeds. This database was curated by analyzing 64 wild sheep samples and 1052 domestic sheep samples and also provides two ways to view SNPs and indels, one is interactive tables and geographical maps, and the other is in Gbrowse format. | |
| Quantitative Trait Locus (QTL) Database | Sheep QTLdb | https://www.animalgenome.org/cgi-bin/QTLdb/OA/index (accessed on 2 November 2022) | The Sheep QTLdb is valuable for population genetic research. The frequency of these tools used for searching by chromosomes, traits, breeds, publications, and candidate genes. Sheep QTLdb contains 4416 QTLs/associations from 226 publications. Those QTLs/associations represent 266 different traits. | [76] | |
| International Sheep Genomics Consortium | ISheep | https://www.sheephapmap.org/ (accessed on 2 November 2022) | The ISGC helps researchers identify genetic areas and genes that influence sheep characteristics. This database serves as a backbone for ruminant species when coupled with data from other ruminant genome sequences. The database contains sheep genome assemblies and variants of 935 sheep representing 69 breeds from 21 countries. In addition to providing a genetic resource for animal biomedical research models, this assembly is a genomic resource for humans. | [77] | |
| Sheep Genomes Database | Sheep Genomes DB | https://sheepgenomesdb.org/ (accessed on 2 November 2022) | The USDA AFRI-funded Sheep Genomes Database is a project of the International Sheep Genomics Consortium that builds on the consortium’s recent achievement of creating and sharing the Oar rambouillet v1.0 genome. It gathers and facilitates sheep genomic data, detects variants, and downloads SNP and CNV data from sheep genomes. | ||
| Pig | Pig Pan-genome Database | PIGPAN | http://222.90.83.22:88/code/index.php/panPig (accessed on 2 November 2022) | Third-generation sequencing technology was used to assemble the 2.4 Gb Duroc genome (Sscrofa11.1) and 72.5 Mb pan-sequences from 11 significant local European and Chinese pig varieties. The pan-genome offers a rich data set for the scientific community, which would support the pig genome’s development. | [78] |
| Swine genome sequencing data | SGSC | https://www.igb.illinois.edu/labs/schook/sgsc/index.php (accessed on 2 November 2022) | It was established in September 2003 to promote biomedical research for animal health. It supports creating DNA-based technologies and products from swine genome sequencing data. | [79] | |
| Pig Expression Data Explorer | PEDE | https://pede.dna.affrc.go.jp/ (accessed on 2 November 2022) | The Animal Genome Research Program in Japan, which is operated by the JATAFF-Institute and National Institute of Agrobiological Sciences, maintains this database website. In conjunction with the NIAS DNA bank, the Animal Genome Database, the SNP Linkage map, and the RH map are resources that include PEDE. Pig cSNPs (SNPs in cDNA) were found using the PolyPhred program on the PEDE EST assembly. | [80,81] | |
| Pig Quantitative Trait Locus (QTL) Database | Pig QTLdb | https://www.animalgenome.org/cgi-bin/QTLdb/SS/index (accessed on 2 November 2022) | The Pig QTL (35,846 QTLs/associations) association data is curated from published data (773 publications). Those QTLs/associations represent 693 different traits. | [70] | |
| Chicken and duck | Chicken SNP Database | ChickenSD | https://ngdc.cncb.ac.cn/chickensd/ (accessed on 2 November 2022) | A total of 865 samples were used to identify approximately 33 million whole genome non-redundant SNPs in ChickenSD (167 wild, 697 domesticated, and 1 hybrids). A total of 865 samples were used to identify approximately 33 million whole genome non-redundant SNPs in ChickenSD (167 wild, 697 domesticated, and 1 hybrid). The Chinese Academy of Sciences BIG Data Center, Beijing Institute of Genomics (BIG), is in charge of creating and maintaining this database (CAS). The Kunming Institute of Zoology (KIZ), part of the Chinese Academy of Sciences, was tasked with gathering and curating the data (CAS). | [82,83] |
| Chicken Quantitative Trait Locus (QTL) Database | Chicken QTLdb | https://www.animalgenome.org/cgi-bin/QTLdb/GG/index (accessed on 2 November 2022) | The Pig QTL (16,656 QTLs/associations) association data is curated from published data (376 publications). Those QTLs/associations represent 370 different traits. | [70] | |
| Gene expression | GEISHA | http://geisha.arizona.edu (accessed on 2 November 2022) | GEISHA is a chicken embryo in situ hybridization gene expression database and genomics resource. More than 36,000 pictures of whole-embryo in situ hybridizations and embryo portions from embryonic days 0–5, as well as some older embryo data focusing on late-developing tissues, are currently available in the GEISHA database. | [84] | |
| Horse | Genetic variation annotation | EquCab2.0 and 3.0 | https://www.ncbi.nlm.nih.gov/assembly/GCF_000002305.2/ (accessed on 2 November 2022) https://www.ncbi.nlm.nih.gov/assembly/GCF_002863925.1/ (accessed on 2 November 2022) | EquCab2.0 is a publicly available genetic variation annotation reference genome assembly for the domesticated horse, assembled in 2007. EquCab3.0 is the updated reference genome assembly. EquCab2.0 was compiled by sequencing the whole genomes of six horses from six different breeds. One thousand three hundred million reads with coverage between 15× to 24× were generated for these six horse breeds. After rigorous filtration, 17,514,723 SNPs and 1923,693 indels, as well as an average of 1540 CNVs and 3321 structural variations per horse, were identified and functionally annotated. | [85,86] |
| Methylated regions | HEpd | http://www.primate.or.kr/hepd (accessed on 2 November 2022) | The HEpd database contains information on differentially methylated regions and epigenetic changes between two horse subspecies. It employs a gene index to compare the methylation status in a gene area. Users can filter highly methylated sites beyond a user-defined threshold using this database. | [87,88] | |
| Common database | Animal metagenomes | AnimalMetagenome DB | https://github.com/boyNextDooooor/AnimalMetagenomeDB (accessed on 2 November 2022) https://doi.org/10.6084/m9.figshare.19728619 (accessed on 2 November 2022) | AnimalMetagenomeDB combines metagenomic sequencing data with host information to help users discover relevant data. Animal metagenomic data may be seen, searched for, and downloaded by users. Metadata for 82,097 metagenomes from four domestic animals (bovines, sheep, horses, and pigs) and 540 wild species are included in the AnimalMetagenome DB version 1.0. These metagenomes span 15 years of research, 73 nations, 1044 investigations, 63,214 amplicon sequencing data points, and 10,672 whole genome sequencing data points. | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suminda, G.G.D.; Ghosh, M.; Son, Y.-O. The Innovative Informatics Approaches of High-Throughput Technologies in Livestock: Spearheading the Sustainability and Resiliency of Agrigenomics Research. Life 2022, 12, 1893. https://doi.org/10.3390/life12111893
Suminda GGD, Ghosh M, Son Y-O. The Innovative Informatics Approaches of High-Throughput Technologies in Livestock: Spearheading the Sustainability and Resiliency of Agrigenomics Research. Life. 2022; 12(11):1893. https://doi.org/10.3390/life12111893
Chicago/Turabian StyleSuminda, Godagama Gamaarachchige Dinesh, Mrinmoy Ghosh, and Young-Ok Son. 2022. "The Innovative Informatics Approaches of High-Throughput Technologies in Livestock: Spearheading the Sustainability and Resiliency of Agrigenomics Research" Life 12, no. 11: 1893. https://doi.org/10.3390/life12111893