
Third-Generation Sequencing: The Spearhead towards the Radical Transformation of Modern Genomics

 , ,
, ,
Abstract
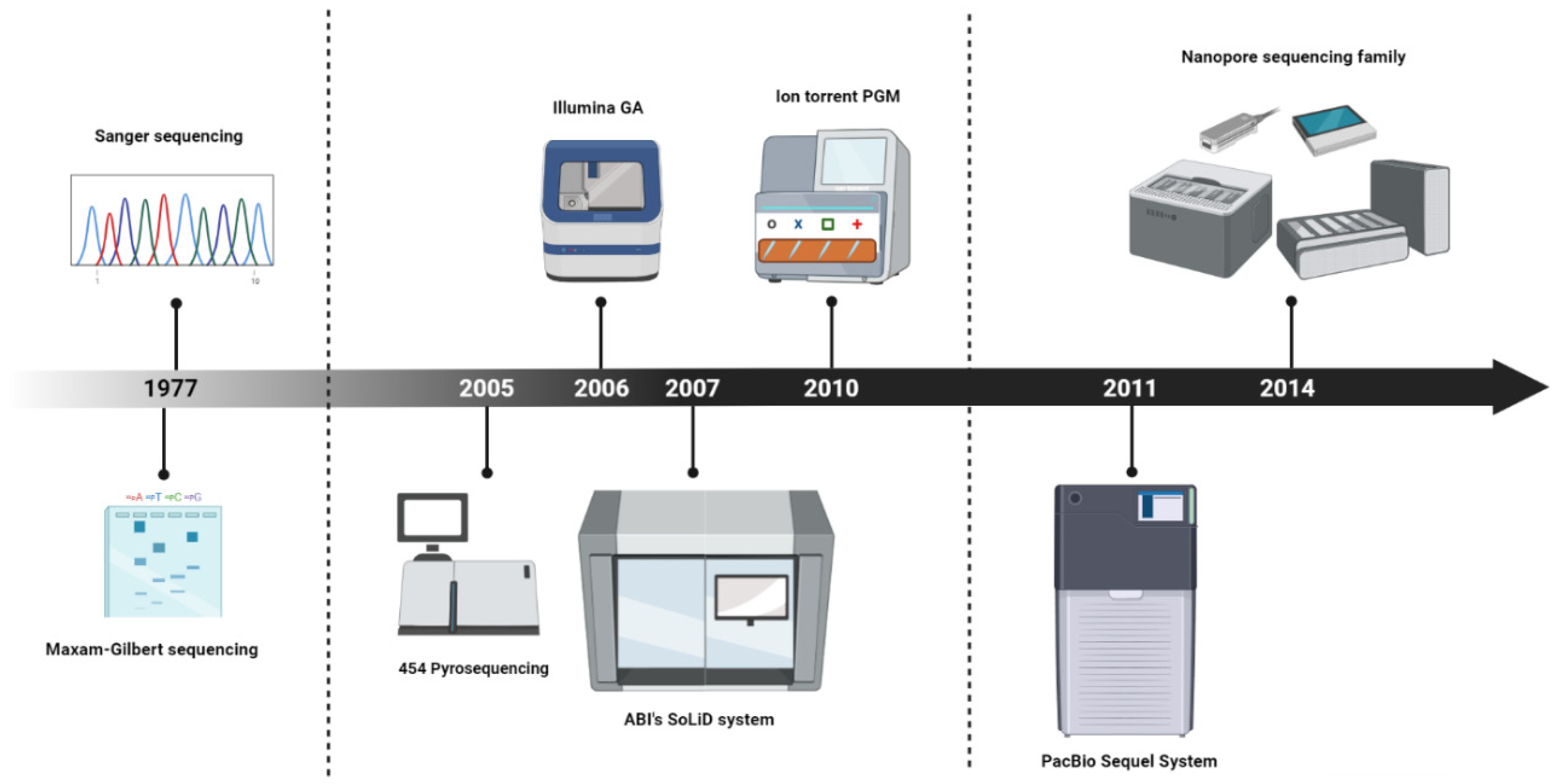
:1. Introduction
2. Overcoming the limitations of NGS
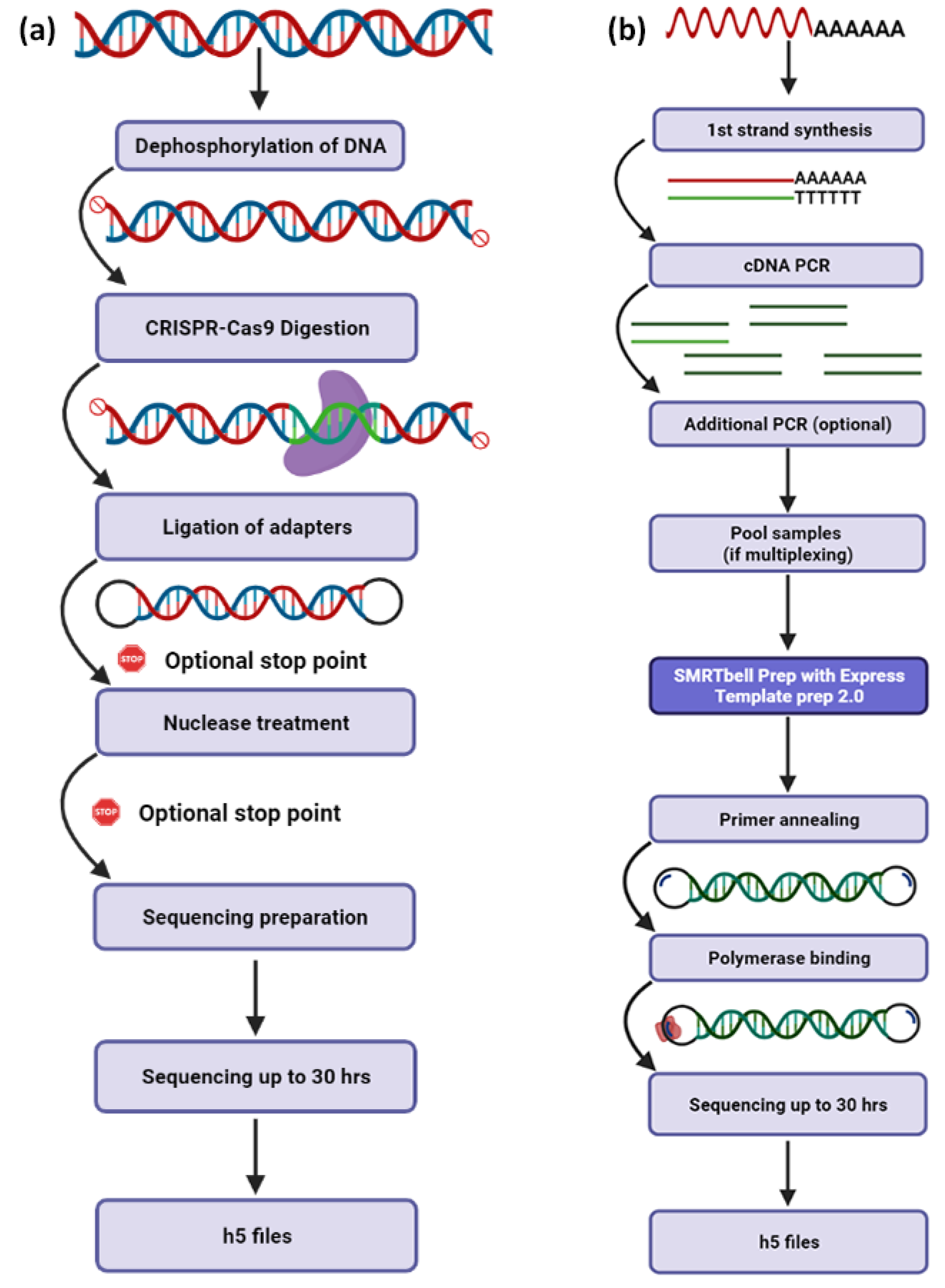
3. The Revolution of PacBio Sequencing
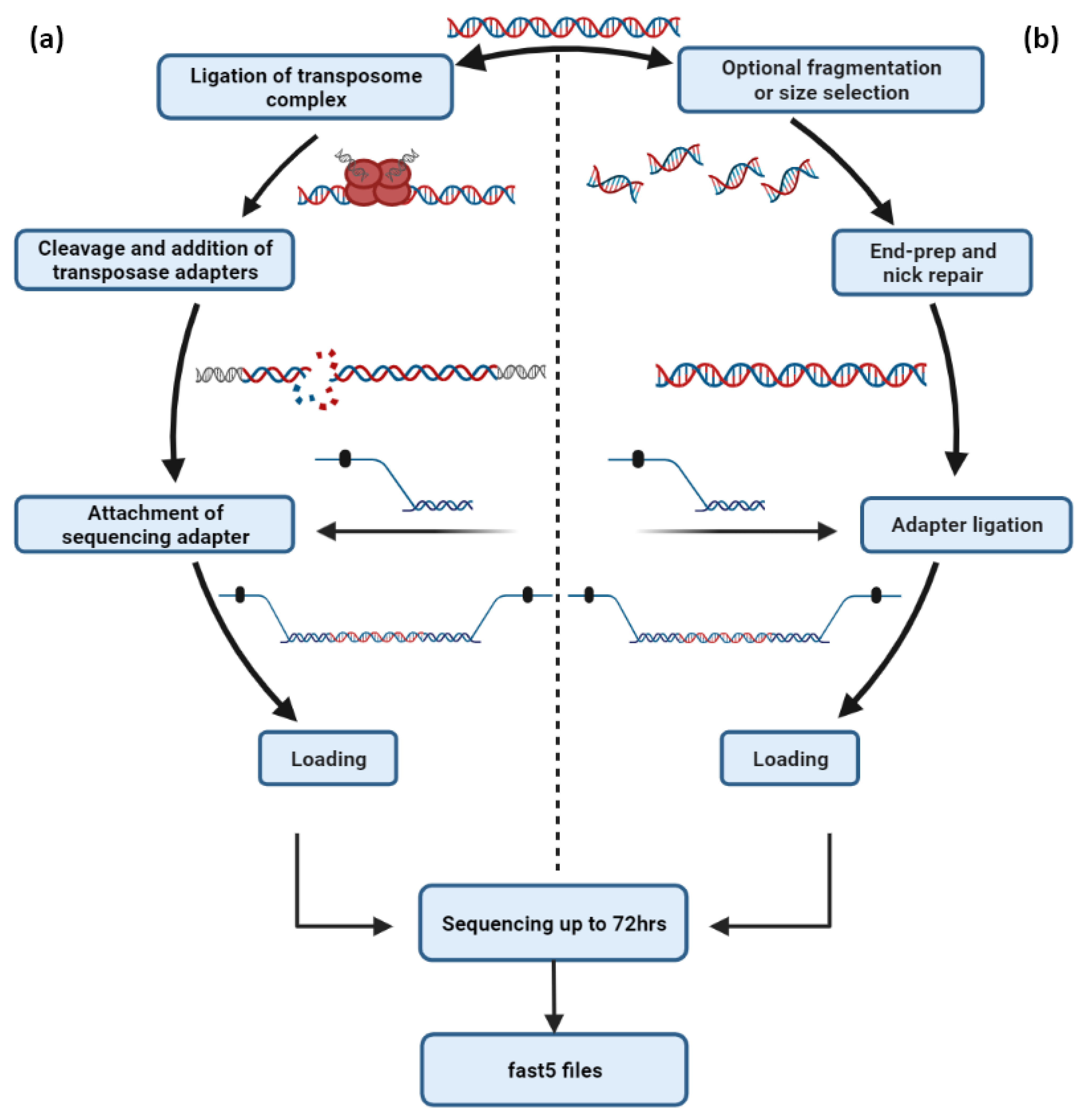
4. The Concept behind Nanopore Sequencing
5. Bioinformatics Tools for Downstream Analysis
6. Applications
6.1. DNA Sequencing
6.1.1. Whole-Exome Sequencing (WES)
6.1.2. Targeted Sequencing
6.1.3. cDNA Sequencing
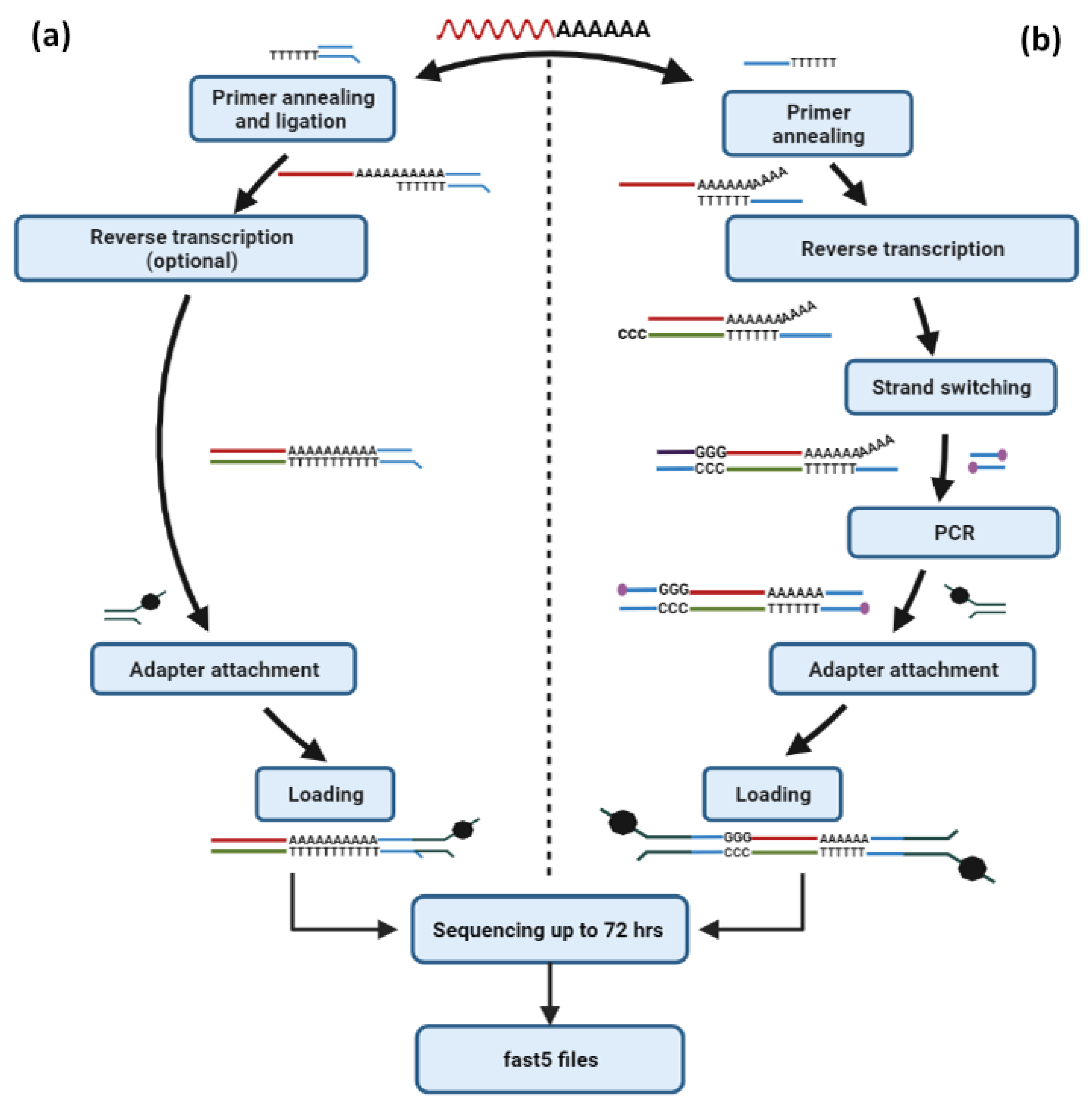
6.2. Nanopore-Based Direct RNA Sequencing
6.2.1. mRNA Sequencing
6.2.2. Small RNA Sequencing
6.3. Epigenetics and Metagenomics
7. Third-Generation Sequencing in Clinical Diagnostics
8. Limitations of Third-Generation Sequencing and Future Challenges
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef] [PubMed]
- Valouev, A.; Ichikawa, J.; Tonthat, T.; Stuart, J.; Ranade, S.; Peckham, H.; Zeng, K.; Malek, J.A.; Costa, G.; McKernan, K.; et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008, 18, 1051–1063. [Google Scholar] [CrossRef] [Green Version]
- Pushkarev, D.; Neff, N.F.; Quake, S.R. Single-molecule sequencing of an individual human genome. Nat. Biotechnol. 2009, 27, 847–850. [Google Scholar] [CrossRef]
- Thompson, J.F.; Steinmann, K.E. Single molecule sequencing with a HeliScope genetic analysis system. Curr. Protoc. Mol. Biol. 2010, 92, 7.10.1–7.10.14. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.Y.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion torrent, pacific biosciences and illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [Green Version]
- Kingan, S.B.; Urban, J.; Lambert, C.C.; Baybayan, P.; Childers, A.K.; Coates, B.; Scheffler, B.; Hackett, K.; Korlach, J.; Geib, S.M. A high-quality genome assembly from a single, field-collected spotted lanternfly (Lycorma delicatula) using the PacBio Sequel II system. GigaScience 2019, 8, giz122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 2015, 12, 351–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deamer, D.; Akeson, M.; Branton, D. Three decades of nanopore sequencing. Nat. Biotechnol. 2016, 34, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Michael, T.P.; Jupe, F.; Bemm, F.; Motley, S.T.; Sandoval, J.P.; Lanz, C.; Loudet, O.; Weigel, D.; Ecker, J.R. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 2018, 9, 541. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef]
- Sharon, D.; Tilgner, H.; Grubert, F.; Snyder, M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [Google Scholar] [CrossRef]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Midha, M.K.; Wu, M.; Chiu, K.-P. Long-read sequencing in deciphering human genetics to a greater depth. Hum. Genet. 2019, 138, 1201–1215. [Google Scholar] [CrossRef]
- Pomerantz, A.; Peñafiel, N.; Arteaga, A.; Bustamante, L.; Pichardo, F.; Coloma, L.A.; Barrio-Amorós, C.L.; Salazar-Valenzuela, D.; Prost, S. Real-time DNA barcoding in a rainforest using nanopore sequencing: Opportunities for rapid biodiversity assessments and local capacity building. GigaScience 2018, 7, giy033. [Google Scholar] [CrossRef] [Green Version]
- Korlach, J.; Bjornson, K.P.; Chaudhuri, B.P.; Cicero, R.L.; Flusberg, B.A.; Gray, J.J.; Holden, D.; Saxena, R.; Wegener, J.; Turner, S.W. Real-time DNA sequencing from single polymerase molecules. Methods Enzymol. 2010, 472, 431–455. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Ambardar, S.; Gupta, R.; Trakroo, D.; Lal, R.; Vakhlu, J. High throughput sequencing: An overview of sequencing chemistry. Indian J. Microbiol. 2016, 56, 394–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Cardenas, J.A.; Garcia-Maroto, F.; Alvarez-Bermejo, J.A.; Manzano-Agugliaro, F. DNA sequencing sensors: An overview. Sensors 2017, 17, 588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of next-generation sequencing technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef] [PubMed]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef] [Green Version]
- Tyler, A.D.; Mataseje, L.; Urfano, C.J.; Schmidt, L.; Antonation, K.S.; Mulvey, M.R.; Corbett, C.R. Evaluation of Oxford Nanopore’s MinION sequencing device for microbial whole genome sequencing applications. Sci. Rep. 2018, 8, 10931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leggett, R.M.; Clark, M.D. A world of opportunities with nanopore sequencing. J. Exp. Bot. 2017, 68, 5419–5429. [Google Scholar] [CrossRef]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION sequencing and genome assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, E.W.; Ng, P.Y. Minion: A novel tool for predicting drug hypersensitivity? Front Pharmacol. 2016, 7, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [Green Version]
- Grädel, C.; Miani, M.A.T.; Barbani, M.T.; Leib, S.L.; Suter-Riniker, F.; Ramette, A. Rapid and cost-efficient enterovirus genotyping from clinical samples using flongle flow cells. Genes 2019, 10, 659. [Google Scholar] [CrossRef] [Green Version]
- Runtuwene, L.R.; Tuda, J.S.B.; Mongan, A.E.; Suzuki, Y. On-site MinION sequencing. Adv. Exp. Med. Biol. 2019, 1129, 143–150. [Google Scholar] [CrossRef]
- Eisenstein, M. Oxford Nanopore announcement sets sequencing sector abuzz. Nat. Biotechnol. 2012, 30, 295–296. [Google Scholar] [CrossRef]
- Suzuki, Y. Informatics for PacBio long reads. Single Mol. Single Cell Seq. 2019, 1129, 119–129. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [Green Version]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukasawa, Y.; Ermini, L.; Wang, H.; Carty, K.; Cheung, M.-S. LongQC: A quality control tool for third generation sequencing long read data. G3 Genes Genomes Genet. 2020, 10, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Nguyen, L.T.; Hayes, B.J.; Ross, E. Prowler: A novel trimming algorithm for Oxford Nanopore sequence data. Bioinformatics 2021, 37, 3936–3937. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Sović, I.; Šikić, M.; Wilm, A.; Fenlon, S.N.; Chen, S.; Nagarajan, N. Fast and sensitive mapping of nanopore sequencing reads with GraphMap. Nat. Commun. 2016, 7, 11307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Wee, Y.; Bhyan, S.B.; Liu, Y.; Lu, J.; Li, X.; Zhao, M. The bioinformatics tools for the genome assembly and analysis based on third-generation sequencing. Brief. Funct. Genom. 2019, 18, 1–12. [Google Scholar] [CrossRef]
- Jung, H.; Winefield, C.; Bombarely, A.; Prentis, P.; Waterhouse, P. Tools and strategies for long-read sequencing and de novo assembly of plant genomes. Trends Plant Sci. 2019, 24, 700–724. [Google Scholar] [CrossRef]
- Miclotte, G.; Heydari, M.; Demeester, P.; Rombauts, S.; Van De Peer, Y.; Audenaert, P.; Fostier, J. Jabba: Hybrid error correction for long sequencing reads. Algorithms Mol. Biol. 2016, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Salmela, L.; Walve, R.; Rivals, E.; Ukkonen, E. Accurate self-correction of errors in long reads using de Bruijn graphs. Bioinformatics 2017, 33, 799–806. [Google Scholar] [CrossRef]
- Goodwin, S.; Gurtowski, J.; Ethe-Sayers, S.; Deshpande, P.; Schatz, M.C.; McCombie, W.R. Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 2015, 25, 1750–1756. [Google Scholar] [CrossRef] [Green Version]
- Loman, N.J.; Quick, J.; Simpson, J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 2015, 12, 733–735. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef] [Green Version]
- Lavezzo, E.; Barzon, L.; Toppo, S.; Palù, G. Third generation sequencing technologies applied to diagnostic microbiology: Benefits and challenges in applications and data analysis. Expert Rev. Mol. Diagn. 2016, 16, 1011–1023. [Google Scholar] [CrossRef]
- Audano, P.A.; Sulovari, A.; Graves-Lindsay, T.A.; Cantsilieris, S.; Sorensen, M.; Welch, A.E.; Dougherty, M.L.; Nelson, B.J.; Shah, A.; Dutcher, S.K.; et al. Characterizing the major structural variant alleles of the human genome. Cell 2019, 176, 663–675.e19. [Google Scholar] [CrossRef] [Green Version]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A.; et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef]
- Chaisson, M.J.P.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M.; et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Ameur, A.; Kloosterman, W.P.; Hestand, M.S. Single-molecule sequencing: Towards clinical applications. Trends Biotechnol. 2019, 37, 72–85. [Google Scholar] [CrossRef]
- van Beek, J.; Haanperä, M.; Smit, P.W.; Mentula, S.; Soini, H. Evaluation of whole genome sequencing and software tools for drug susceptibility testing of Mycobacterium tuberculosis. Clin. Microbiol. Infect. 2019, 25, 82–86. [Google Scholar] [CrossRef] [Green Version]
- Ozsolak, F. Third-generation sequencing techniques and applications to drug discovery. Expert Opin. Drug Discov. 2012, 7, 231–243. [Google Scholar] [CrossRef]
- Xiao, T.; Zhou, W. The third generation sequencing: The advanced approach to genetic diseases. Transl. Pediatr. 2020, 9, 163–173. [Google Scholar] [CrossRef]
- Mannarapu, M.; Dariya, B.; Bandapalli, O.R. Application of single-cell sequencing technologies in pancreatic cancer. Mol. Cell. Biochem. 2021, 476, 2429–2437. [Google Scholar] [CrossRef]
- Need, A.C.; Shashi, V.; Hitomi, Y.; Schoch, K.; Shianna, K.V.; McDonald, M.T.; Meisler, M.H.; Goldstein, D.B. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 2012, 49, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Gilpatrick, T.; Lee, I.; Graham, J.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 2020, 38, 433–438. [Google Scholar] [CrossRef]
- Giesselmann, P.; Brändl, B.; Raimondeau, E.; Bowen, R.; Rohrandt, C.; Tandon, R.; Kretzmer, H.; Assum, G.; Galonska, C.; Siebert, R.; et al. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat. Biotechnol. 2019, 37, 1478–1481. [Google Scholar] [CrossRef] [Green Version]
- Magi, A.; Semeraro, R.; Mingrino, A.; Giusti, B.; D’Aurizio, R. Nanopore sequencing data analysis: State of the art, applications and challenges. Brief. Bioinform. 2018, 19, 1256–1272. [Google Scholar] [CrossRef]
- Ciuffreda, L.; Rodríguez-Pérez, H.; Flores, C. Nanopore sequencing and its application to the study of microbial communities. Comput. Struct. Biotechnol. J. 2021, 19, 1497–1511. [Google Scholar] [CrossRef]
- Lin, B.; Hui, J.; Mao, H. Nanopore technology and its applications in gene sequencing. Biosensors 2021, 11, 214. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Zhao, S. Alternative splicing, RNA-seq and drug discovery. Drug Discov. Today 2019, 24, 1258–1267. [Google Scholar] [CrossRef]
- Hussain, S. Native RNA-sequencing throws its hat into the transcriptomics ring. Trends Biochem. Sci. 2018, 43, 225–227. [Google Scholar] [CrossRef]
- Oikonomopoulos, S.; Bayega, A.; Fahiminiya, S.; Djambazian, H.; Berube, P.; Ragoussis, J. Methodologies for transcript profiling using long-read technologies. Front. Genet. 2020, 11, 606. [Google Scholar] [CrossRef]
- Lorenz, D.A.; Sathe, S.; Einstein, J.M.; Yeo, G.W. Direct RNA sequencing enables m6A detection in endogenous transcript isoforms at base-specific resolution. RNA 2020, 26, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef]
- Soneson, C.; Yao, Y.; Bratus-Neuenschwander, A.; Patrignani, A.; Robinson, M.D.; Hussain, S. A comprehensive examination of Nanopore native RNA sequencing for characterization of complex transcriptomes. Nat. Commun. 2019, 10, 3359. [Google Scholar] [CrossRef] [Green Version]
- Maynard, A.; McCoach, C.E.; Rotow, J.K.; Harris, L.; Haderk, F.; Kerr, D.L.; Yu, E.A.; Schenk, E.L.; Tan, W.; Zee, A.; et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell 2020, 182, 1232–1251.e22. [Google Scholar] [CrossRef]
- Ding, S.; Chen, X.; Shen, K. Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. 2020, 40, 329–344. [Google Scholar] [CrossRef]
- Leigh, D.M.; Schefer, C.; Cornejo, C. Determining the suitability of MinION’s direct RNA and DNA amplicon sequencing for viral subtype identification. Viruses 2020, 12, 801. [Google Scholar] [CrossRef]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef]
- Matsumura, H.; Nirasawa, S.; Terauchi, R. Technical advance: Transcript profiling in rice (Oryza sativa L.) seedlings using serial analysis of gene expression (SAGE). Plant J. 1999, 20, 719–726. [Google Scholar] [CrossRef]
- Hihara, Y.; Kamei, A.; Kanehisa, M.; Kaplan, A.; Ikeuchi, M. DNA microarray analysis of cyanobacterial gene expression during acclimation to high light. Plant Cell 2001, 13, 793–806. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Chen, G.; Chen, F.; Hu, E.; Dong, X.; Li, Z.; He, L.; Sun, Y.; Qiu, L.; Xu, H.; et al. Accurate transcriptome assembly by Nanopore RNA sequencing reveals novel functional transcripts in hepatocellular carcinoma. Cancer Sci. 2021, 112, 3555–3568. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [Green Version]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.; Gu, L.; Reddy, A.S.N. Analysis of transcriptome and epitranscriptome in plants using PacBio Iso-Seq and Nanopore-based direct RNA sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef] [Green Version]
- Depledge, D.P.; Srinivas, K.P.; Sadaoka, T.; Bready, D.; Mori, Y.; Placantonakis, D.G.; Mohr, I.; Wilson, A.C. Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat. Commun. 2019, 10, 754. [Google Scholar] [CrossRef] [Green Version]
- Ozsolak, F.; Milos, P.M. Transcriptome profiling using single-molecule direct rna sequencing. Methods Mol. Biol. 2011, 733, 51–61. [Google Scholar]
- Price, A.M.; Hayer, K.E.; McIntyre, A.B.R.; Gokhale, N.S.; Abebe, J.S.; Della Fera, A.N.; Mason, C.E.; Horner, S.M.; Wilson, A.C.; Depledge, D.P.; et al. Direct RNA sequencing reveals m6A modifications on adenovirus RNA are necessary for efficient splicing. Nat. Commun. 2020, 11, 6016. [Google Scholar] [CrossRef]
- Suzuki, T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol. 2021, 22, 375–392. [Google Scholar] [CrossRef]
- Sloan, K.E.; Warda, A.S.; Sharma, S.; Entian, K.-D.; Lafontaine, D.; Bohnsack, M.T. Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017, 14, 1138–1152. [Google Scholar] [CrossRef]
- Gilbert, W.V.; Bell, T.A.; Schaening, C. Messenger RNA modifications: Form, distribution, and function. Science 2016, 352, 1408–1412. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, S.; Xiao, W.; Zhao, Y.-L.; Yang, Y.-G. m6A: Signaling for mRNA splicing. RNA Biol. 2016, 13, 756–759. [Google Scholar] [CrossRef] [Green Version]
- Slobodin, B.; Han, R.; Calderone, V.; Vrielink, J.; Loayza-Puch, F.; Elkon, R.; Agami, R. Transcription impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine methylation. Cell 2017, 169, 326–337.e12. [Google Scholar] [CrossRef] [Green Version]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA modifications in gene expression regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.-Q.; Wanunu, M.; Wang, M.X.; McReynolds, L.; Wang, Y. Detection of miRNAs with a nanopore single-molecule counter. Expert Rev. Mol. Diagn. 2012, 12, 573–584. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, S.; Chang, L.; Guo, W.; Wang, Y.; Wang, Y.; Zhang, P.; Chen, H.-Y.; Huang, S. Direct microRNA sequencing using nanopore-induced phase-shift sequencing. iScience 2020, 23, 100916. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Chang, H.Y. Epigenomics: Technologies and applications. Circ. Res. 2018, 122, 1191–1199. [Google Scholar] [CrossRef]
- Simpson, J.T.; Workman, R.E.; Zuzarte, P.C.; David, M.; Dursi, L.J.; Timp, W. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef]
- Gigante, S.; Gouil, Q.; Lucattini, A.; Keniry, A.; Beck, T.; Tinning, M.; Gordon, L.; Woodruff, C.; Speed, T.P.; Blewitt, M.E.; et al. Using long-read sequencing to detect imprinted DNA methylation. Nucleic Acids Res. 2019, 47, e46. [Google Scholar] [CrossRef] [Green Version]
- Ewing, A.D.; Smits, N.; Sanchez-Luque, F.J.; Faivre, J.; Brennan, P.M.; Richardson, S.R.; Cheetham, S.W.; Faulkner, G.J. Nanopore sequencing enables comprehensive transposable element epigenomic profiling. Mol. Cell 2020, 80, 915–928.e5. [Google Scholar] [CrossRef]
- Heikema, A.; Horst-Kreft, D.; Boers, S.; Jansen, R.; Hiltemann, S.; De Koning, W.; Kraaij, R.; De Ridder, M.; Van Houten, C.; Bont, L.; et al. Comparison of Illumina versus nanopore 16S rRNA gene sequencing of the human nasal microbiota. Genes 2020, 11, 1105. [Google Scholar] [CrossRef]
- Callahan, B.J.; Wong, J.; Heiner, C.; Oh, S.; Theriot, C.M.; Gulati, A.S.; McGill, S.K.; Dougherty, M.K. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 2019, 47, e103. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, S.; Beka, L.; Graf, J.; Klassen, J.L. Evaluation of strategies for the assembly of diverse bacterial genomes using MinION long-read sequencing. BMC Genom. 2019, 20, 23. [Google Scholar] [CrossRef] [Green Version]
- Kai, S.; Matsuo, Y.; Nakagawa, S.; Kryukov, K.; Matsukawa, S.; Tanaka, H.; Iwai, T.; Imanishi, T.; Hirota, K. Rapid bacterial identification by direct PCR amplification of 16S rRNA genes using the MinION™ nanopore sequencer. FEBS Open Bio 2019, 9, 548–557. [Google Scholar] [CrossRef] [Green Version]
- Nygaard, A.B.; Tunsjø, H.S.; Meisal, R.; Charnock, C. A preliminary study on the potential of Nanopore MinION and Illumina MiSeq 16S rRNA gene sequencing to characterize building-dust microbiomes. Sci. Rep. 2020, 10, 3209. [Google Scholar] [CrossRef] [Green Version]
- Petersen, L.M.; Martin, I.W.; Moschetti, W.E.; Kershaw, C.M.; Tsongalis, G.J. Third-generation sequencing in the clinical laboratory: Exploring the advantages and challenges of nanopore sequencing. J. Clin. Microbiol. 2019, 58, e01315-19. [Google Scholar] [CrossRef]
- Brinkmann, A.; Ulm, S.L.; Uddin, S.; Forster, S.; Seifert, D.; Oehme, R.; Corty, M.; Schaade, L.; Michel, J.; Nitsche, A. Amplicov: Rapid whole-genome sequencing using multiplex pcr amplification and real-time oxford nanopore minion sequencing enables rapid variant identification of SARS-CoV-2. Front. Microbiol. 2021, 12, 651151. [Google Scholar] [CrossRef]
- Morsli, M.; Anani, H.; Brechard, L.; Delerce, J.; Bedotto, M.; Fournier, P.E.; Drancourt, M. Lampore SARS-CoV-2 diagnosis and genotyping: A preliminary report. J. Clin. Virol. 2021, 138, 104815. [Google Scholar] [CrossRef]
- Peto, L.; Rodger, G.; Carter, D.P.; Osman, K.L.; Yavuz, M.; Johnson, K.; Raza, M.; Parker, M.D.; Wyles, M.D.; Andersson, M.; et al. Diagnosis of SARS-CoV-2 infection with LamPORE, a high-throughput platform combining loop-mediated isothermal amplification and nanopore sequencing. J. Clin. Microbiol. 2021, 59, e03271-20. [Google Scholar] [CrossRef]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platform | Sequencer | Average Read Length | Error Rate Per Read | Run Time | Maximum Throughput |
|---|---|---|---|---|---|
| PacBio | PacBio RS II | 10–15 kb | 10–15% | 0.5–4 h | 10 Gb |
| Sequel System | 10–15 kb | 10–15% | ≤20 h | 10 Gb | |
| Sequel II System | 10–15 kb | 10–15% | ≤30 h | 500 Gb | |
| Sequel IIe System | 10–15 kb | 10–15% | ≤30 h | 500 Gb | |
| ONT | MinION Mk1B | >4 Mb | ~13% | 1 min–72 h | 50 Gb |
| MinION Mk1C | >4 Mb | ~13% | 1 min–72 h | 50 Gb | |
| GridION Mk1 | >4 Mb | ~13% | 1 min–72 h | 250 Gb | |
| PromethION 24 | >4 Mb | ~13% | 1 min–72 h | 7 Tb | |
| PromethION 48 | >4 Mb | ~13% | 1 min–72 h | 14 Tb |
| Method | Application | Research Interest | Optimal Technology |
|---|---|---|---|
| DNA sequencing | Whole-genome sequencing | De novo genome assembly | PacBio/ONT |
| Mutational analysis | NGS | ||
| Extended structural variations | PacBio/ONT | ||
| Haplotyping | PacBio/ONT | ||
| DNA modifications | PacBio/ONT | ||
| Whole-exome sequencing | Detection of small indels | NGS | |
| Detection of SNPs | NGS | ||
| Detection of CNVs | PacBio/ONT | ||
| Mutational analysis | NGS | ||
| Targeted sequencing | Construction of gene panel for SNPs | NGS | |
| Construction of gene panel for small indels | NGS | ||
| Construction of gene panel for extended structural variations | PacBio/ONT | ||
| Identification of novel mRNA isoforms | PacBio/ONT | ||
| Detection of infectious diseases | PacBio/ONT | ||
| Whole-transcriptome sequencing | Identification of full-length transcripts | PacBio/ONT | |
| Characterization of fusion transcripts | PacBio/ONT | ||
| Direct RNA sequencing | Direct mRNA sequencing | Detection of RNA modifications | ONT |
| Detection of full-length transcripts | ONT | ||
| Characterization of fusion transcripts | ONT | ||
| Characterization of RNA viruses | ONT | ||
| Direct miRNA sequencing | Identification of novel miRNAs | ONT | |
| Detection of specific RNA modifications | ONT |
| Platform | Approach | Average Cost | Preparation Time | Input Amount | PCR Required |
|---|---|---|---|---|---|
| PacBio | SMRTbell Express for large DNA inserts | ~$730 | 4 h | 1–5 μg | No |
| SMRTbell Express for ultra-low DNA inputs | ~$850 | 5–8 h | 5–20 ng gDNA | Yes | |
| CRISPR-Cas 9 system | * | >10 h | 5 μg gDNA | No | |
| Iso-seq | ~$500 | 8 h | 300 ng total RNA | Yes | |
| 16S amplicon sequencing | ~$450 | PCR + 4 h | 25 pg–2.5 ng gDNA | Yes | |
| ONT | Rapid DNA sequencing | ~$200 | 10 min | 400 ng gDNA | No |
| DNA ligation sequencing | ~$350 | 60 min | 1000 ng dsDNA | No | |
| Cas9 Sequencing Kit | * | 110 min | 1–10 µg dsDNA | No | |
| PCR-cDNA sequencing | ~$430 | 165 min | 1 ng poly-A+ RNA (or 50 ng total RNA) | Yes | |
| Direct cDNA sequencing | ~$400 | 275 min | 100 ng poly-A+ RNA | No | |
| Direct RNA sequencing | ~$350 | 105 min | 500 ng poly-A+ RNA | No | |
| 16S sequencing | ~$350 | PCR + 10 min | 10 ng gDNA | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Athanasopoulou, K.; Boti, M.A.; Adamopoulos, P.G.; Skourou, P.C.; Scorilas, A. Third-Generation Sequencing: The Spearhead towards the Radical Transformation of Modern Genomics. Life 2022, 12, 30. https://doi.org/10.3390/life12010030
Athanasopoulou K, Boti MA, Adamopoulos PG, Skourou PC, Scorilas A. Third-Generation Sequencing: The Spearhead towards the Radical Transformation of Modern Genomics. Life. 2022; 12(1):30. https://doi.org/10.3390/life12010030
Chicago/Turabian StyleAthanasopoulou, Konstantina, Michaela A. Boti, Panagiotis G. Adamopoulos, Paraskevi C. Skourou, and Andreas Scorilas. 2022. "Third-Generation Sequencing: The Spearhead towards the Radical Transformation of Modern Genomics" Life 12, no. 1: 30. https://doi.org/10.3390/life12010030