
Mitochondrial Dynamics, ROS, and Cell Signaling: A Blended Overview

 , and
, and
Abstract
:1. Introduction
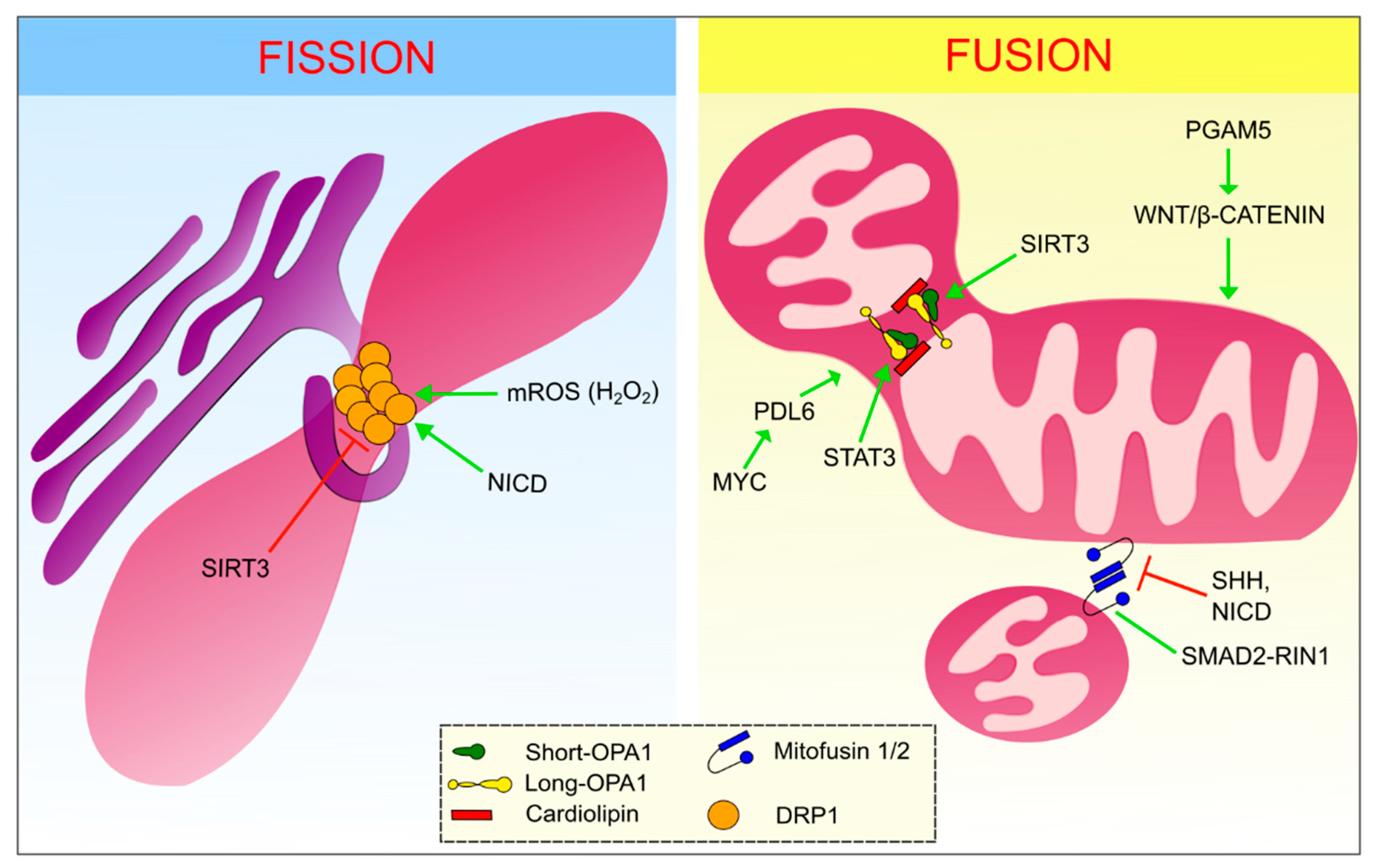
2. Mitochondrial Dynamics: A Multiplayer Regulation
3. Mitochondrial Plasticity and Cell Signaling: A Two-Way Interaction
4. Mitochondrial ROS in the Modulation of Cell Signaling
5. Mitochondrial ROS Involvement in Cancer
6. Hints for Anticancer Therapy: Exploitation of Mitochondrial ROS
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Dyall, S.D.; Brown, M.T.; Johnson, P.J. Ancient Invasions: From Endosymbionts to Organelles. Science (80-. ) 2004, 304, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J. Mitochondria-Striking a balance between host and endosymbiont. Science 2019, 365, eaaw9855. [Google Scholar] [CrossRef]
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Managò, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct Pharmacological Targeting of a Mitochondrial Ion Channel Selectively Kills Tumor Cells In Vivo. Cancer Cell 2017, 31, 516–531.e10. [Google Scholar] [CrossRef]
- Xia, M.; Zhang, Y.; Jin, K.; Lu, Z.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci. 2019, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Mishara, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Bennett, B.; Zielonka, J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: Mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol. 2018, 15, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Willems, P.H.G.M.; Rossignol, R.; Dieteren, C.E.J.; Murphy, M.P.; Koopman, W.J.H. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, N.; Jofuku, A.; Eura, Y.; Mihara, K. Regulation of mitochondrial morphology by membrane potential, and DRP1-dependent division and FZO1-dependent fusion reaction in mammalian cells. Biochem. Biophys. Res. Commun. 2003, 301, 891–898. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef]
- Kashatus, D.F. The regulation of tumor cell physiology by mitochondrial dynamics. Biochem. Biophys. Res. Commun. 2018, 500, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Kalia, R.; Wang, R.Y.R.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–16. [Google Scholar] [CrossRef]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Peruzzo, R.; Costa, R.; Bachmann, M.; Leanza, L.; Szabò, I. Mitochondrial metabolism, contact sites and cellular calcium signaling: Implications for tumorigenesis. Cancers (Basel) 2020, 12, 2574. [Google Scholar] [CrossRef]
- D’Eletto, M.; Rossin, F.; Occhigrossi, L.; Farrace, M.G.; Faccenda, D.; Desai, R.; Marchi, S.; Refolo, G.; Falasca, L.; Antonioli, M.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573–3581.e4. [Google Scholar] [CrossRef] [Green Version]
- Rossin, F.; Costa, R.; Bordi, M.; D’Eletto, M.; Occhigrossi, L.; Farrace, M.G.; Barlev, N.; Ciccosanti, F.; Muccioli, S.; Chieregato, L.; et al. Transglutaminase Type 2 regulates the Wnt/β-catenin pathway in vertebrates. Cell Death Dis. 2021, 12, 249. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Griffoin, J.M.; Kaplan, J.; Dollfus, H.; Lorenz, B.; Faivre, L.; Lenaers, G.; Belenguer, P.; Hamel, C.P. Mutation spectrum and splicing variants in the OPA1 gene. Hum. Genet. 2001, 109, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Malhotra, A.; Dey, A.; Prasad, N.; Kenney, A.M. Sonic Hedgehog signaling drives mitochondrial fragmentation by suppressing mitofusins in cerebellar granule neuron precursors and medulloblastoma. Mol. Cancer Res. 2016, 14, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrázola, M.S.; Silva-Alvarez, C.; Inestrosa, N.C. How the Wnt signaling pathway protects from neurodegeneration: The mitochondrial scenario. Front. Cell. Neurosci. 2015, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengoa-Vergniory, N.; Kypta, R.M. Canonical and noncanonical Wnt signaling in neural stem/progenitor cells. Cell. Mol. Life Sci. 2015, 72, 4157–4172. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Luo, Y.; Chen, L.; Zhang, Z.; Shen, C.; Li, Y.; Xu, R. Sirt3 inhibits cerebral ischemia-reperfusion injury through normalizing Wnt/β-catenin pathway and blocking mitochondrial fission. Cell Stress Chaperones 2018, 23, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Bernkopf, D.B.; Jalal, K.; Brückner, M.; Knaup, K.X.; Gentzel, M.; Schambony, A.; Behrens, J. Pgam5 released from damaged mitochondria induces mitochondrial biogenesis via Wnt signaling. J. Cell Biol. 2018, 217, 1383–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.; Lee, N.Y. Regulation of gene expression and mitochondrial dynamics by SMAD. Mol. Cell. Oncol. 2016, 3, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhang, J.; Lyu, Z.; Chen, Y.; Ji, X.; Cao, H.; Jin, M.; Zhu, J.; Yang, J.; Ling, R.; et al. Positive feedback loop between mitochondrial fission and Notch signaling promotes survivin-mediated survival of TNBC cells. Cell Death Dis. 2018, 9, 1050. [Google Scholar] [CrossRef]
- Von Eyss, B.; Jaenicke, L.A.; Kortlever, R.M.; Royla, N.; Wiese, K.E.; Letschert, S.; McDuffus, L.A.; Sauer, M.; Rosenwald, A.; Evan, G.I.; et al. A MYC-Driven Change in Mitochondrial Dynamics Limits YAP/TAZ Function in Mammary Epithelial Cells and Breast Cancer. Cancer Cell 2015, 28, 743–757. [Google Scholar] [CrossRef] [Green Version]
- Quan, Y.; Park, W.; Jin, J.; Kim, W.; Park, S.K.; Kang, K.P. Sirtuin 3 Activation by Honokiol Decreases Unilateral Ureteral Obstruction-Induced Renal Inflammation and Fibrosis via Regulation of Mitochondrial Dynamics and the Renal NF-κB-TGF-β1/Smad Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; He, L.; Dong, Y.; Fei, Y.; Wen, J.; Li, X.; Guan, J.; Liu, F.; Zhou, T.; Li, Z.; et al. Sitagliptin ameliorates renal tubular injury in diabetic kidney disease via STAT3-dependent mitochondrial homeostasis through SDF-1α/CXCR4 pathway. FASEB J. 2020, 34, 7500–7519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Liu, Z.; Zhao, M.; Zhang, F.; Wang, K.; Feng, N.; Fu, F.; Li, J.; Li, J.; Liu, Y.; et al. κ-opioid receptor activation promotes mitochondrial fusion and enhances myocardial resistance to ischemia and reperfusion injury via STAT3-OPA1 pathway. Eur. J. Pharmacol. 2020, 874, 172987. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Wong, H.-S.; Benoit, B.; Brand, M.D. Mitochondrial and cytosolic sources of hydrogen peroxide in resting C2C12 myoblasts. Free Radic. Biol. Med. 2019, 130, 140–150. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Treberg, J.R.; Perevoshchikova, I.V.; Orr, A.L.; Brand, M.D. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic. Biol. Med. 2012, 53, 1807–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mazat, J.-P.; Devin, A.; Ransac, S. Modelling mitochondrial ROS production by the respiratory chain. Cell. Mol. Life Sci. 2020, 77, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Brandt, U. Molecular Mechanisms of Superoxide Production by the Mitochondrial Respiratory Chain. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2012; pp. 145–169. ISBN 9781461435723. [Google Scholar]
- Burtenshaw, D.; Hakimjavadi, R.; Redmond, E.; Cahill, P. Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants 2017, 6, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aviello, G.; Knaus, U.G. NADPH oxidases and ROS signaling in the gastrointestinal tract. Mucosal Immunol. 2018, 11, 1011–1023. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Conteh, A.M.; Reissaus, C.A.; Hernandez-Perez, M.; Nakshatri, S.; Anderson, R.M.; Mirmira, R.G.; Tersey, S.A.; Linnemann, A.K. Platelet-type 12-lipoxygenase deletion provokes a compensatory 12/15-lipoxygenase increase that exacerbates oxidative stress in mouse islet β cells. J. Biol. Chem. 2019, 294, 6612–6620. [Google Scholar] [CrossRef]
- Zhao, R.; Jiang, S.; Zhang, L.; Yu, Z. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Delierneux, C.; Kouba, S.; Shanmughapriya, S.; Potier-Cartereau, M.; Trebak, M.; Hempel, N. Mitochondrial Calcium Regulation of Redox Signaling in Cancer. Cells 2020, 9, 432. [Google Scholar] [CrossRef] [Green Version]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef]
- Zou, X.; Ratti, B.A.; O’Brien, J.G.; Lautenschlager, S.O.; Gius, D.R.; Bonini, M.G.; Zhu, Y. Manganese superoxide dismutase (SOD2): Is there a center in the universe of mitochondrial redox signaling? J. Bioenerg. Biomembr. 2017, 49, 325–333. [Google Scholar] [CrossRef]
- Arteel, G.E.; Sies, H. The biochemistry of selenium and the glutathione system. Environ. Toxicol. Pharmacol. 2001, 10, 153–158. [Google Scholar] [CrossRef]
- Cao, Z.; Lindsay, J.G. The Peroxiredoxin Family: An Unfolding Story. Subcell. Biochem. 2017, 83, 127–147. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Zafar, A.; Naseem, I. Copper-redox cycling by coumarin-di(2-picolyl)amine hybrid molecule leads to ROS-mediated DNA damage and apoptosis: A mechanism for cancer chemoprevention. Chem. Biol. Interact. 2018, 290, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Mishra, J.P.N.; Singh, R.P. Usnic acid induces apoptosis in human gastric cancer cells through ROS generation and DNA damage and causes up-regulation of DNA-PKcs and γ-H2A.X phosphorylation. Chem. Biol. Interact. 2020, 315, 108898. [Google Scholar] [CrossRef]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Del Pilar SosaIdelchik, M.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. Semin. Cancer Biol. 2017, 47, 57–66. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jendrach, M.; Mai, S.; Pohl, S.; Vöth, M.; Bereiter-Hahn, J. Short- and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion 2008, 8, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Hussien, R.; Brooks, G.A. H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free Radic. Biol. Med. 2010, 49, 1646–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, S.; Hood, D.A. Oxidative stress-induced mitochondrial fragmentation and movement in skeletal muscle myoblasts. Am. J. Physiol. Physiol. 2014, 306, C1176–C1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distelmaier, F.; Valsecchi, F.; Liemburg-Apers, D.C.; Lebiedzinska, M.; Rodenburg, R.J.; Heil, S.; Keijer, J.; Fransen, J.; Imamura, H.; Danhauser, K.; et al. Mitochondrial dysfunction in primary human fibroblasts triggers an adaptive cell survival program that requires AMPK-α. Biochim. Biophys. Acta - Mol. Basis Dis. 2015, 1852, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Trewin, A.; Berry, B.; Wojtovich, A. Exercise and Mitochondrial Dynamics: Keeping in Shape with ROS and AMPK. Antioxidants 2018, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Liesa, M.; Borda-d’Água, B.; Medina-Gómez, G.; Lelliott, C.J.; Paz, J.C.; Rojo, M.; Palacín, M.; Vidal-Puig, A.; Zorzano, A. Mitochondrial Fusion Is Increased by the Nuclear Coactivator PGC-1β. PLoS ONE 2008, 3, e3613. [Google Scholar] [CrossRef] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Rendra, E.; Riabov, V.; Mossel, D.M.; Sevastyanova, T.; Harmsen, M.C.; Kzhyshkowska, J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology 2019, 224, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/ RNS generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell. Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shao, L.; Pan, C.; Ye, J.; Ding, Z.; Wu, J.; Du, Q.; Ren, Y.; Zhu, C. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial–mesenchymal transition. Stem Cell Res. Ther. 2019, 10, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Li, P.; Xuan, J.; Zhu, C.; Liu, J.; Shan, L.; Du, Q.; Ren, Y.; Ye, J. Cholesterol Enhances Colorectal Cancer Progression via ROS Elevation and MAPK Signaling Pathway Activation. Cell. Physiol. Biochem. 2017, 42, 729–742. [Google Scholar] [CrossRef]
- Kim, B.-H.; Yi, E.H.; Ye, S.-K. Signal transducer and activator of transcription 3 as a therapeutic target for cancer and the tumor microenvironment. Arch. Pharm. Res. 2016, 39, 1085–1099. [Google Scholar] [CrossRef]
- Kim, D.H.; Park, K.W.; Chae, I.G.; Kundu, J.; Kim, E.H.; Kundu, J.K.; Chun, K.S. Carnosic acid inhibits STAT3 signaling and induces apoptosis through generation of ROS in human colon cancer HCT116 cells. Mol. Carcinog. 2016. [Google Scholar] [CrossRef]
- Meier, J.A.; Hyun, M.; Cantwell, M.; Raza, A.; Mertens, C.; Raje, V.; Sisler, J.; Tracy, E.; Torres-Odio, S.; Gispert, S.; et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci. Signal. 2017, 10, eaag2588. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-K.; Kwon, S.M.; Lee, E.; Kim, G.-H.; Min, S.; Hong, S.-M.; Wang, H.-J.; Lee, D.M.; Choi, K.S.; Park, T.J.; et al. Mitochondrial Respiratory Defect Enhances Hepatoma Cell Invasiveness via STAT3/NFE2L1/STX12 Axis. Cancers (Basel) 2020, 12, 2632. [Google Scholar] [CrossRef]
- Kim, B.; Song, Y.S. Mitochondrial dynamics altered by oxidative stress in cancer. Free Radic. Res. 2016, 50, 1065–1070. [Google Scholar] [CrossRef]
- Han, Y.; Kim, B.; Cho, U.; Park, I.S.; Kim, S.I.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Mitochondrial fission causes cisplatin resistance under hypoxic conditions via ROS in ovarian cancer cells. Oncogene 2019, 38, 7089–7105. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1–NRF2 System in Cancer. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Lacher, S.E.; Levings, D.C.; Freeman, S.; Slattery, M. Identification of a functional antioxidant response element at the HIF1A locus. Redox Biol. 2018, 19, 401–411. [Google Scholar] [CrossRef]
- Schroedl, C.; McClintock, D.S.; Budinger, G.R.S.; Chandel, N.S. Hypoxic but not anoxic stabilization of HIF-1α requires mitochondrial reactive oxygen species. Am. J. Physiol. Cell. Mol. Physiol. 2002, 283, L922–L931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.S.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Pastukh, V.; Roberts, J.T.; Clark, D.W.; Bardwell, G.C.; Patel, M.; Al-Mehdi, A.-B.; Borchert, G.M.; Gillespie, M.N. An oxidative DNA “damage” and repair mechanism localized in the VEGF promoter is important for hypoxia-induced VEGF mRNA expression. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L1367–L1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Liu, W.-Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Yang, R.; Piperdi, S.; Gorlick, R. Activation of the RAF/Mitogen-Activated Protein/Extracellular Signal-Regulated Kinase Kinase/Extracellular Signal-Regulated Kinase Pathway Mediates Apoptosis Induced by Chelerythrine in Osteosarcoma. Clin. Cancer Res. 2008, 14, 6396–6404. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Wang, J.; Tian, H.; Fu, G.-H. Mitochondrial ROS accumulation inhibiting JAK2/STAT3 pathway is a critical modulator of CYT997-induced autophagy and apoptosis in gastric cancer. J. Exp. Clin. Cancer Res. 2020, 39, 119. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Verzella, D.; Di Francesco, B.; Alesse, E.; Franzoso, G.; Zazzeroni, F. NF-κB and mitochondria cross paths in cancer: Mitochondrial metabolism and beyond. Semin. Cell Dev. Biol. 2020, 98, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, S.; Liu, P. Deferoxamine Enhanced Mitochondrial Iron Accumulation and Promoted Cell Migration in Triple-Negative MDA-MB-231 Breast Cancer Cells via a ROS-Dependent Mechanism. Int. J. Mol. Sci. 2019, 20, 4952. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Yuan, Z.; Zhang, Q.; Long, Z.; Chen, J.; Tang, Z.; Zhu, Y.; Chen, S.; Xu, J.; Yan, M.; et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy 2012, 8, 1798–1810. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.; Liu, L.; Zhu, Y.; Chen, Q. Molecular signaling toward mitophagy and its physiological significance. Exp. Cell Res. 2013, 319, 1697–1705. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Pallichankandy, S.; Rahman, A.; Thayyullathil, F.; Galadari, S. ROS-dependent activation of autophagy is a critical mechanism for the induction of anti-glioma effect of sanguinarine. Free Radic. Biol. Med. 2015, 89, 708–720. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Dewangan, J.; Tandon, D.; Srivastava, S.; Verma, A.K.; Yapuri, A.; Rath, S.K. Novel combination of salinomycin and resveratrol synergistically enhances the anti-proliferative and pro-apoptotic effects on human breast cancer cells. Apoptosis 2017, 22, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Miao, Y.; Liu, S. Withaferin A induces apoptosis by ROS-dependent mitochondrial dysfunction in human colorectal cancer cells. Biochem. Biophys. Res. Commun. 2018, 503, 2363–2369. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.Q.; Feng, Y.Y.; Luo, Y.H.; Zhai, Y.Q.; Ju, X.Y.; Feng, Y.C.; Sheng, Y.N.; Wang, J.R.; Yu, C.Q.; Jin, C.H. Quinalizarin induces ROS-mediated apoptosis via the MAPK, STAT3 and NF-κB signaling pathways in human breast cancer cells. Mol. Med. Rep. 2019, 20, 4576–4586. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Liu, C.; Luo, Y.; Piao, X.; Wang, Y.; Zhang, Y.; Wang, J.; Wang, H.; Xu, W.; Liu, Y.; et al. Quinalizarin exerts an anti-tumour effect on lung cancer A549 cells by modulating the Akt, MAPK, STAT3 and p53 signalling pathways. Mol. Med. Rep. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.-S.; Lee, K.; Ku, J.M.; Choi, Y.-J.; Mok, K.; Kim, D.; Cheon, C.; Ko, S.-G. Cucurbitacin D Induces G2/M Phase Arrest and Apoptosis via the ROS/p38 Pathway in Capan-1 Pancreatic Cancer Cell Line. Evid.-Based Complement. Altern. Med. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Chuang, K.-C.; Chang, C.-R.; Chang, S.-H.; Huang, S.-W.; Chuang, S.-M.; Li, Z.-Y.; Wang, S.-T.; Kao, J.-K.; Chen, Y.-J.; Shieh, J.-J. Imiquimod-induced ROS production disrupts the balance of mitochondrial dynamics and increases mitophagy in skin cancer cells. J. Dermatol. Sci. 2020, 98, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, Y.; Jiang, X.; Zhang, H.; Gao, Z.; Li, Y.; Fu, R.; Li, L.; Li, J.; Cui, H.; et al. ROS-mediated activation and mitochondrial translocation of CaMKII contributes to Drp1-dependent mitochondrial fission and apoptosis in triple-negative breast cancer cells by isorhamnetin and chloroquine. J. Exp. Clin. Cancer Res. 2019, 38, 225. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Cao, S.; Jiang, B.; Zhang, Q.; Donkor, P.O.; Zhu, Y.; Qiu, F.; Gao, X. Cetuximab enhances oridonin-induced apoptosis through mitochondrial pathway and endoplasmic reticulum stress in laryngeal squamous cell carcinoma cells. Toxicol. In Vitro 2020, 67, 104885. [Google Scholar] [CrossRef]
- Gilardini Montani, M.S.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; D’Orazi, G.; Faggioni, A.; Cirone, M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell. Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schexnayder, C.; Broussard, K.; Onuaguluchi, D.; Poché, A.; Ismail, M.; McAtee, L.; Llopis, S.; Keizerweerd, A.; McFerrin, H.; Williams, C. Metformin Inhibits Migration and Invasion by Suppressing ROS Production and COX2 Expression in MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3692. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Lanza-Jacoby, S. Metformin decreases growth of pancreatic cancer cells by decreasing reactive oxygen species: Role of NOX4. Biochem. Biophys. Res. Commun. 2015, 465, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.-Q.; Chen, X.; Cai, Q.; Yang, Z.-H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The role of pyroptosis in cancer: Pro-cancer or pro-“host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Zhang, J.; Liu, X.; Chen, H.; Ai, Y.; Cheng, K.; Sun, R.; Zhou, D.; Han, J.; Wu, Q. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018, 28, 1171–1185. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef]
- Ray, T.; Chakrabarti, M.K.; Pal, A. Hemagglutinin protease secreted by V. cholerae induced apoptosis in breast cancer cells by ROS mediated intrinsic pathway and regresses tumor growth in mice model. Apoptosis 2016, 21, 143–154. [Google Scholar] [CrossRef]
- Dolmans, D.E.J.G.J.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA. Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Van Straten, D.; Mashayekhi, V.; de Bruijn, H.; Oliveira, S.; Robinson, D. Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers (Basel) 2017, 9, 19. [Google Scholar] [CrossRef]
- Buytaert, E.; Dewaele, M.; Agostinis, P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim. Biophys. Acta - Rev. Cancer 2007, 1776, 86–107. [Google Scholar] [CrossRef]
- Vanlangenakker, N.; Berghe, T.; Krysko, D.; Festjens, N.; Vandenabeele, P. Molecular Mechanisms and Pathophysiology of Necrotic Cell Death. Curr. Mol. Med. 2008, 8, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Reiners, J.J.; Agostinis, P.; Berg, K.; Oleinick, N.L.; Kessel, D.H. Assessing autophagy in the context of photodynamic therapy. Autophagy 2010, 6, 7–18. [Google Scholar] [CrossRef]
- Inguscio, V.; Panzarini, E.; Dini, L. Autophagy Contributes to the Death/Survival Balance in Cancer PhotoDynamic Therapy. Cells 2012, 1, 464–491. [Google Scholar] [CrossRef]
- Nowis, D.; Szokalska, A.; Makowski, M.; Winiarska, M.; Golab, J. Improvement of anti-tumor activity of photodynamic therapy through inhibition of cytoprotective mechanism in tumor cells. In Proceedings of the Photodynamic Therapy: Back to the Future, Seattle, WA, USA, 13 July 2009; p. 73804F. [Google Scholar]
- Oleinick, N.L.; Morris, R.L.; Belichenko, I. The role of apoptosis in response to photodynamic therapy: What, where, why, and how. Photochem. Photobiol. Sci. 2002, 1, 1–21. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Weijer, R.; van Gulik, T.M.; Hamblin, M.R.; Heger, M. Tumor cell survival pathways activated by photodynamic therapy: A molecular basis for pharmacological inhibition strategies. Cancer Metastasis Rev. 2015, 34, 643–690. [Google Scholar] [CrossRef] [Green Version]
- Matroule, J.-Y.; Bonizzi, G.; Morlière, P.; Paillous, N.; Santus, R.; Bours, V.; Piette, J. Pyropheophorbide-a Methyl Ester-mediated Photosensitization Activates Transcription Factor NF-κB through the Interleukin-1 Receptor-dependent Signaling Pathway. J. Biol. Chem. 1999, 274, 2988–3000. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Li, L. Photodynamic combinational therapy in cancer treatment. J. BUON. 2018, 23, 561–567. [Google Scholar] [PubMed]
- Sun, C.-Y.; Cao, Z.; Zhang, X.-J.; Sun, R.; Yu, C.-S.; Yang, X. Cascade-amplifying synergistic effects of chemo-photodynamic therapy using ROS-responsive polymeric nanocarriers. Theranostics 2018, 8, 2939–2953. [Google Scholar] [CrossRef]
- Rengeng, L.; Qianyu, Z.; Yuehong, L.; Zhongzhong, P.; Libo, L. Sonodynamic therapy, a treatment developing from photodynamic therapy. Photodiagnosis Photodyn. Ther. 2017, 19, 159–166. [Google Scholar] [CrossRef]
- Yang, Y.; Tu, J.; Yang, D.; Raymond, J.L.; Roy, R.A.; Zhang, D. Photo- and Sono-Dynamic Therapy: A Review of Mechanisms and Considerations for Pharmacological Agents Used in Therapy Incorporating Light and Sound. Curr. Pharm. Des. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gao, W.; Yang, Y.; Guo, S.; Wang, H.; Wang, W.; Zhang, S.; Zhou, Q.; Xu, H.; Yao, J.; et al. Inhibition of VDAC1 prevents Ca2+-mediated oxidative stress and apoptosis induced by 5-aminolevulinic acid mediated sonodynamic therapy in THP-1 macrophages. Apoptosis 2014, 19, 1712–1726. [Google Scholar] [CrossRef] [PubMed]
- Ming, L.; Cheng, K.; Chen, Y.; Yang, R.; Chen, D. Enhancement of tumor lethality of ROS in photodynamic therapy. Cancer Med. 2021, 10, 257–268. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Pharmacological Treatments | Cancer Types | Cell Lines | Mechanism of Action | Reference |
|---|---|---|---|---|
| Resveratrol + salinomycin | Breast cancer | MCF-7 | ↑ ROS impairs mitochondrial membrane potential; decreased Bcl2 expression, activation of caspases 7,8,9, chromatin condensation, PARP cleavage, apoptosis | [100] |
| Resveratrol + salinomycin | Breast cancer | MCF-7 | ↑ ROS activates MAPK pathway, phosphorylates JNK and p38, leading to apoptosis | [100] |
| Withaferin A (WA) | Colorectal cancer | HCT-116, RKO | ↑ ROS reduces mitochondrial membrane potential, decreasing Bcl-2/Bax ratio, activating caspase 3–9, leading to apoptosis, and activating JNK pathway | [101] |
| Carnosic Acid (CA) | Colon cancer | HCT-116 | ↑ ROS diminishes STAT3 phosphorylation, decreasing STAT3 gene products | [76] |
| Quinalizarin | Breast cancer | MCF-7 | ↑ ROS affects MAPK, STAT3, and NF-κB signaling pathways, inducing cell-cycle arrest and apoptosis | [102] |
| Quinalizarin | Lung cancer | A549 | ↑ ROS affects MAPK, STAT3, and NF-κB signaling pathways, inducing cell-cycle arrest and apoptosis | [103] |
| Cucurbitacin (CuD) | Pancreatic cancer | Capan-1 | ↑ ROS induces G2/M cell-cycle arrest and mediates p38/MAPK pathway, promoting cell death | [104] |
| Imiquimod (IMQ) | Skin cancer | BCC/KMC-1, B16F10 and A375 | ↑ ROS causes mitochondrial membrane potential loss, mitochondrial fission, and mitophagy | [105] |
| Isorhamnetin (IH) + chloroquine (CQ) | Breast cancer | MDA-MB-231, MCF-7, BT549, MCF-10A | ROS-mediated phosphorylation of CaMKII/Drp1 promotes Bax translocation and release of cytochrome c, mitochondrial fission, caspase activation, and apoptosis | [106] |
| Cetuximab + oridonin | Laryngeal cancer | Hep-2, Tu212 | ↑ ROS, through NF-κB, PI3K/Akt, and JAK2/STAT3, induces apoptosis | [107] |
| Valproic acid (VPA) + Trichostatin A (TSA) | Pancreatic cancer | PANC1, PaCa44 | ↑ ROS triggers autophagy | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brillo, V.; Chieregato, L.; Leanza, L.; Muccioli, S.; Costa, R. Mitochondrial Dynamics, ROS, and Cell Signaling: A Blended Overview. Life 2021, 11, 332. https://doi.org/10.3390/life11040332
Brillo V, Chieregato L, Leanza L, Muccioli S, Costa R. Mitochondrial Dynamics, ROS, and Cell Signaling: A Blended Overview. Life. 2021; 11(4):332. https://doi.org/10.3390/life11040332
Chicago/Turabian StyleBrillo, Valentina, Leonardo Chieregato, Luigi Leanza, Silvia Muccioli, and Roberto Costa. 2021. "Mitochondrial Dynamics, ROS, and Cell Signaling: A Blended Overview" Life 11, no. 4: 332. https://doi.org/10.3390/life11040332