
Automated Exploration of Prebiotic Chemical Reaction Space: Progress and Perspectives




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
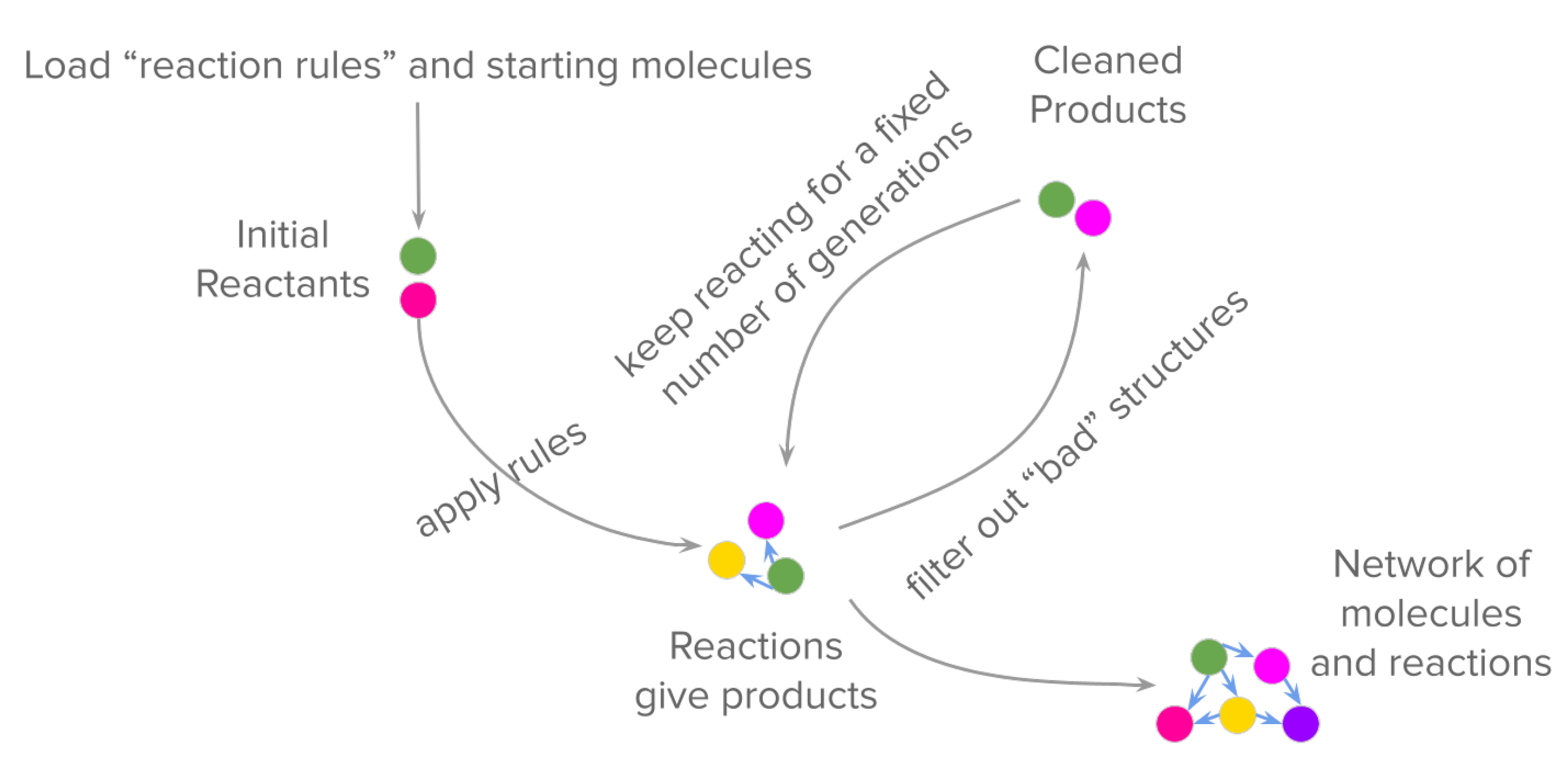
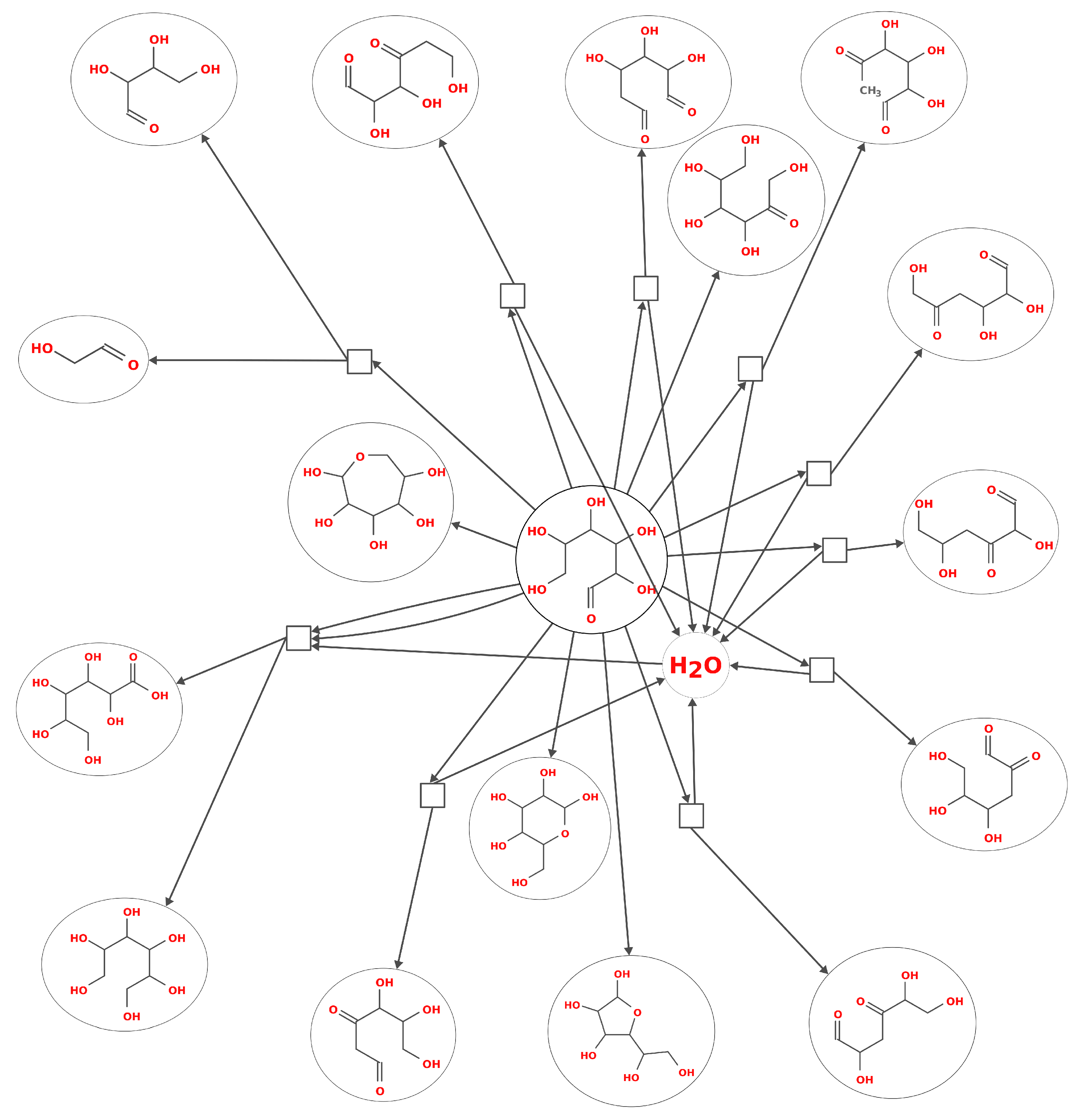
2. Modelling Prebiotic Chemistry: From Individual Reactions to a Network
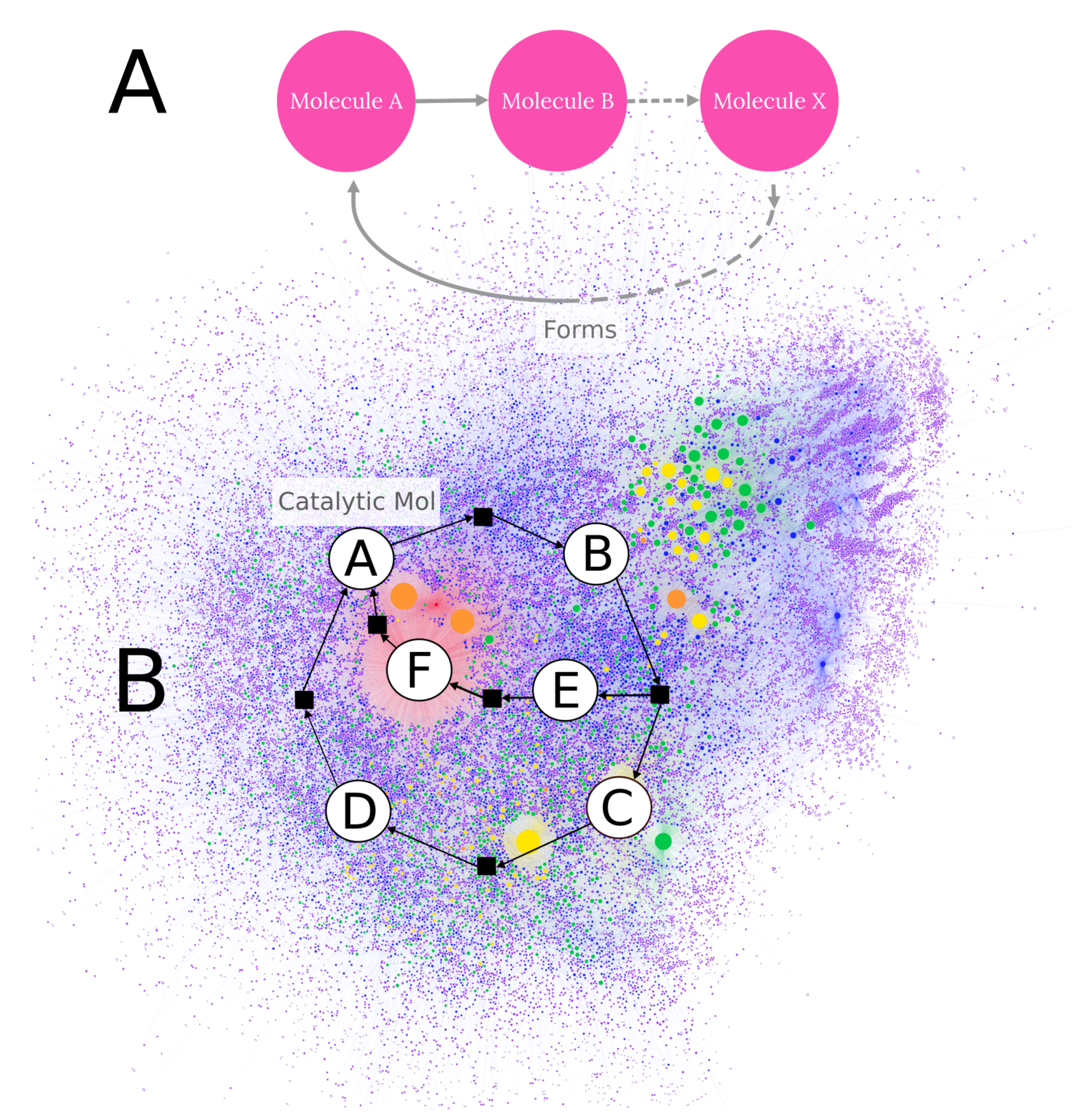
3. Detection of Autocatalytic Motifs in Computed Chemical Networks
4. Use of Machine Learning (ML) for Understanding CRNs
5. Problem-Specific Cheminformatic Tools and Approaches
5.1. Computing Molecular Descriptors
5.2. Broad Functionality Chemoinformatics Tools
5.3. Handling Isomerism
5.4. Miscellaneous Tools
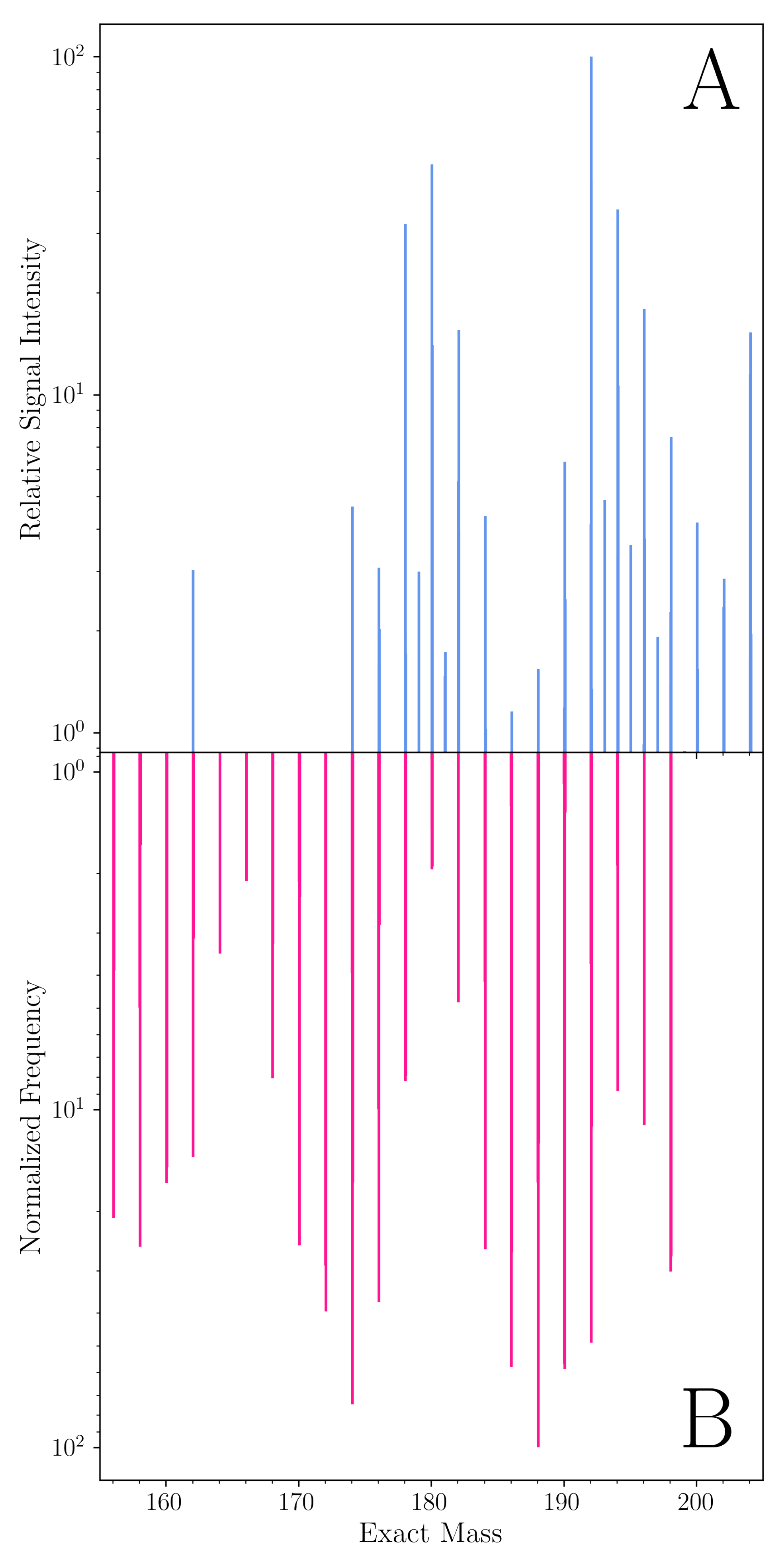
6. Experimental Vetting of the Computational Methods
7. Visualization of Chemically Relevant Datasets
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robinson, W.; Daines, E.; van Duppen, P.; de Jong, T.; Huck, W. Environmental Conditions Drive Self-Organisation of Reaction Pathways in a Prebiotic Reaction Network; 2021; Available online: https://www.researchsquare.com/article/rs-775456/v1 (accessed on 20 October 2021).
- Cleaves, H.J. Prebiotic chemistry: What we know, what we don’t. Evol. Edu. Outreach 2012, 5, 342–360. [Google Scholar] [CrossRef] [Green Version]
- Cleaves, H.J., II. Prebiotic chemistry: Geochemical context and reaction screening. Life 2013, 3, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Mirazo, K.; Briones, C.; de la Escosura, A. Prebiotic systems chemistry: New perspectives for the origins of life. Chem. Rev. 2014, 2014, 1. [Google Scholar] [CrossRef]
- Islam, S.; Powner, M.W. Prebiotic systems chemistry: Complexity overcoming clutter. Chem 2017, 2, 470–501. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Villa, A.; Pietrucci, F.; Saitta, A.M. Prebiotic chemistry and origins of life research with atomistic computer simulations. Phys. Life Rev. 2020, 34–35, 105–135. [Google Scholar] [CrossRef]
- Cheng, G.J.; Zhang, X.; Chung, L.W.; Xu, L.; Wu, Y.D. Computational organic chemistry: Bridging theory and experiment in establishing the mechanisms of chemical reactions. J. Am. Chem. Soc. 2015, 137, 1706–1725. [Google Scholar] [CrossRef]
- Andersen, J.L.; Andersen, T.; Flamm, C.; Hanczyc, M.M.; Merkle, D.; Stadler, P.F. Navigating the chemical space of HCN polymerization and hydrolysis: Guiding graph grammars by mass spectrometry data. Entropy 2013, 15, 4066–4083. [Google Scholar] [CrossRef] [Green Version]
- Tran, Q.P.; Adam, Z.R.; Fahrenbach, A.C. Prebiotic reaction networks in water. Life 2020, 10, 352. [Google Scholar] [CrossRef]
- Yi, R.; Tran, Q.P.; Ali, S.; Yoda, I.; Adam, Z.R.; Cleaves, H.J.; Fahrenbach, A.C. A continuous reaction network that produces RNA precursors. Proc. Natl. Acad. Sci. USA 2020, 117, 13267–13274. [Google Scholar] [CrossRef] [PubMed]
- Vasas, V.; Szathmáry, E.; Santos, M. Lack of evolvability in self-sustaining autocatalytic networks constraints metabolism-first scenarios for the origin of life. Proc. Natl. Acad. Sci. USA 2010, 107, 1470–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butch, C.J.; Meringer, M.; Gagnon, J.S.; Cleaves, H.J. Open questions in understanding life’s origins. Commun. Chem. 2021, 4, 1–4. [Google Scholar] [CrossRef]
- Shapiro, R. Small molecule interactions were central to the origin of life. Q. Rev. Biol. 2006, 81, 105–125. [Google Scholar] [CrossRef] [Green Version]
- Cronin, L.; Walker, S.I. Origin of life. Beyond prebiotic chemistry. Science 2016, 352, 1174–1175. [Google Scholar] [CrossRef] [Green Version]
- Meringer, M.; Cleaves, H.J. Exploring astrobiology using in silico molecular structure generation. Philos. Trans. R. Soc. A 2017, 375, 20160344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, C.; Rimmer, P.B.; Williams, H.; Shorttle, O. Prebiotic chemistry in the wild: How geology interferes with the origins of life. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Surman, A.J.; Rodriguez-Garcia, M.; Abul-Haija, Y.M.; Cooper, G.J.T.; Gromski, P.S.; Turk-MacLeod, R.; Mullin, M.; Mathis, C.; Walker, S.I.; Cronin, L. Environmental control programs the emergence of distinct functional ensembles from unconstrained chemical reactions. Proc. Natl. Acad. Sci. USA 2019, 116, 5387–5392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromski, P.S.; Henson, A.B.; Granda, J.M.; Cronin, L. How to explore chemical space using algorithms and automation. Nat. Rev. Chem. 2019, 3, 119–128. [Google Scholar] [CrossRef]
- Wołos, A.; Roszak, R.; Żądło Dobrowolska, A.; Beker, W.; Mikulak-Klucznik, B.; Spólnik, G.; Dygas, M.; Szymkuć, S.; Grzybowski, B.A. Synthetic connectivity, emergence, and self-regeneration in the network of prebiotic chemistry. Science 2020, 369, eaaw1955. [Google Scholar] [CrossRef]
- Dewyer, A.L.; Argüelles, A.J.; Zimmerman, P.M. Methods for exploring reaction space in molecular systems. WIREs Comput. Mol. Sci. 2018, 8, e1354. [Google Scholar] [CrossRef]
- Coley, C.W. Defining and exploring chemical spaces. Trends Chem. 2021, 3, 133–145. [Google Scholar] [CrossRef]
- Walker, S.I.; Mathis, C. Network teory in prebiotic evolution. In Prebiotic Chemistry and Chemical Evolution of Nucleic Acids; Springer: Cham, Switzerland, 2018; pp. 263–291. [Google Scholar] [CrossRef]
- Smith, H.B.; Kim, H.; Walker, S.I. Scarcity of scale-free topology is universal across biochemical networks. Sci. Rep. 2021, 11, 6542. [Google Scholar] [CrossRef]
- Das, T.; Ghule, S.; Vanka, K. Insights into the origin of life: Did it begin from HCN and H2O? ACS Cent. Sci. 2019, 5, 1532–1540. [Google Scholar] [CrossRef] [Green Version]
- Magrino, T.; Pietrucci, F.; Saitta, A.M. Step by step strecker amino acid synthesis from ab initio prebiotic chemistry. J. Phys. Chem. Lett. 2021, 12, 2630–2637. [Google Scholar] [CrossRef]
- Marshall, S.M.; Mathis, C.; Carrick, E.; Keenan, G.; Cooper, G.J.T.; Graham, H.; Craven, M.; Gromski, P.S.; Moore, D.G.; Walker, S.I.; et al. Identifying molecules as biosignatures with assembly theory and mass spectrometry. Nat. Commun. 2021, 12, 3033. [Google Scholar] [CrossRef]
- Liu, Y.; Mathis, C.; Bajczyk, M.D.; Marshall, S.M.; Wilbraham, L.; Cronin, L. Exploring and mapping chemical space with molecular assembly trees. Sci. Adv. 2021, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- López-López, E.; Bajorath, J.; Medina-Franco, J.L. Informatics for chemistry, biology, and biomedical sciences. J. Chem. Inf. Model. 2021, 61, 26–35. [Google Scholar] [CrossRef]
- Pirhadi, S.; Sunseri, J.; Koes, D.R. Open source molecular modeling. J. Mol. Graph. Model. 2016, 69, 127–143. [Google Scholar] [CrossRef] [Green Version]
- González-Medina, M.; Naveja, J.J.; Sánchez-Cruz, N.; Medina-Franco, J.L. Open chemoinformatic resources to explore the structure, properties and chemical space of molecules. RSC Adv. 2017, 7, 54153–54163. [Google Scholar] [CrossRef] [Green Version]
- Medina-Franco, J.L.; Sánchez-Cruz, N.; López-López, E.; Díaz-Eufracio, B.I. Progress on open chemoinformatic tools for expanding and exploring the chemical space. J. Comput.-Aided Mol. Des. 2021. [Google Scholar] [CrossRef] [PubMed]
- Guttenberg, N.; Chen, H.; Mochizuki, T.; Cleaves, H.J. Classification of the biogenicity of complex organic mixtures for the detection of extraterrestrial life. Life 2021, 11, 234. [Google Scholar] [CrossRef] [PubMed]
- Rimmer, P.B.; Helling, C. A chemical kinetics network for lightning and life in planetary atmospheres. Astrophys. J. Suppl. Ser. 2016, 224, 9. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Kim, J.W.; Kim, W.Y. Efficient construction of a chemical reaction network guided by a Monte Carlo tree search. ChemSystemsChem 2020, 2, e1900057. [Google Scholar] [CrossRef]
- Barone, V.; Biczysko, M.; Puzzarini, C. Quantum chemistry meets spectroscopy for astrochemistry: Increasing complexity toward prebiotic molecules. Acc. Chem. Res. 2015, 48, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Simm, G.N.; Reiher, M. Context-driven exploration of complex chemical reaction networks. J. Chem. Theory Comput. 2017, 13, 6108–6119. [Google Scholar] [CrossRef] [Green Version]
- Simm, G.; Vaucher, A.C.; Reiher, M. Exploration of reaction pathways and chemical transformation networks. J. Phys. Chem. A 2019, 123, 385–399. [Google Scholar] [CrossRef] [Green Version]
- Nghe, P.; Hordijk, W.; Kauffman, S.A.; Walker, S.I.; Schmidt, F.J.; Kemble, H.; Yeates, J.A.M.; Lehman, N. Prebiotic network evolution: Six key parameters. Mol. Biosyst. 2015, 11, 3206–3217. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, J.W.; Kim, Z.; Kim, W.Y. Efficient prediction of reaction paths through molecular graph and reaction network analysis. Chem. Sci. 2018, 9, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Blau, S.M.; Patel, H.D.; Spotte-Smith, E.W.C.; Xie, X.; Dwaraknath, S.; Persson, K.A. A chemically consistent graph architecture for massive reaction networks applied to solid-electrolyte interphase formation. Chem. Sci. 2021, 12, 4931–4939. [Google Scholar] [CrossRef]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. A software package for chemically inspired graph transformation. arXiv 2016, arXiv:1603.02481. [Google Scholar]
- Ratkiewicz, A.; Truong, T.N. Application of chemical graph theory for automated mechanism generation. J. Chem. Inf. Comput. Sci. 2003, 43, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temkin, O.N.; Zeigarnik, A.V.; Bonchev, D.G. Chemical Reaction Networks; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- Pearce, B.K.D.; Ayers, P.W.; Pudritz, R.E. A consistent reduced network for HCN chemistry in early earth and Titan atmospheres: Quantum calculations of reaction rate coefficients. J. Phys. Chem. A 2019. [Google Scholar] [CrossRef] [Green Version]
- St. John, P.C.; Guan, Y.; Kim, Y.; Etz, B.D.; Kim, S.; Paton, R.S. Quantum chemical calculations for over 200,000 organic radical species and 40,000 associated closed-shell molecules. Sci. Data 2020, 7, 244. [Google Scholar] [CrossRef] [PubMed]
- Kolska, Z.; Za’bransky, M.; Randova, A. Group contribution methods for estimation of selected physico-chemical properties of organic compounds. In Thermodynamics—Fundamentals and Its Application in Science; IntechOpen: Rijeka, Croatia, 2012. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Borchardt, T.B. JRgui: A Python program of Joback and Reid method. ACS Omega 2017, 2, 8682–8688. [Google Scholar] [CrossRef]
- Beber, M.E.; Gollub, M.G.; Mozaffari, D.; Shebek, K.M.; Noor, E. eQuilibrator 3.0—A Platform for the Estimation of Thermodynamic Constants. 2021. Available online: http://xxx.lanl.gov/abs/2103.00621 (accessed on 20 October 2021).
- Python Group Additivity (pgradd) Documentation. Available online: https://vlachosgroup.github.io/PythonGroupAdditivity/ (accessed on 20 October 2021).
- Noor, E.; Haraldsdóttir, H.S.; Milo, R.; Fleming, R.M.T. Consistent estimation of Gibbs energy using component contributions. PLoS Comput. Biol. 2013, 9, e1003098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.J.P. MOPAC: A semiempirical molecular orbital program. J. Comput. Aided Mol. Des. 1990, 4, 1–103. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; Rodríguez-Fernández, R.; Vázquez, S.A.; Barnes, G.L.; Stewart, J.J.P.; Martínez-Núñez, E. tsscds2018: A code for automated discovery of chemical reaction mechanisms and solving the kinetics. J. Comput. Chem. 2018, 39, 1922–1930. [Google Scholar] [CrossRef]
- Kaur, S.; Sharma, P.; Wetmore, S.D. Can cyanuric acid and 2,4,6-Triaminopyrimidine containing ribonucleosides be components of prebiotic RNA? Insights from QM calculations and MD simulations. ChemPhysChem 2019, 20, 1425–1436. [Google Scholar] [CrossRef]
- Kahana, A.; Lancet, D. Protobiotic systems chemistry analyzed by molecular dynamics. Life 2019, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Kua, J.; Hernandez, A.L.; Velasquez, D.N. Thermodynamics of potential CHO metabolites in a reducing environment. Life 2021, 11, 1025. [Google Scholar] [CrossRef]
- Hoksza, D.; Škoda, P.; Voršilák, M.; Svozil, D. Molpher: A software framework for systematic chemical space exploration. J. Cheminf. 2014, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Cao, L.; Chin, C.H.; Ren, H.; Zhang, J.Z.H.; Zhu, T. ReacNetGenerator: An automatic reaction network generator for reactive molecular dynamics simulations. Phys. Chem. Chem. Phys. 2020, 22, 683–691. [Google Scholar] [CrossRef]
- Gao, C.W.; Allen, J.W.; Green, W.H.; West, R.H. Reaction Mechanism Generator: Automatic construction of chemical kinetic mechanisms. Comput. Phys. Commun. 2016, 203, 212–225. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Grinberg Dana, A.; Johnson, M.S.; Goldman, M.J.; Jocher, A.; Payne, A.M.; Grambow, C.A.; Han, K.; Yee, N.W.; Mazeau, E.J.; et al. Reaction Mechanism Generator v3.0: Advances in Automatic Mechanism Generation. J. Chem. Inf. Model. 2021, 61, 2686–2696. [Google Scholar] [CrossRef]
- Nugmanov, R.I.; Mukhametgaleev, R.N.; Akhmetshin, T.; Gimadiev, T.R.; Afonina, V.A.; Madzhidov, T.I.; Varnek, A. CGRtools: Python library for molecule, reaction, and condensed graph of reaction processing. J. Chem. Inf. Model. 2019, 59, 2516–2521. [Google Scholar] [CrossRef] [PubMed]
- Gupta, U.; Le, T.; Hu, W.S.; Bhan, A.; Daoutidis, P. Automated network generation and analysis of biochemical reaction pathways using RING. Metab. Eng. 2018, 49, 84–93. [Google Scholar] [CrossRef]
- Gupta, U.; Vlachos, D.G. Learning chemistry of complex reaction systems via a python first-principles Reaction rule Stencil (pReSt) generator. J. Chem. Inf. Model. 2021, 61, 3431–3441. [Google Scholar] [CrossRef]
- Kazeroonian, A.; Fröhlich, F.; Raue, A.; Theis, F.J.; Hasenauer, J. CERENA: ChEmical REaction Network Analyzer—A toolbox for the simulation and analysis of stochastic chemical kinetics. PLoS ONE 2016, 11, e0146732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. Chemical graph transformation with stereo-information. In Graph Transformation; Springer: Cham, Switzerland, 2017; pp. 54–69. [Google Scholar] [CrossRef]
- Laurent, G.; Lacoste, D.; Gaspard, P. Emergence of homochirality in large molecular systems. Proc. Natl. Acad. Sci. USA 2021, 118, e2012741118. [Google Scholar] [CrossRef]
- Coley, C.; Green, W.H.; Jensen, K.F. RDChiral: An RDKit wrapper for handling stereochemistry in retrosynthetic template extraction and application. J. Chem. Inf. Model. 2019, 59, 2529–2537. [Google Scholar] [CrossRef] [PubMed]
- rdkit.Chem.EnumerateStereoisomers Module—The RDKit 2021.03.1 Documentation. Available online: https://www.rdkit.org/docs/source/rdkit.Chem.EnumerateStereoisomers.html (accessed on 20 October 2021).
- Yirik, M.A.; Sorokina, M.; Steinbeck, C. MAYGEN: An open-source chemical structure generator for constitutional isomers based on the orderly generation principle. J. Cheminf. 2021, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- Gánti, T. Organization of chemical reactions into dividing and metabolizing units: The chemotons. Biosystems 1975, 7, 15–21. [Google Scholar] [CrossRef]
- Kauffman, S.A. Autocatalytic sets of proteins. J. Theor. Biol. 1986, 119, 1–24. [Google Scholar] [CrossRef]
- Wächtershäuser, G. Before enzymes and templates: Theory of surface metabolism. Microbiol. Rev. 1988, 52, 452. [Google Scholar] [CrossRef]
- Kauffman, S.A. The Origins of Order; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Vaidya, N.; Manapat, M.L.; Chen, I.A.; Xulvi-Brunet, R.; Hayden, E.J.; Lehman, N. Spontaneous network formation among cooperative RNA replicators. Nature 2012, 491, 72–77. [Google Scholar] [CrossRef]
- Tjhung, K.F.; Shokhirev, M.N.; Horning, D.P.; Joyce, G.F. An RNA polymerase ribozyme that synthesizes its own ancestor. Proc. Natl. Acad. Sci. USA 2020, 117, 2906–2913. [Google Scholar] [CrossRef]
- Kauffman, S.; Steel, M. The expected number of viable autocatalytic sets in chemical reaction systems. Artif. Life 2021, 27, 1–14. [Google Scholar] [CrossRef]
- Virgo, N.; Ikegami, T.; McGregor, S. Complex autocatalysis in simple chemistries. Artif. Life 2016, 22, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Jeilani, Y.A.; Nguyen, M.T. Autocatalysis in formose reaction and formation of RNA nucleosides. J. Phys. Chem. B 2020, 124, 11324–11336. [Google Scholar] [CrossRef]
- Schwartz, A.W.; Goverde, M. Acceleration of HCN oligomerization by formaldehyde and related compounds: Implications for prebiotic syntheses. J. Mol. Evol. 1982, 18, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, W.; Steel, M. Detecting autocatalytic, self-sustaining sets in chemical reaction systems. J. Theor. Biol. 2004, 227, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Kun, Á.; Papp, B.; Szathmáry, E. Computational identification of obligatorily autocatalytic replicators embedded in metabolic networks. Genome Biol. 2008, 9, R51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preiner, M.; Xavier, J.C.; do Nascimento Vieira, A.; Kleinermanns, K.; Allen, J.F.; Martin, W.F. Catalysts, autocatalysis and the origin of metabolism. Interface Focus 2019, 9, 20190072. [Google Scholar] [CrossRef]
- Steel, M.; Hordijk, W.; Xavier, J.C. Autocatalytic networks in biology: Structural theory and algorithms. J. R. Soc. Interface 2019, 16, 20180808. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Linderoth, J.; Baum, D. A mechanism of abiogenesis based on complex reaction networks organized by seed-dependent autocatalytic systems. ChemRxiv 2021. [Google Scholar] [CrossRef]
- Luo, Y.; Epstein, I.R. Feedback analysis of mechanisms for chemical oscillators. In Advances in Chemical Physics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1990; pp. 269–299. [Google Scholar] [CrossRef]
- Xavier, J.C.; Hordijk, W.; Kauffman, S.; Steel, M.; Martin, W.F. Autocatalytic chemical networks at the origin of metabolism. Proc. R. Soc. B 2020, 287, 20192377. [Google Scholar] [CrossRef]
- Adam, Z.R.; Fahrenbach, A.C.; Kacar, B.; Aono, M. Prebiotic geochemical automata at the intersection of radiolytic chemistry, physical complexity, and systems biology. Complexity 2018, 2018, 9376183. [Google Scholar] [CrossRef]
- Adam, Z.R.; Fahrenbach, A.C.; Jacobson, S.M.; Kacar, B.; Zubarev, D.Y. Radiolysis generates a complex organosynthetic chemical network. Sci. Rep. 2021, 11, 1743. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Arriaga, A.; Meléndez-López, A.; Heredia, A.; Cruz-Castañeda, J.; Negrón-Mendoza, A.; Ramos-Bernal, S. Role of Na+-montmorillonite in the stability of guanine exposed to high-radiation energy in primitive environments: Heterogeneous models. Radiat. Phys. Chem. 2021, 186, 109509. [Google Scholar] [CrossRef]
- Pastorek, A.; Ferus, M.; Čuba, V.; Šrámek, O.; Ivanek, O.; Civiš, S. Primordial radioactivity and prebiotic chemical evolution: Effect of γ radiation on formamide-based synthesis. J. Phys. Chem. B 2020, 124, 8951–8959. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Orgel, L.E. The Origins of Life on the Earth; Prentice-Hall: Upper Saddle River, NJ, USA, 1974. [Google Scholar]
- Cafferty, B.; Wong, A.S.Y.; Semenov, S.N.; Belding, L.; Gmür, S.; Huck, W.T.S.; Whitesides, G.M. Robustness, entrainment, and hybridization in dissipative molecular networks, and the origin of life. J. Am. Chem. Soc. 2019, 141, 8289–8295. [Google Scholar] [CrossRef] [PubMed]
- Semenov, S.N.; Kraft, L.J.; Ainla, A.; Zhao, M.; Baghbanzadeh, M.; Campbell, V.E.; Kang, K.; Fox, J.M.; Whitesides, G.M. Autocatalytic, bistable, oscillatory networks of biologically relevant organic reactions. Nature 2016, 537, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. Defining autocatalysis in chemical reaction networks. arXiv 2021, arXiv:2107.03086. [Google Scholar]
- Zubarev, D.Y.; Rappoport, D.; Aspuru-Guzik, A. Uncertainty of prebiotic scenarios: The case of the non-enzymatic reverse tricarboxylic acid cycle. Sci. Rep. 2015, 5, 8009. [Google Scholar] [CrossRef] [Green Version]
- Meringer, M.; Cleaves, H.J. Computational exploration of the chemical structure space of possible reverse tricarboxylic acid cycle constituents. Sci. Rep. 2017, 7, 17540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. In silico support for Eschenmoser’s glyoxylate scenario. Isr. J. Chem. 2015, 55, 919–933. [Google Scholar] [CrossRef]
- Strieth-Kalthoff, F.; Sandfort, F.; Segler, M.H.S.; Glorius, F. Machine learning the ropes: Principles, applications and directions in synthetic chemistry. Chem. Soc. Rev. 2020, 49, 6154–6168. [Google Scholar] [CrossRef] [PubMed]
- Keith, J.A.; Vassilev-Galindo, V.; Cheng, B.; Chmiela, S.; Gastegger, M.; Müller, K.R.; Tkatchenko, A. Combining machine learning and computational chemistry for predictive insights Into chemical systems. Chem. Rev. 2021, 121, 9816–9872. [Google Scholar] [CrossRef]
- Stocker, S.; Csányi, G.; Reuter, K.; Margraf, J.T. Machine learning in chemical reaction space. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Plehiers, P.P.; Marin, G.B.; Stevens, C.V.; Van Geem, K.M. Automated reaction database and reaction network analysis: Extraction of reaction templates using cheminformatics. J. Cheminf. 2018, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.P.; Barzilay, R. Applications of deep learning in molecule generation and molecular property prediction. Acc. Chem. Res. 2021, 54, 263–270. [Google Scholar] [CrossRef]
- Kayala, M.A.; Baldi, P. ReactionPredictor: Prediction of complex chemical reactions at the mechanistic level using machine learning. J. Chem. Inf. Model. 2012, 52, 2526–2540. [Google Scholar] [CrossRef] [PubMed]
- Coley, C.W.; Barzilay, R.; Jaakkola, T.S.; Green, W.H.; Jensen, K.F. Prediction of organic reaction outcomes using machine learning. ACS Cent. Sci. 2017, 3, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.N.; Duvenaud, D.; Aspuru-Guzik, A. Neural networks for the prediction of organic chemistry reactions. ACS Cent. Sci. 2016, 2, 725–732. [Google Scholar] [CrossRef]
- Pathak, Y.; Mehta, S.; Priyakumar, U.D. Learning atomic interactions through solvation free energy prediction using graph neural networks. J. Chem. Inf. Model. 2021, 61, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Skoraczyński, G.; Dittwald, P.; Miasojedow, B.; Szymkuć, S.; Gajewska, E.P.; Grzybowski, B.A.; Gambin, A. Predicting the outcomes of organic reactions via machine learning: Are current descriptors sufficient? Sci. Rep. 2017, 7, 3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Struble, T.J.; Coley, C.W.; Wang, Y.; Green, W.H.; Jensen, K.F. Using machine learning to predict suitable conditions for organic reactions. ACS Cent. Sci. 2018, 4, 1465–1476. [Google Scholar] [CrossRef] [Green Version]
- Sandfort, F.; Strieth-Kalthoff, F.; Kühnemund, M.; Beecks, C.; Glorius, F. A structure-based platform for predicting chemical reactivity. Chem 2020, 6, 1379–1390. [Google Scholar] [CrossRef]
- Fooshee, D.; Mood, A.; Gutman, E.; Tavakoli, M.; Urban, G.; Liu, F.; Huynh, N.; Van Vranken, D.; Baldi, P. Deep learning for chemical reaction prediction. Mol. Syst. Des. Eng. 2018, 3, 442–452. [Google Scholar] [CrossRef]
- Schwaller, P.; Vaucher, A.C.; Laino, T.; Reymond, J.L. Prediction of chemical reaction yields using deep learning. Mach. Learn. Sci. Technol. 2021, 2, 015016. [Google Scholar] [CrossRef]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C.A.; Bekas, C.; Lee, A.A. Molecular transformer: A model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 2019, 5, 1572–1583. [Google Scholar] [CrossRef] [Green Version]
- Schwaller, P.; Petraglia, R.; Zullo, V.; Nair, V.H.; Haeuselmann, R.A.; Pisoni, R.; Bekas, C.; Iuliano, A.; Laino, T. Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy. Chem. Sci. 2020, 11, 3316–3325. [Google Scholar] [CrossRef] [Green Version]
- Kreutter, D.; Schwaller, P.; Reymond, J.L. Predicting enzymatic reactions with a molecular transformer. Chem. Sci. 2021, 12, 8648–8659. [Google Scholar] [CrossRef]
- Meuwly, M. Machine learning for chemical reactions. Chem. Rev. 2021, 121, 10218–10239. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Jorner, K.; Tomberg, A.; Bauer, C.; Sköld, C.; Norrby, P.O. Organic reactivity from mechanism to machine learning. Nat. Rev. Chem. 2021, 5, 240–255. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- RDKit: Open-Source Cheminformatics. Available online: https://rdkit.org/ (accessed on 20 October 2021).
- Dong, J.; Cao, D.S.; Miao, H.Y.; Liu, S.; Deng, B.C.; Yun, Y.H.; Wang, N.N.; Lu, A.P.; Zeng, W.B.; Chen, A.F. ChemDes: An integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminf. 2015, 7, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.S.; Xu, Q.S.; Hu, Q.N.; Liang, Y.Z. ChemoPy: Freely available python package for computational biology and chemoinformatics. Bioinformatics 2013, 29, 1092–1094. [Google Scholar] [CrossRef]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminf. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willighagen, E.L.; Mayfield, J.W.; Alvarsson, J.; Berg, A.; Carlsson, L.; Jeliazkova, N.; Kuhn, S.; Pluskal, T.; Rojas-Chertó, M.; Spjuth, O.; et al. The Chemistry Development Kit (CDK) v2.0: Atom typing, depiction, molecular formulas, and substructure searching. J. Cheminf. 2017, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Yao, Z.J.; Zhang, L.; Luo, F.; Lin, Q.; Lu, A.P.; Chen, A.F.; Cao, D.S. PyBioMed: A python library for various molecular representations of chemicals, proteins and DNAs and their interactions. J. Cheminf. 2018, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Bento, A.P.; Hersey, A.; Félix, E.; Landrum, G.; Gaulton, A.; Atkinson, F.; Bellis, L.J.; De Veij, M.; Leach, A.R. An open source chemical structure curation pipeline using RDKit. J. Cheminf. 2020, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- Indigo Toolkit. Available online: https://lifescience.opensource.epam.com/indigo/ (accessed on 20 October 2021).
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Hutchison, G.R. Cinfony – combining open source cheminformatics toolkits behind a common interface. Chem. Cent. J. 2008, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Capuzzi, S.J.; Kim, I.S.J.; Lam, W.I.; Thornton, T.E.; Muratov, E.N.; Pozefsky, D.; Tropsha, A. Chembench: A publicly accessible, integrated cheminformatics portal. J. Chem. Inf. Model. 2017, 57, 105–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delocalization-Induced Molecular Equality. Available online: https://depth-first.com/articles/2021/06/17/delocalization-induced-molecular-equality/ (accessed on 20 October 2021).
- Dhaked, D.K.; Ihlenfeldt, W.D.; Patel, H.; Delannée, V.; Nicklaus, M.C. Toward a comprehensive treatment of tautomerism in chemoinformatics including in InChI v2. J. Chem. Inf. Model. 2020, 60, 1253–1275. [Google Scholar] [CrossRef] [PubMed]
- Kochev, N.T.; Paskaleva, V.H.; Jeliazkova, N. Ambit-Tautomer: An open source tool for tautomer generation. Mol. Inf. 2013, 32, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Harańczyk, M.; Gutowski, M. Quantum mechanical energy-based screening of combinatorially generated library of tautomers. TauTGen: A tautomer generator program. J. Chem. Inf. Model. 2006, 47, 686–694. [Google Scholar] [CrossRef]
- MolVS: Molecule Validation and Standardization—MolVS 0.1.1 Documentation. Available online: https://molvs.readthedocs.io/en/latest/ (accessed on 20 October 2021).
- Ropp, P.J.; Spiegel, J.O.; Walker, J.L.; Green, H.; Morales, G.A.; Milliken, K.A.; Ringe, J.J.; Durrant, J.D. Gypsum-DL: An open-source program for preparing small-molecule libraries for structure-based virtual screening. J. Cheminf. 2019, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- OpenEye Scientific. Available online: https://www.eyesopen.com/ (accessed on 20 October 2021).
- ChemAxon—Software Solutions and Services for Chemistry & Biology. Available online: https://chemaxon.com/ (accessed on 20 October 2021).
- Sitzmann, M.; Ihlenfeldt, W.D.; Nicklaus, M.C. Tautomerism in large databases. J. Comput. Aided Mol. Des. 2010, 24, 521–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, O.; Sander, T. Tautobase: An open tautomer database. J. Chem. Inf. Model. 2020, 60, 1085–1089. [Google Scholar] [CrossRef]
- Sobez, J.G.; Reiher, M. Molassembler: Molecular graph construction, modification, and conformer generation for inorganic and organic molecules. J. Chem. Inf. Model. 2020, 60, 3884–3900. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Vainio, M.J.; Johnson, M.S. Generating conformer ensembles using a multiobjective genetic algorithm. J. Chem. Inf. Model. 2007, 47, 2462–2474. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Vandermeersch, T.; Flynn, C.J.; Maguire, A.R.; Hutchison, G.R. Confab—Systematic generation of diverse low-energy conformers. J. Cheminf. 2011, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Miteva, M.A.; Guyon, F.; Tuffery, P. Frog2: Efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res. 2010, 38, W622–W627. [Google Scholar] [CrossRef] [Green Version]
- Ebejer, J.P.; Morris, G.M.; Deane, C.M. Freely available conformer generation methods: How good are they? J. Chem. Inf. Model. 2012, 52, 1146–1158. [Google Scholar] [CrossRef]
- Lewis-Atwell, T.; Townsend, P.A.; Grayson, M.N. Comparisons of different force fields in conformational analysis and searching of organic molecules: A review. Tetrahedron 2021, 79, 131865. [Google Scholar] [CrossRef]
- Rackers, J.A.; Wang, Z.; Lu, C.; Laury, M.L.; Lagardère, L.; Schnieders, M.J.; Piquemal, J.P.; Ren, P.; Ponder, J.W. Tinker 8: Software tools for molecular design. J. Chem. Theory Comput. 2018, 14, 5273–5289. [Google Scholar] [CrossRef]
- Folmsbee, D.; Hutchison, G. Assessing conformer energies using electronic structure and machine learning methods. Int. J. Quantum Chem. 2020, 121, e26381. [Google Scholar] [CrossRef]
- Tanemura, K.A.; Das, S.; Merz, K.M. AutoGraph: Autonomous graph-based clustering of small-molecule conformations. J. Chem. Inf. Model. 2021, 61, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, B. ChemPy: A package useful for chemistry written in Python. Open Source Softw. 2018, 3, 565. [Google Scholar] [CrossRef]
- Rackauckas, C.; Nie, Q. DifferentialEquations.jl—A performant and feature-rich ecosystem for solving differential equations in Julia. J. Open Res. Software 2017, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Backman, T.W.H.; Cao, Y.; Girke, T. ChemMine tools: An online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef]
- Cao, Y.; Charisi, A.; Cheng, L.C.; Jiang, T.; Girke, T. ChemmineR: A compound mining framework for R. Bioinformatics 2008, 24, 1733–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voicu, A.; Duteanu, N.; Voicu, M.; Vlad, D.; Dumitrascu, V. The rcdk and cluster R packages applied to drug candidate selection. J. Cheminf. 2020, 12, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, S.; Holy, T.; TagBot, J.; Richard. mojaie/MolecularGraph.jl: v0.9.0. Zenodo 2021. [Google Scholar] [CrossRef]
- Dask: Scalable Analytics in Python. Available online: https://dask.org/ (accessed on 20 October 2021).
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef]
- Dalke, A. The chemfp project. J. Cheminf. 2019, 11, 76. [Google Scholar] [CrossRef] [Green Version]
- Rajan, K.; Hein, J.M.; Steinbeck, C.; Zielesny, A. Molecule set comparator (MSC): A CDK-based open rich-client tool for molecule set similarity evaluations. J. Cheminf. 2021, 13, 5. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminf. 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Scott, O.B.; Chan, A.W.E. ScaffoldGraph: An open-source library for the generation and analysis of molecular scaffold networks and scaffold trees. Bioinformatics 2020, 36, 3930–3931. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Li, X.; Wang, Y.; Yin, S.; Zhou, J.; Liu, Z. AIScaffold: A web-based tool for scaffold diversification using deep learning. J. Chem. Inf. Model. 2021, 61, 1–6. [Google Scholar] [CrossRef]
- Yang, Q.; Li, Y.; Yang, J.D.; Liu, Y.; Zhang, L.; Luo, S.; Cheng, J.P. Holistic prediction of the pKa in diverse solvents based on a machine-learning approach. Angew. Chem. Int. Ed. 2020, 59, 19282–19291. [Google Scholar] [CrossRef]
- Mansouri, K.; Cariello, N.F.; Korotcov, A.; Tkachenko, V.; Grulke, C.M.; Sprankle, C.S.; Allen, D.; Casey, W.M.; Kleinstreuer, N.C.; Williams, A.J. Open-source QSAR models for pKa prediction using multiple machine learning approaches. J. Cheminf. 2019, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Roszak, R.; Beker, W.; Molga, K.; Grzybowski, B.A. Rapid and accurate prediction of pKa Values of C–H acids using graph convolutional neural networks. J. Am. Chem. Soc. 2019, 141, 17142–17149. [Google Scholar] [CrossRef]
- Pan, X.; Wang, H.; Li, C.; Zhang, J.Z.H.; Ji, C. MolGpka: A web server for small molecule pKa prediction using a graph-convolutional neural network. J. Chem. Inf. Model. 2021, 61, 3159–3165. [Google Scholar] [CrossRef]
- Tomar, N.; De, R.K. Comparing methods for metabolic network analysis and an application to metabolic engineering. Gene 2013, 521, 1–14. [Google Scholar] [CrossRef]
- Radojković, V.; Schreiber, I. Constrained stoichiometric network analysis. Phys. Chem. Chem. Phys. 2018, 20, 9910–9921. [Google Scholar] [CrossRef]
- Ruf, A.; d’Hendecourt, L.; Schmitt-Kopplin, P. Data-driven astrochemistry: One step further within the origin of life puzzle. Life 2018, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisberger, T.; Diederich, P.; Steiner, T.; Eisenreich, W.; Schmitt-Kopplin, P.; Huber, C. Evolutionary steps in the analytics of primordial metabolic evolution. Life 2019, 9, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheubert, K.; Hufsky, F.; Böcker, S. Computational mass spectrometry for small molecules. J. Cheminf. 2013, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, S.; Colreavy-Donnelly, S.; de Souza, J.S.; Borges, R.M. An integrated approach for mixture analysis using MS and NMR techniques. Faraday Discuss. 2019, 218, 339–353. [Google Scholar] [CrossRef]
- Howarth, A.; Ermanis, K.; Goodman, J.M. DP4-AI automated NMR data analysis: Straight from spectrometer to structure. Chem. Sci. 2020, 11, 4351–4359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kew, W.; Blackburn, J.W.; Clarke, D.J.; Uhrín, D. Interactive van Krevelen diagrams—Advanced visualisation of mass spectrometry data of complex mixtures. Rapid Commun. Mass Spectrom. 2017, 31, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Brockman, S.A.; Roden, E.V.; Hegeman, A.D. van Krevelen diagram visualization of high resolution-mass spectrometry metabolomics data with OpenVanKrevelen. Metabolomics 2018, 14, 48. [Google Scholar] [CrossRef] [PubMed]
- Hughey, C.A.; Hendrickson, C.L.; Rodgers, R.P.; Marshall, A.G.; Qian, K. Kendrick Mass Defect spectrum: A compact visual analysis for ultrahigh-resolution broadband mass mpectra. Anal. Chem. 2001, 73, 4676–4681. [Google Scholar] [CrossRef] [PubMed]
- Bramer, L.M.; White, A.M.; Stratton, K.G.; Thompson, A.M.; Claborne, D.; Hofmockel, K.; McCue, L.A. ftmsRanalysis: An R package for exploratory data analysis and interactive visualization of FT-MS data. PLoS Comput. Biol. 2020, 16, e1007654. [Google Scholar] [CrossRef]
- Reaxys. Available online: https://www.reaxys.com/ (accessed on 20 October 2021).
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2017, 46, D608–D617. [Google Scholar] [CrossRef]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- biodb: An R Package for Accessing Biological and Chemical Databases and Developing or Extending New Connectors. Available online: https://github.com/pkrog/biodb (accessed on 20 October 2021).
- Szöcs, E.; Stirling, T.; Scott, E.R.; Scharmüller, A.; Schäfer, R.B. webchem: An R Package to retrieve chemical information from the web. J. Stat. Softw. 2020, 93, 13. [Google Scholar] [CrossRef]
- PubChemPy. Available online: https://pubchempy.readthedocs.io/en/latest/ (accessed on 20 October 2021).
- Awale, M.; Visini, R.; Probst, D.; Arús-Pous, J.; Reymond, J.L. Chemical space: Big data challenge for molecular diversity. Chim. Int. J. Chem. 2017, 71, 661–666. [Google Scholar] [CrossRef]
- Probst, D.; Reymond, J.L. Visualization of very large high-dimensional data sets as minimum spanning trees. J. Cheminf. 2020, 12, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awale, M.; Probst, D.; Reymond, J.L. WebMolCS: A web-based interface for visualizing molecules in three-dimensional chemical spaces. J. Chem. Inf. Model. 2017, 57, 643–649. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Oftware for Exploring and Manipulating Networks. In Proceedings of the Third International Conference on Weblogs and Social Media, ICWSM 2009, San Jose, CA, USA, 17–20 May 2009. [Google Scholar] [CrossRef]
- Shannon, P. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference, Pasadena, CA, USA, 19–24 August 2008; pp. 11–15. [Google Scholar]
- Peixoto, T.P. The graph-Tool Python Ibrary; 2014; Available online: https://figshare.com/articles/dataset/graph_tool/1164194/14 (accessed on 20 October 2021).
- Gupta, U.; Vlachos, D.G. Reaction Network Viewer (ReNView): An open-source framework for reaction path visualization of chemical reaction systems. SoftwareX 2020, 11, 100442. [Google Scholar] [CrossRef]
- Neo4j Graph platform—The Leader in Graph Databases. Available online: https://neo4j.com/ (accessed on 20 October 2021).
- Probst, D.; Reymond, J.L. SmilesDrawer: Parsing and Drawing SMILES-Encoded molecular structures using client-side JavaScript. J. Chem. Inf. Model. 2018, 58, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Leruli. Available online: https://www.leruli.com/ (accessed on 20 October 2021).






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, S.; Arya, A.; Cruz, R.; Cleaves II, H.J. Automated Exploration of Prebiotic Chemical Reaction Space: Progress and Perspectives. Life 2021, 11, 1140. https://doi.org/10.3390/life11111140
Sharma S, Arya A, Cruz R, Cleaves II HJ. Automated Exploration of Prebiotic Chemical Reaction Space: Progress and Perspectives. Life. 2021; 11(11):1140. https://doi.org/10.3390/life11111140
Chicago/Turabian StyleSharma, Siddhant, Aayush Arya, Romulo Cruz, and Henderson James Cleaves II. 2021. "Automated Exploration of Prebiotic Chemical Reaction Space: Progress and Perspectives" Life 11, no. 11: 1140. https://doi.org/10.3390/life11111140