Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
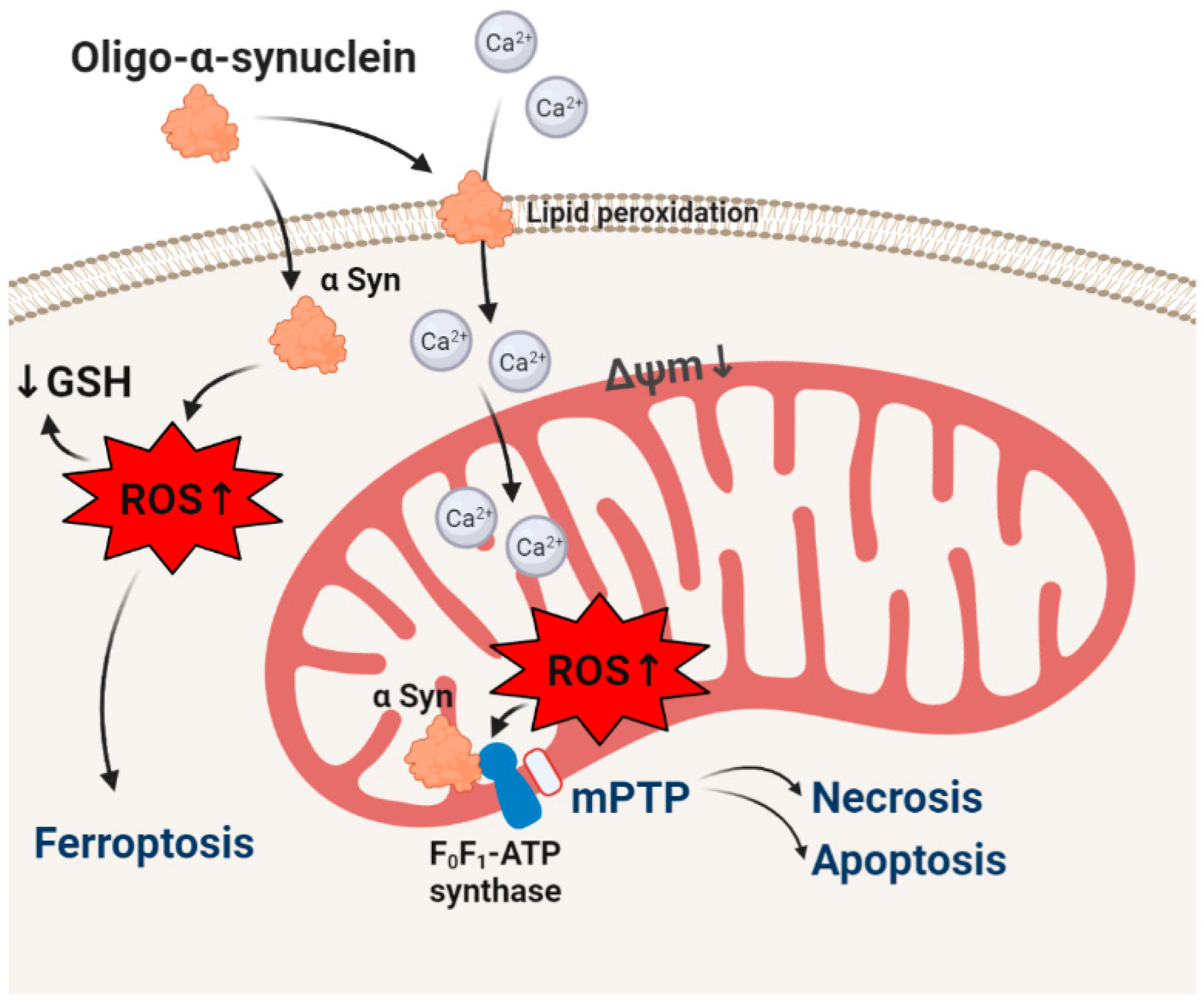
2. α-synuclein and Oxidative Stress
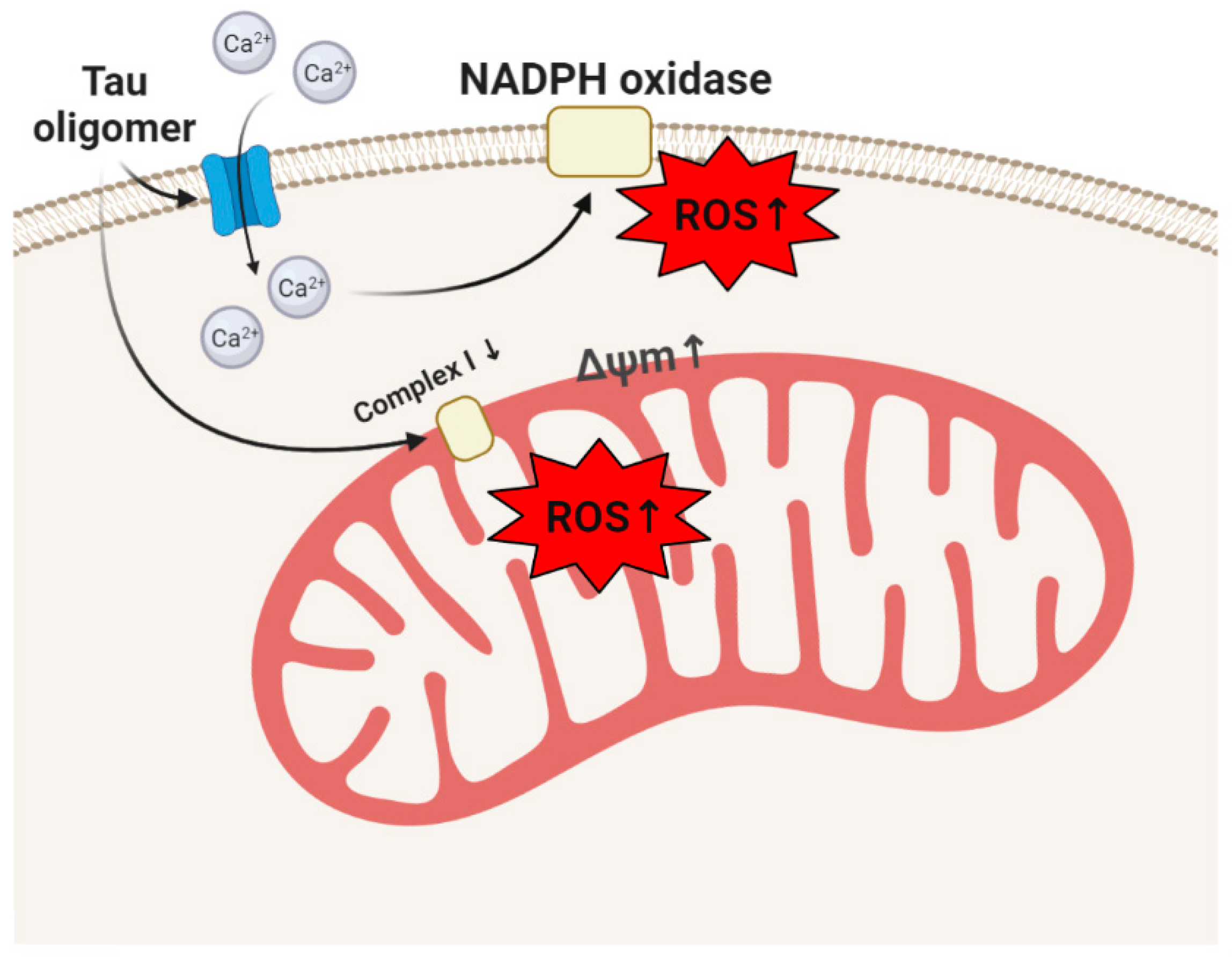
3. Tau, Tauopathies, and Oxidative Stress
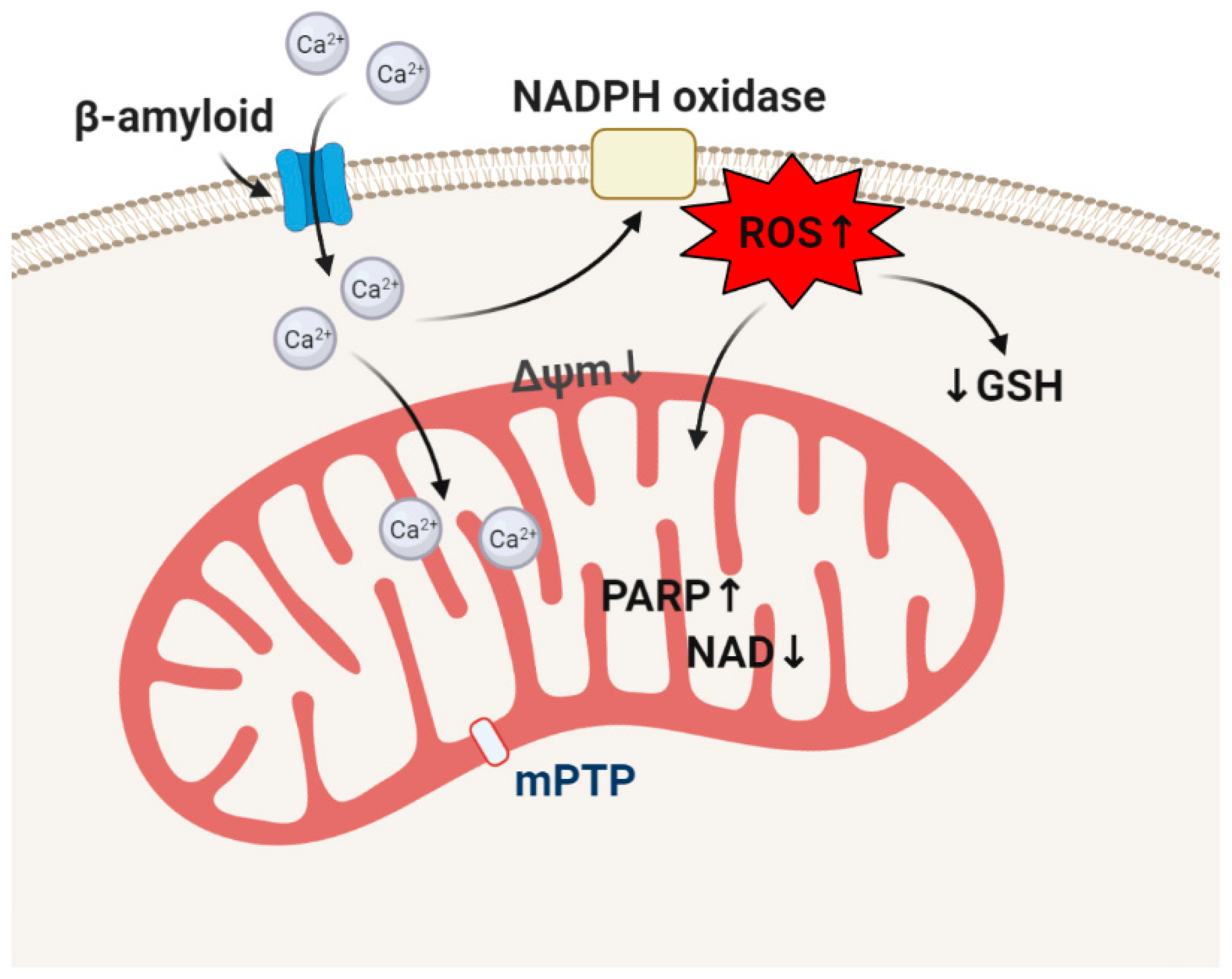
4. β-amyloid and Oxidative Stress in the Mechanism of Neurodegeneration
5. Huntingtin and Oxidative Damage
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Abramov, A.Y.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Dolgacheva, L.P. Interaction of Misfolded Proteins and Mitochondria in Neurodegenerative Disorders. Biochem. Soc. Trans. 2017, 45, 1025–1033. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Abramov, A.Y. Mechanism of Oxidative Stress in Neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Functional Role of Mitochondrial Reactive Oxygen Species in Physiology. Free Radic. Biol. Med. 2016, 100, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 Years of Lewy Pathology. Nat. Rev. Neurol. 2013, 9, 13. [Google Scholar] [CrossRef]
- Choi, M.L.; Gandhi, S. Crucial Role of Protein Oligomerization in the Pathogenesis of Alzheimer’s and Parkinson’s Diseases. FEBS J. 2018, 285, 3631–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, G.G. Tauopathies. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 145, pp. 355–368. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Tau Pathology and Neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Gustafson, D.R.; Hardy, J. The Genetic Architecture of Alzheimer’s Disease: Beyond APP, PSENs and APOE. Neurobiol. Aging 2012, 33, 437–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, R.; Wood-Kaczmar, A.; Dobson, L.; Smith, E.J.; Sirinathsinghji, E.C.; Kriston-Vizi, J.; Hargreaves, I.P.; Heaton, R.; Herrmann, F.; Abramov, A.Y. Expression of Mutant Exon 1 Huntingtin Fragments in Human Neural Stem Cells and Neurons Causes Inclusion Formation and Mitochondrial Dysfunction. FASEB J. 2020. [Google Scholar] [CrossRef] [Green Version]
- Uemura, N.; Uemura, M.T.; Luk, K.C.; Lee, V.M.-Y.; Trojanowski, J.Q. Cell-to-Cell Transmission of Tau and α-Synuclein. Trends Mol. Med. 2020. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science. 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Ludtmann, M.H.R.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef] [PubMed]
- Ninkina, N.N.; Tarasova, T.V.; Chaprov, K.D.; Goloborshcheva, V.V.; Bachurin, S.O.; Buchman, V.L. Synuclein Deficiency Decreases the Efficiency of Dopamine Uptake by Synaptic Vesicles. In Doklady Biochemistry and Biophysics; Springer: Berlin/Heidelberg, Germany, 2019; Volume 486, pp. 168–170. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Mandelkow, E. Tau in Physiology and Pathology. Nat. Rev. Neurosci. 2016, 17, 22. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative Stress and the Amyloid Beta Peptide in Alzheimer’s Disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Alpha-Synuclein and Beta-Amyloid–Different Targets, Same Players: Calcium, Free Radicals and Mitochondria in the Mechanism of Neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, J.N.; Adams, J.D.; Schuller, H.M.; Bacon, J.P.; Markey, S.P. 1-Methyl-4-Phenylpyridine (MPP+) Induces Oxidative Stress in the Rodent. Life Sci. 1986, 38, 743–749. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Lipid Peroxidation as Cause of Nigral Cell Death in Parkinson’s Disease. Lancet 1986, 328, 639–640. [Google Scholar] [CrossRef]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A. Direct Observation of the Interconversion of Normal and Toxic Forms of α-Synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.R.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J. Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Nemani, V.M.; Wallender, E.K.; Kaehlcke, K.; Ott, M.; Edwards, R.H. Optical Reporters for the Conformation of α-Synuclein Reveal a Specific Interaction with Mitochondria. J. Neurosci. 2008, 28, 12305–12317. [Google Scholar] [CrossRef]
- Reeve, A.K.; Ludtmann, M.H.R.; Angelova, P.R.; Simcox, E.M.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Turnbull, D.M.; Abramov, A.Y. Aggregated α-Synuclein and Complex I Deficiency: Exploration of Their Relationship in Differentiated Neurons. Cell Death Dis. 2015, 6, e1820. [Google Scholar] [CrossRef]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J. Structural Characterization of Toxic Oligomers That Are Kinetically Trapped during α-Synuclein Fibril Formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B. α-Synuclein Oligomers Interact with ATP Synthase and Open the Permeability Transition Pore in Parkinson’s Disease. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-Triggered Structural Transformations, Aggregation, and Fibrillation of Human α-Synuclein a Possible Molecular Link between Parkinson′ s Disease and Heavy Metal Exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramis, R.; Ortega-Castro, J.; Vilanova, B.; Adrover, M.; Frau, J. A Systematic DFT Study of Some Plausible Zn(II) and Al(III) Interaction Sites in N-Terminally Acetylated α-Synuclein. J. Phys. Chem. A 2018, 122, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Lothian, A.; Lago, L.; Mukherjee, S.; Connor, A.R.; Fowler, C.; McLean, C.A.; Horne, M.; Masters, C.L.; Cappai, R.; Roberts, B.R. Characterization of the Metal Status of Natively Purified Alpha-Synuclein from Human Blood, Brain Tissue, or Recombinant Sources Using Size Exclusion ICP-MS Reveals No Significant Binding of Cu, Fe or Zn. Metallomics 2019, 11, 118–140. [Google Scholar] [CrossRef] [PubMed]
- Zakharov, S.D.; Hulleman, J.D.; Dutseva, E.A.; Antonenko, Y.N.; Rochet, J.C.; Cramer, W.A. Helical α-Synuclein Forms Highly Conductive Ion Channels. Biochemistry 2007, 46, 14369–14379. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Ludtmann, M.H.R.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ Is a Key Factor in α-Synuclein-Induced Neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801. [Google Scholar] [CrossRef] [Green Version]
- Angelova, P.R.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Abramov, A.Y.; Shchepinov, M.S. Lipid Peroxidation Is Essential for α-Synuclein-Induced Cell Death. J. Neurochem. 2015, 133, 582–589. [Google Scholar] [CrossRef] [Green Version]
- Angelova, P.R.; Choi, M.L.; Berezhnov, A.V.; Horrocks, M.H.; Hughes, C.D.; De, S.; Rodrigues, M.; Yapom, R.; Little, D.; Dolt, K.S.; et al. Alpha Synuclein Aggregation Drives Ferroptosis: An Interplay of Iron, Calcium and Lipid Peroxidation. Cell Death Differ. 2020. [Google Scholar] [CrossRef]
- Zhang, W.; Dallas, S.; Zhang, D.; Guo, J.P.; Pang, H.; Wilson, B.; Miller, D.S.; Chen, B.; Zhang, W.; Mcgeer, P.L.; et al. Microglial PHOX and Mac-1 Are Essential to the Enhanced Dopaminergic Neurodegeneration Elicited by A30P and A53T Mutant Alpha-Synuclein. Glia 2007, 55, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qian, L.; Chen, S.-H.; Chu, C.-H.; Wilson, B.; Oyarzabal, E.; Ali, S.; Robinson, B.; Rao, D.; Hong, J.-S. Post-Treatment with an Ultra-Low Dose of NADPH Oxidase Inhibitor Diphenyleneiodonium Attenuates Disease Progression in Multiple Parkinson’s Disease Models. Brain 2015, 138 Pt 5, 1247–1262. [Google Scholar] [CrossRef]
- Hou, L.; Sun, F.; Huang, R.; Sun, W.; Zhang, D.; Wang, Q. Inhibition of NADPH Oxidase by Apocynin Prevents Learning and Memory Deficits in a Mouse Parkinson’s Disease Model. Redox Biol. 2019, 22, 101134. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Hoekstra, J.; Heng, X.; Kang, W.; Ding, J.; Liu, J.; Chen, S.; Zhang, J. P2X7 Receptor Is Critical in α-Synuclein–Mediated Microglial NADPH Oxidase Activation. Neurobiol. Aging 2015, 36, 2304–2318. [Google Scholar] [CrossRef] [PubMed]
- Whiten, D.R.; Cox, D.; Horrocks, M.H.; Taylor, C.G.; De, S.; Flagmeier, P.; Tosatto, L.; Kumita, J.R.; Ecroyd, H.; Dobson, C.M. Single-Molecule Characterization of the Interactions between Extracellular Chaperones and Toxic α-Synuclein Oligomers. Cell Rep. 2018, 23, 3492–3500. [Google Scholar] [CrossRef] [PubMed]
- Hinault, M.-P.; Cuendet, A.F.H.; Mattoo, R.U.H.; Mensi, M.; Dietler, G.; Lashuel, H.A.; Goloubinoff, P. Stable α-Synuclein Oligomers Strongly Inhibit Chaperone Activity of the Hsp70 System by Weak Interactions with J-Domain Co-Chaperones. J. Biol. Chem. 2010, 285, 38173–38182. [Google Scholar] [CrossRef] [Green Version]
- Aprile, F.A.; Källstig, E.; Limorenko, G.; Vendruscolo, M.; Ron, D.; Hansen, C. The Molecular Chaperones DNAJB6 and Hsp70 Cooperate to Suppress α-Synuclein Aggregation. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Camprubi, M.; Esteras, N.; Soutar, M.P.M.; Plun-Favreau, H.; Abramov, A.Y. Deficiency of Parkinson’s Disease-Related Gene Fbxo7 Is Associated with Impaired Mitochondrial Metabolism by PARP Activation. Cell Death Differ. 2017, 24, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Kam, T.-I.; Mao, X.; Park, H.; Chou, S.-C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R. Poly (ADP-Ribose) Drives Pathologic α-Synuclein Neurodegeneration in Parkinson’s Disease. Science 2018, 362. [Google Scholar] [CrossRef] [Green Version]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.-Y.; Kirschner, M.W. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [Green Version]
- Orr, M.E.; Sullivan, A.C.; Frost, B. A Brief Overview of Tauopathy: Causes, Consequences, and Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 637–648. [Google Scholar] [CrossRef]
- Kumar, P.; Jha, N.K.; Jha, S.K.; Ramani, K.; Ambasta, R.K. Tau Phosphorylation, Molecular Chaperones, and Ubiquitin E3 Ligase: Clinical Relevance in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 43, 341–361. [Google Scholar] [CrossRef] [PubMed]
- Kundel, F.; Hong, L.; Falcon, B.; McEwan, W.A.; Michaels, T.C.T.; Meisl, G.; Esteras, N.; Abramov, A.Y.; Knowles, T.J.P.; Goedert, M.; et al. Measurement of Tau Filament Fragmentation Provides Insights into Prion-like Spreading. ACS Chem. Neurosci. 2018, 9, 1276–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteras, N.; Kundel, F.; Amodeo, G.F.; Pavlov, E.V.; Klenerman, D.; Abramov, A.Y. Insoluble Tau Aggregates Induce Neuronal Death through Modification of Membrane Ion Conductance, Activation of Voltage-Gated Calcium Channels and NADPH Oxidase. FEBS J. 2020. [Google Scholar] [CrossRef]
- Olguín-Albuerne, M.; Morán, J. ROS Produced by NOX2 Control in Vitro Development of Cerebellar Granule Neurons Development. ASN Neuro 2015, 7, 1759091415578712. [Google Scholar] [CrossRef]
- Esteras, N.; Rohrer, J.D.; Hardy, J.; Wray, S.; Abramov, A.Y. Mitochondrial Hyperpolarization in IPSC-Derived Neurons from Patients of FTDP-17 with 10+16 MAPT Mutation Leads to Oxidative Stress and Neurodegeneration. Redox Biol. 2017, 12, 410–422. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Angelova, P.R. Cellular Mechanisms of Complex I-Associated Pathology. Biochem. Soc. Trans. 2019, 47, 1963–1969. [Google Scholar] [CrossRef] [PubMed]
- Kopach, O.; Esteras, N.; Wray, S.; Rusakov, D.A.; Abramov, A.Y. Maturation and Phenotype of Pathophysiological Neuronal Excitability of Human Cells in Tau-Related Dementia. J. Cell Sci. 2020, jcs.241687. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Barilani, M.; Lovejoy, C.; Dossena, M.; Viganò, M.; Seresini, A.; Piga, D.; Gandhi, S.; Pezzoli, G.; Abramov, A.Y.; et al. Mitochondrial Dysfunction in Parkinsonian Mesenchymal Stem Cells Impairs Differentiation. Redox Biol. 2018, 14, 474–484. [Google Scholar] [CrossRef]
- Kulic, L.; Wollmer, M.A.; Rhein, V.; Pagani, L.; Kuehnle, K.; Cattepoel, S.; Tracy, J.; Eckert, A.; Nitsch, R.M. Combined Expression of Tau and the Harlequin Mouse Mutation Leads to Increased Mitochondrial Dysfunction, Tau Pathology and Neurodegeneration. Neurobiol. Aging 2011, 32, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal Phosphorylated Tau Induced Cognitive Decline, Dendritic Spine Loss and Mitochondrial Abnormalities in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2018, 27, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 Impacts Cellular Bioenergetics by Controlling Substrate Availability for Mitochondrial Respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Esteras, N.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 Activation in the Treatment of Neurodegenerative Diseases: A Focus on Its Role in Mitochondrial Bioenergetics and Function. Biol. Chem. 2016, 397, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Lee, H.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic Oxidative Stress Causes Increased Tau Phosphorylation in M17 Neuroblastoma Cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Jeandel, C.; Nicolas, M.B.; Dubois, F.; Nabet-Belleville, F.; Penin, F.; Cuny, G. Lipid Peroxidation and Free Radical Scavengers in Alzheimer’s Disease. Gerontology 1989, 35, 275–282. [Google Scholar] [CrossRef]
- Subbarao, K.V.; Richardson, J.S.; Ang, L.C. Autopsy Samples of Alzheimer’s Cortex Show Increased Peroxidation In Vitro. J. Neurochem. 1990, 55, 342–345. [Google Scholar] [CrossRef]
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal Ions in Alzheimer’s Disease: A Key Role or Not? Acc. Chem. Res. 2019, 52, 2026–2035. [Google Scholar] [CrossRef]
- Huang, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; Rogers, J.T. Redox-Active Metals, Oxidative Stress, and Alzheimer’s Disease Pathology. Ann. N. Y. Acad. Sci. 2004, 1012, 153–163. [Google Scholar] [CrossRef]
- Smith, D.G.; Cappai, R.; Barnham, K.J. The Redox Chemistry of the Alzheimer’s Disease Amyloid β Peptide. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1976–1990. [Google Scholar] [CrossRef] [Green Version]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Changes in Intracellular Calcium and Glutathione in Astrocytes as the Primary Mechanism of Amyloid Neurotoxicity. J. Neurosci. 2003, 23, 5088–5095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramov, A.Y.; Ionov, M.; Pavlov, E.; Duchen, M.R. Membrane Cholesterol Content Plays a Key Role in the Neurotoxicity of β-Amyloid: Implications for Alzheimer’s Disease. Aging Cell 2011, 10, 595–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-Amyloid Peptides Induce Mitochondrial Dysfunction and Oxidative Stress in Astrocytes and Death of Neurons through Activation of NADPH Oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramov, A.Y.; Duchen, M.R. The Role of an Astrocytic NADPH Oxidase in the Neurotoxicity of Amyloid Beta Peptides. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2005, 360, 2309–2314. [Google Scholar] [CrossRef] [Green Version]
- Abeti, R.; Abramov, A.Y.; Duchen, M.R. β-Amyloid Activates PARP Causing Astrocytic Metabolic Failure and Neuronal Death. Brain 2011, 134, 1658–1672. [Google Scholar] [CrossRef] [Green Version]
- Narayan, P.; Holmström, K.M.; Kim, D.-H.; Whitcomb, D.J.; Wilson, M.R.; St George-Hyslop, P.; Wood, N.W.; Dobson, C.M.; Cho, K.; Abramov, A.Y.; et al. Rare Individual Amyloid-β Oligomers Act on Astrocytes to Initiate Neuronal Damage. Biochemistry 2014, 53, 2442–2453. [Google Scholar] [CrossRef]
- Ionov, M.; Burchell, V.; Klajnert, B.; Bryszewska, M.; Abramov, A.Y. Mechanism of Neuroprotection of Melatonin against Beta-Amyloid Neurotoxicity. Neuroscience 2011, 180, 229–237. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, S.; Zhao, X.; Wei, T. Melatonin Impairs NADPH Oxidase Assembly and Decreases Superoxide Anion Production in Microglia Exposed to Amyloid-Β1–42. J. Pineal Res. 2008, 45, 157–165. [Google Scholar] [CrossRef]
- Giacovazzi, R.; Ciofini, I.; Rao, L.; Amatore, C.; Adamo, C. Copper–Amyloid-β Complex May Catalyze Peroxynitrite Production in Brain: Evidence from Molecular Modeling. Phys. Chem. Chem. Phys. 2014, 16, 10169–10174. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Kasymov, V.A.; Zinchenko, V.P. β-Amyloid Activates Nitric Oxide Synthesis and Causes Neuronal Death in Hippocampal Astrocytes. Biochem. Suppl. Ser. A Membr. Cell Biol. 2008, 2, 8–13. [Google Scholar] [CrossRef]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and Oxidative Stress Analysis in Human Brain Samples of Huntington Disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johri, A.; Beal, M.F. Antioxidants in Huntington’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 664–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Ratan, R.R. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J. Huntingtons. Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell. Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Winderickx, J.; Franssens, V.; Liu, B. A Mitochondria-Associated Oxidative Stress Perspective on Huntington’s Disease. Front. Mol. Neurosci. 2018, 11, 329. [Google Scholar] [CrossRef] [PubMed]
- Essa, M.M.; Moghadas, M.; Ba-Omar, T.; Walid Qoronfleh, M.; Guillemin, G.J.; Manivasagam, T.; Justin-Thenmozhi, A.; Ray, B.; Bhat, A.; Chidambaram, S.B.; et al. Protective Effects of Antioxidants in Huntington’s Disease: An Extensive Review. Neurotox. Res. 2019, 35, 739–774. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front. Mol. Neurosci. 2019, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Stoy, N.; Mackay, G.M.; Forrest, C.M.; Christofides, J.; Egerton, M.; Stone, T.W.; Darlington, L.G. Tryptophan Metabolism and Oxidative Stress in Patients with Huntington’s Disease. J. Neurochem. 2005, 93, 611–623. [Google Scholar] [CrossRef]
- Browne, S.E.; Beal, M.F. Oxidative Damage in Huntington’s Disease Pathogenesis. Antioxid. Redox Signal. 2006, 8, 2061–2073. [Google Scholar] [CrossRef]
- Túnez, I.; Sánchez-López, F.; Agüera, E.; Fernández-Bolaños, R.; Sánchez, F.M.; Tasset-Cuevas, I. Important Role of Oxidative Stress Biomarkers in Huntington’s Disease. J. Med. Chem. 2011, 54, 5602–5606. [Google Scholar] [CrossRef]
- Hands, S.; Sajjad, M.U.; Newton, M.J.; Wyttenbach, A. In Vitro and in Vivo Aggregation of a Fragment of Huntingtin Protein Directly Causes Free Radical Production. J. Biol. Chem. 2011, 286, 44512–44520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Ward, W.F. PGC-1α: A Key Regulator of Energy Metabolism. Am. J. Physiol. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional Repression of PGC-1α by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fão, L.; Rego, A.C. Mitochondrial and Redox-Based Therapeutic Strategies in Huntington’s Disease. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef]
- Bertoni, A.; Giuliano, P.; Galgani, M.; Rotoli, D.; Ulianich, L.; Adornetto, A.; Santillo, M.R.; Porcellini, A.; Avvedimento, V.E. Early and Late Events Induced by PolyQ-Expanded Proteins: Identification of a Common Pathogenic Property of PolYQ-Expanded Proteins. J. Biol. Chem. 2011, 286, 4727–4741. [Google Scholar] [CrossRef] [Green Version]
- Chumakov, P.M. Versatile Functions of P53 Protein in Multicellular Organisms. Biochemistry (Mosc.) 2007, 72, 1399–1421. [Google Scholar] [CrossRef] [Green Version]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.-Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s Disease Protein Interacts with P53 and CREB-Binding Protein and Represses Transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Sbodio, J.I.; Xu, R.; Vandiver, M.S.; Cha, J.Y.; Snowman, A.M.; Snyder, S.H. Cystathionine γ-Lyase Deficiency Mediates Neurodegeneration in Huntington’s Disease. Nature 2014, 509, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Transcriptional Control of Amino Acid Homeostasis Is Disrupted in Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2016, 113, 8843–8848. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, M.; Rosenstock, T.R.; Cunha-Oliveira, T.; Ferreira, I.L.; Oliveira, C.R.; Rego, A.C. Glutathione Redox Cycle Dysregulation in Huntington’s Disease Knock-in Striatal Cells. Free Radic. Biol. Med. 2012, 53, 1857–1867. [Google Scholar] [CrossRef]
- Reijonen, S.; Kukkonen, J.P.; Hyrskyluoto, A.; Kivinen, J.; Kairisalo, M.; Takei, N.; Lindholm, D.; Korhonen, L. Downregulation of NF-ΚB Signaling by Mutant Huntingtin Proteins Induces Oxidative Stress and Cell Death. Cell. Mol. Life Sci. 2010, 67, 1929–1941. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Baranov, S.V.; Baranova, O.V.; Kim, J.; Pan, Y.; Yablonska, S.; Carlisle, D.L.; Ferrante, R.J.; Kim, A.H.; Friedlander, R.M. Inhibition of Mitochondrial Protein Import by Mutant Huntingtin. Nat. Neurosci. 2014, 17, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Yablonska, S.; Ganesan, V.; Ferrando, L.M.; Kim, J.; Pyzel, A.; Baranova, O.V.; Khattar, N.K.; Larkin, T.M.; Baranov, S.V.; Strohlein, C.E.; et al. Mutant Huntingtin Disrupts Mitochondrial Proteostasis by Interacting with TIM23. Proc. Natl. Acad. Sci. USA 2019, 116, 16593–16602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, Y.S.; Johnson, G.V.W.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant Huntingtin Directly Increases Susceptibility of Mitochondria to the Calcium-Induced Permeability Transition and Cytochrome c Release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Tapia, C.; Pérez, M.J. Possible Role of Mitochondrial Permeability Transition Pore in the Pathogenesis of Huntington Disease. Biochem. Biophys. Res. Commun. 2017, 483, 1078–1083. [Google Scholar] [CrossRef]
- Peng, T.-I.; Jou, M.-J. Oxidative Stress Caused by Mitochondrial Calcium Overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef]
- Cherubini, M.; Lopez-Molina, L.; Gines, S. Mitochondrial Fission in Huntington’s Disease Mouse Striatum Disrupts ER-Mitochondria Contacts Leading to Disturbances in Ca2+ Efflux and Reactive Oxygen Species (ROS) Homeostasis. Neurobiol. Dis. 2020, 136, 104741. [Google Scholar] [CrossRef]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant Huntingtin’s Interaction with Mitochondrial Protein Drp1 Impairs Mitochondrial Biogenesis and Causes Defective Axonal Transport and Synaptic Degeneration in Huntington’s Disease. Hum. Mol. Genet. 2011, 21, 406–420. [Google Scholar] [CrossRef]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant Huntingtin Binds the Mitochondrial Fission GTPase Dynamin-Related Protein-1 and Increases Its Enzymatic Activity. Nat. Med. 2011, 17, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Haun, F.; Nakamura, T.; Shiu, A.D.; Cho, D.-H.; Tsunemi, T.; Holland, E.A.; La Spada, A.R.; Lipton, S.A. S-Nitrosylation of Dynamin-Related Protein 1 Mediates Mutant Huntingtin-Induced Mitochondrial Fragmentation and Neuronal Injury in Huntington’s Disease. Antioxid. Redox Signal. 2013, 19, 1173–1184. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. https://doi.org/10.3390/life10070101
Abramov AY, Potapova EV, Dremin VV, Dunaev AV. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life. 2020; 10(7):101. https://doi.org/10.3390/life10070101
Chicago/Turabian StyleAbramov, Andrey Y., Elena V. Potapova, Viktor V. Dremin, and Andrey V. Dunaev. 2020. "Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration" Life 10, no. 7: 101. https://doi.org/10.3390/life10070101