
Uranium(VI) Sorption onto Hardened Cement Paste under High Saline and Alkaline Conditions

Abstract
:1. Introduction
2. Materials and Methods
2.1. Inductively Coupled Plasma Mass Spectrometry
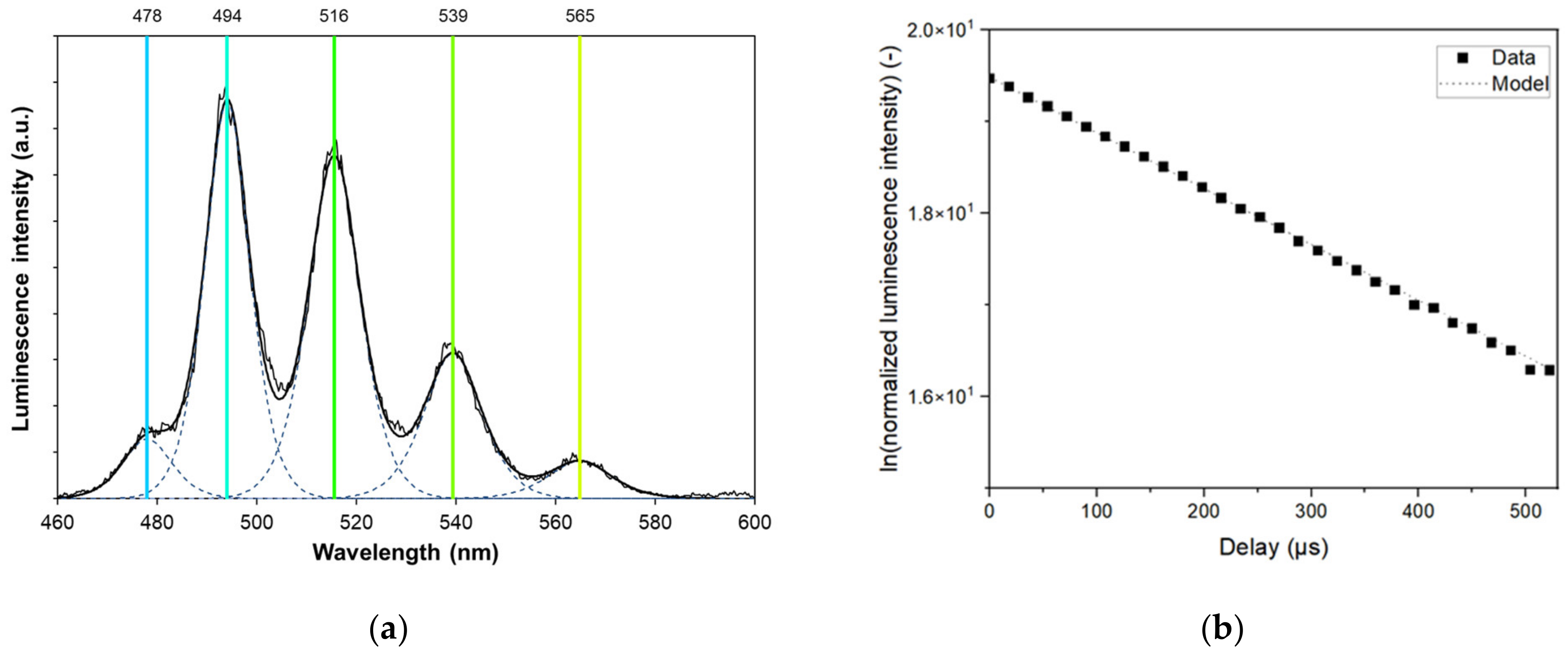
2.2. Time-Resolved Laser Fluorescence Spectroscopy
2.3. Cement-Based Materials Preparation and Characterization
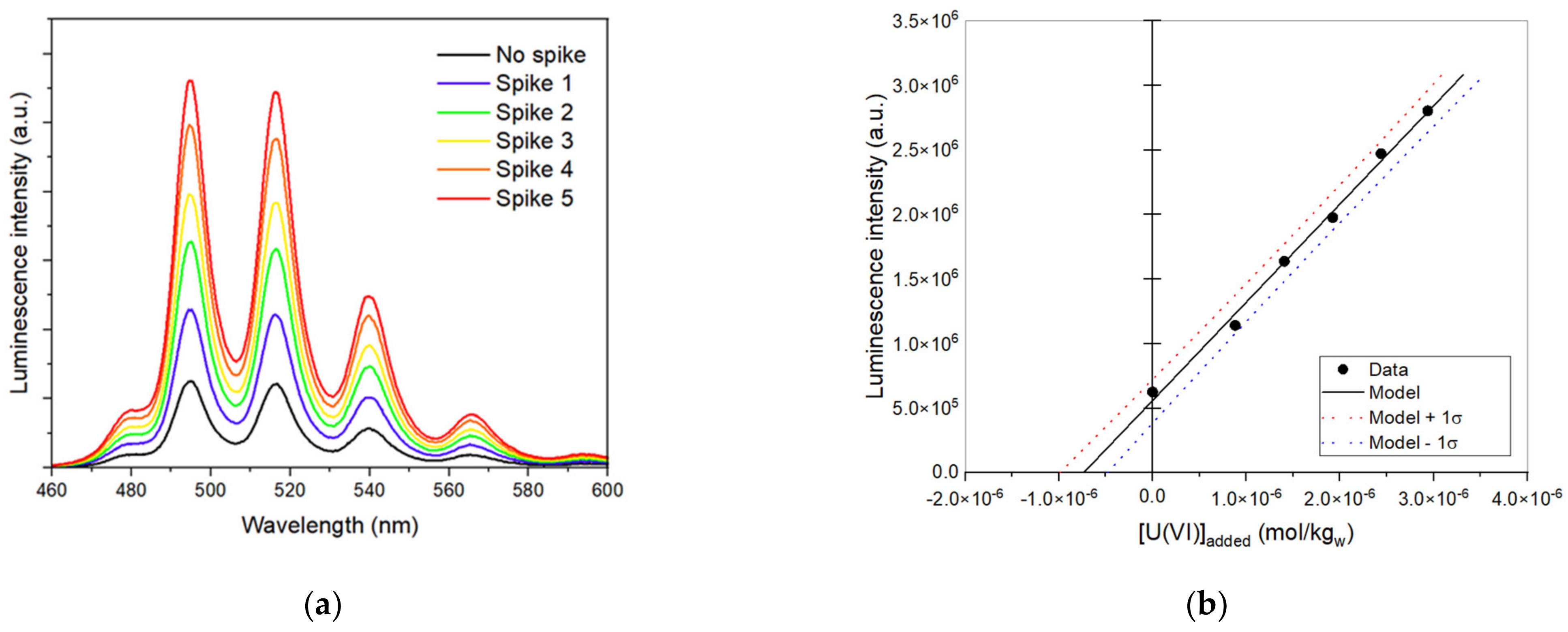
2.4. U(VI) Operational Solubility Determination
2.5. Batch Sorption Determination
3. Results
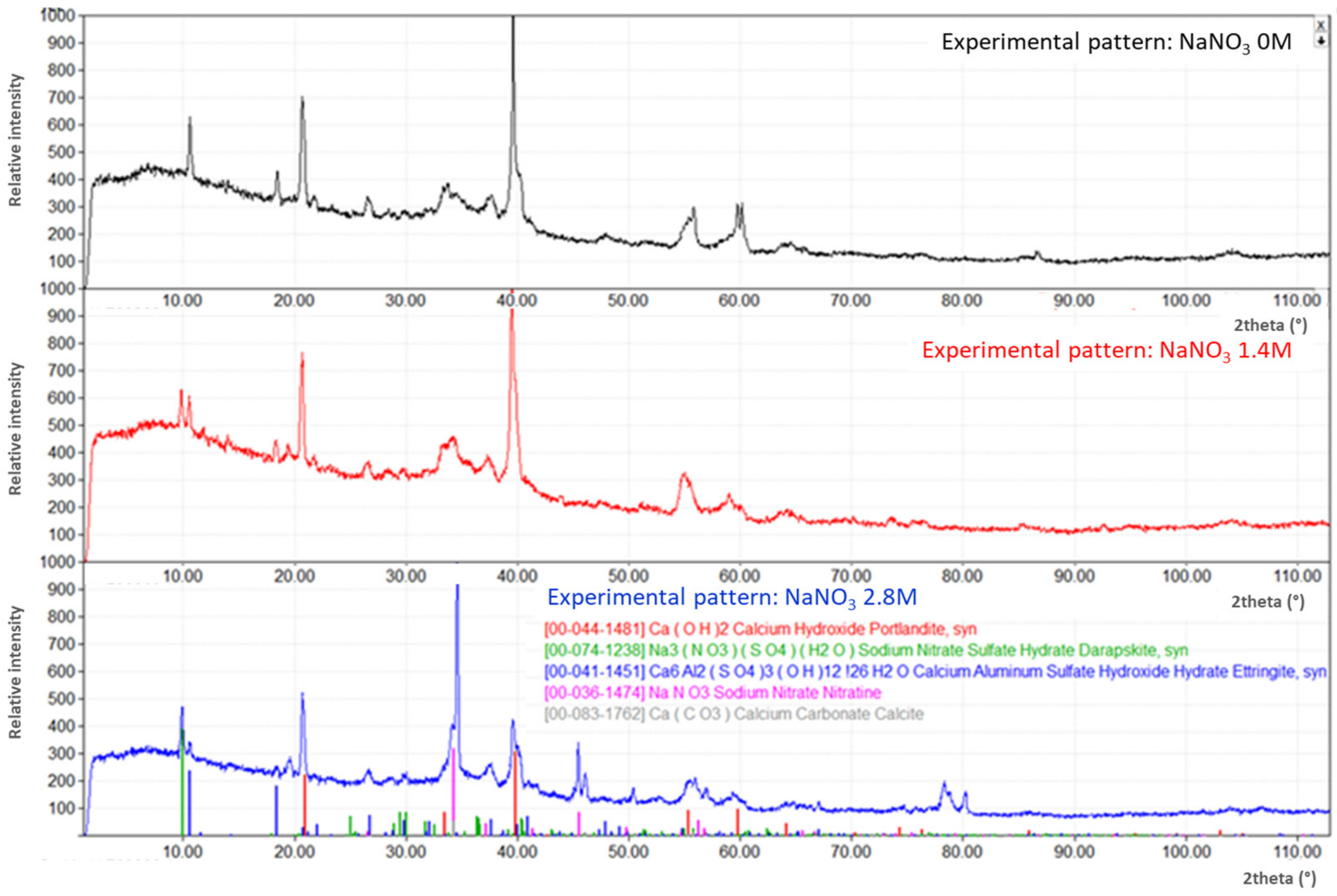
3.1. Evolution of the Cementitious-Phase Assembly as a Function of NaNO3 Content
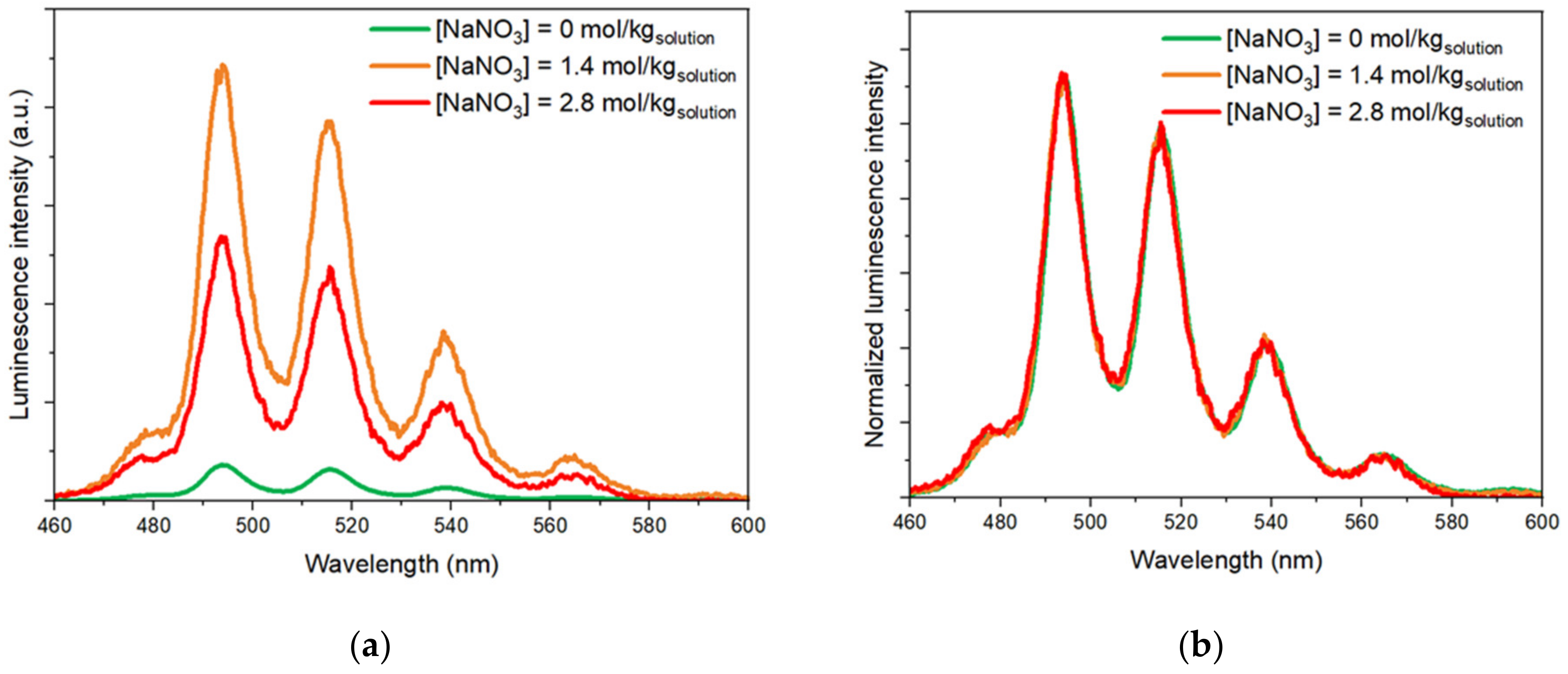
3.2. Evolution of the U(VI) Solubility Limit as a Function of NaNO3 Content
3.3. U(VI) Sorption onto Hardened Cement Paste under High Saline and Alkaline Conditions
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reynolds, J.G.; Cooke, G.A.; Herting, D.L.; Warrant, R.W. Salt mineralogy of Hanford high-level nuclear waste staged for treatment. Ind. Eng. Chem. Res. 2013, 52, 9741–9751. [Google Scholar] [CrossRef]
- Ochs, M.; Mallants, D.; Wang, L. Radionuclide and metal sorption on cement and concrete. In Topics in Safety, Risk, Reliability and Quality; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Guillaumont, R.; Fanghänel, T.; Fuger, J.; Grenthe, I.; Neck, V.; Palmer, D.A.; Rand, M. Chemical Thermodynamics 5. Update on the Chemical Thermodynamics of Uranium, Neptunium, Plutonium, Americium and Technetium; North Holland Elsevier Science Publishers B. V.: Amsterdam, The Netherlands, 2003; Volume 5, p. 918. [Google Scholar]
- Macé, N.; Wieland, E.; Dähn, R.; Tits, J.; Scheinost, A.C. EXAFS investigation on U(VI) immobilization in hardened cement paste: Influence of experimental conditions on speciation. Radiochim. Acta 2013, 101, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Wieland, E.; Macé, N.; Dahn, R.; Kunz, D.; Tits, J. Macro- and micro-scale studies on U(VI) immobilization in hardened cement paste. J. Radioanal. Nucl. Chem. 2010, 286, 793–800. [Google Scholar] [CrossRef]
- Altmaier, M.; Neck, V.; Mueller, R.; Fanghänel, T. Solubility of U(VI) and formation of CaU2O7:3H2O(cr) in alkaline CaCl2 solutions. In Proceedings of the MIGRATION 2005, 10th International Conference on Chemistry and Migration Behaviour of Actinides and Fission Products in the Geosphere, Avignon, France, 18–23 September 2005. [Google Scholar]
- Altmaier, M.; Yalçintaş, E.; Gaona, X.; Neck, V.; Muller, R.; Schlieker, M.; Fanghänel, T. Solubility of U(VI) in chloride solutions. I. The stable oxides/hydroxides in NaCl systems, solubility products, hydrolysis constants and SIT coefficients. J. Chem. Thermodyn. 2017, 114, 2–13. [Google Scholar] [CrossRef]
- Çevirim-Papaioannou, N.; Yalcintas, E.; Gaona, X.; Altmaier, M.; Geckeis, H. Solubility of U(VI) in chloride solutions. II. The stable oxides/hydroxides in alkaline KCl solutions: Thermodynamic description and relevance in cementitious systems. Appl. Geochem. 2018, 98, 237–246. [Google Scholar] [CrossRef]
- Endrizzi, F.; Gaona, X.; Zhang, Z.C.; Xu, C.; Rao, L.F.; Garcia-Perez, C.; Altmaier, M. Thermodynamic description of U(VI) solubility and hydrolysis in dilute to concentrated NaCl solutions at T = 25, 55 and 80 °C. Radiochim. Acta 2019, 107, 663–678. [Google Scholar] [CrossRef]
- Felipe-Sotelo, M.; Hinchliff, J.; Field, L.P.; Milodowski, A.E.; Preedy, O.; Read, D. Retardation of uranium and thorium by a cementitious backfill developed for radioactive waste disposal. Chemosphere 2017, 179, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, J.; Grove, C. The solubility of calcium hydroxide in aqueous salt solutions. J. Am. Chem. Soc. 1931, 53, 3976–3991. [Google Scholar] [CrossRef]
- Malone, P.G.; Poole, T.S.; Wakeley, L.D.; Burkes, J.P. Salt related expansion reactions in Portland-cement-based wasteforms. J. Hazard. Mater. 1997, 52, 237–246. [Google Scholar] [CrossRef]
- Liu, L.; Sun, C.; Geng, G.Q.; Feng, P.; Li, J.Q.; Dähn, R. Influence of decalcification on structural and mechanical properties of synthetic calcium silicate hydrate (C-S-H). Cem. Concr. Res. 2019, 123, 105793. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, Y.X.; Yang, J.; Cui, M.X.; Wang, H.T.; Ma, X. Insights into the deterioration of C-S-H gels in hardened cement pastes with different NaNO3 concentrations. Constr. Build. Mater. 2020, 259, 120423. [Google Scholar] [CrossRef]
- Pitzer, K.S. Activity Coefficients in Electrolyte Solutions; CRC Press: Boca Raton, FL, USA, 1991; p. 542. [Google Scholar]
- Lach, A.; André, L.; Lassin, A. Darapskite solubility in basic solutions at 25 °C: A Pitzer model for the Na-NO3-SO4-OH-H2O system. Appl. Geochem. 2017, 78, 311–320. [Google Scholar] [CrossRef]
- Reynolds, J.G.; Carter, R. A sulfate and darapskite solubility model with Pitzer interaction coefficients for aqueous solutions containing NaNO2, NaNO3, and NaOH. J. Chem. Thermodyn. 2016, 101, 380–386. [Google Scholar] [CrossRef]
- Shang, C.; Reiller, P.E. Effect of temperature on the complexation of triscarbonatouranyl(VI) with calcium and magnesium in NaCl aqueous solution. Dalton Trans. 2021, 50, 17165–17180. [Google Scholar] [CrossRef]
- Tits, J.; Stumpf, T.; Rabung, T.; Wieland, E.; Fanghänel, T. Uptake of Cm(III) and Eu(III) by calcium silicate hydrates: A solution chemistry and time-resolved laser fluorescence spectroscopy study. Environ. Sci. Technol. 2003, 37, 3568–3573. [Google Scholar] [CrossRef]
- Huclier-Markai, S.; Mazza, M.; Alliot, C.; Reiller, P.E. Complexation of europium(III) with exopolysaccharides from a marine bacterium envisaged as luminescent probe in a theranostic approach. Dalton Trans. 2021, 50, 17215–17227. [Google Scholar] [CrossRef]
- Mauchien, P. Dosage de l’Uranium par Spectrofluorimétrie à Source d’Excitation Laser; CEA-R-5300; Commissariat à l’Energie Atomique, CEN Saclay: Gif-sur-Yvette, France, 1985. [Google Scholar]
- Berthoud, T.; Decambox, P.; Kirsch, B.; Mauchien, P.; Moulin, C. Direct uranium trace analysis in plutonium solutions by time-resolved laser-induced spectrofluorometry. Anal. Chem. 1988, 60, 1296–1299. [Google Scholar] [CrossRef]
- Moulin, C.; Beaucaire, C.; Decambox, P.; Mauchien, P. Determination of uranium in solution at the ng.l−1 level by time-resolved laser-induced spectrofluorimetry: Application to geological samples. Anal. Chim. Acta 1990, 238, 291–296. [Google Scholar] [CrossRef]
- Decambox, P.; Mauchien, P.; Moulin, C. Direct and fast determination of uranium in human urine samples by laser-induced time-resolved spectrofluorometry. Appl. Spectrosc. 1991, 45, 116–118. [Google Scholar] [CrossRef]
- Reiller, P.; Moulin, C.; Beaucaire, C.; Lemordant, D. Dual use of micellar enhanced ultrafiltration and time-resolved laser-induced spectrofluorometry for the study of uranyl exchange at the surface of alkylsulfate micelles. J. Colloid Interface Sci. 1994, 163, 81–86. [Google Scholar] [CrossRef]
- Reiller, P.E.; Fromentin, E.; Ferry, M.; Dannoux-Papin, A.; Badji, H.; Tabarant, M.; Vercouter, T. Complexing power of hydro-soluble degradation products from γ-irradiated polyvinylchloride: Influence on Eu(OH)3(s) solubility and Eu(III) speciation in neutral to alkaline environment. Radiochim. Acta 2017, 105, 665–675. [Google Scholar] [CrossRef] [Green Version]
- Brevet, J.; Claret, F.; Reiller, P.E. Spectral and temporal luminescent properties of Eu(III) in humic substance solutions from different origins. Spectrochim. Acta Part A 2009, 74, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouhail, Y.Z.; Benedetti, M.F.; Reiller, P.E. Eu(III)-fulvic acid complexation: Evidence of fulvic acid concentration dependent interactions by time-resolved luminescence spectroscopy. Environ. Sci. Technol. 2016, 50, 3706–3716. [Google Scholar] [CrossRef] [Green Version]
- Fromentin, E.; Reiller, P.E. Influence of adipic acid on the speciation of Eu(III): Review of thermodynamic data in NaCl and NaClO4 media, and a new determination of Eu-adipate complexation constant in 0.5 mol.kgw−1 NaClO4 medium by time-resolved luminescence spectroscopy. Inorg. Chim. Acta 2018, 482, 588–596. [Google Scholar] [CrossRef]
- Kouhail, Y.Z.; Benedetti, M.F.; Reiller, P.E. Formation of mixed Eu(III)-CO3-fulvic acid complex: Spectroscopic evidence and NICA-Donnan modeling. Chem. Geol. 2019, 522, 175–185. [Google Scholar] [CrossRef]
- De Levie, R. Advanced Excel for Scientific Data Analysis; Oxford University Press: Oxford, UK, 2004; p. 638. [Google Scholar]
- Giffaut, E.; Grivé, M.; Blanc, P.; Vieillard, P.; Colàs, E.; Gailhanou, H.; Gaboreau, S.; Marty, N.; Madé, B.; Duro, L. Andra thermodynamic database for performance assessment: ThermoChimie. Appl. Geochem. 2014, 49, 225–236. [Google Scholar] [CrossRef]
- Marion, G.M. A molal-based model for strong acid chemistry at low temperatures (<200 to 298 K). Geochim. Cosmochim. Acta 2002, 66, 2499–2516. [Google Scholar] [CrossRef]
- Toghiani, R.K.; Phillips, V.A.; Smith, L.T.; Lindner, J.S. Solubility in the Na + SO4 + NO3 and Na + SO4 + NO2 systems in water and in sodium hydroxide solutions. J. Chem. Eng. Data 2008, 53, 798–804. [Google Scholar] [CrossRef]
- Yeatts, L.B.; Marshall, W.L. Aqueous systems at high temperature. XVIII. Activity coefficient behavior of calcium hydroxide in aqueous sodium nitrate to critical temperature of water. J. Phys. Chem. 1967, 71, 2641–2650. [Google Scholar] [CrossRef]
- Reardon, E.J. An ion interaction model for the determination of chemical equilibria in cement/water systems. Cem. Concr. Res. 1990, 20, 175–192. [Google Scholar] [CrossRef]
- Grenthe, I.; Gaona, X.; Plyasunov, A.V.; Rao, L.; Runde, W.H.; Grambow, B.; Koning, R.J.M.; Smith, A.L.; Moore, E.E. Chemical Thermodynamics 14. Second Update on the Chemical Thermodynamics of Uranium, Neptunium, Plutonium, Americium and Technetium; Ragoussi, M.-E., Martinez, J.S., Costa, D., Eds.; OECD Publications: Paris, France, 2020; Volume 14, p. 1572.
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC (Version 2)—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; 99-4259; U.S. Geological Survey, Water-Resources Investigations: Lakewood, CO, USA, 1999.
- Pointeau, I.; Landesman, C.; Giffaut, E.; Reiller, P. Reproducibility of the uptake of U(VI) onto degraded cement pastes and calcium silicate hydrate phases. Radiochim. Acta 2004, 92, 645–650. [Google Scholar] [CrossRef] [Green Version]
- Tits, J.; Fujita, T.; Tsukamoto, M.; Wieland, E. Uranium(VI) uptake by synthetic calcium silicate hydrates. MRS Proc. 2008, 1107, 467–474. [Google Scholar] [CrossRef]
- Tits, J.; Geipel, G.; Macé, N.; Eilzer, M.; Wieland, E. Determination of uranium(VI) sorbed species in calcium silicate hydrate phases: A laser-induced luminescence spectroscopy and batch sorption study. J. Colloid Interface Sci. 2011, 359, 248–256. [Google Scholar] [CrossRef]
- Ochs, M.; Vriens, B.; Tachi, Y. Retention of uranium in cement systems: Effects of cement degradation and complexing ligands. Prog. Nucl. Sci. Technol. 2018, 5, 208–2012. [Google Scholar] [CrossRef]
- Pointeau, I.; Reiller, P.; Macé, N.; Landesman, C.; Coreau, N. Measurement and modeling of the surface potential evolution of hydrated cement pastes as a function of degradation. J. Colloid Interface Sci. 2006, 300, 33–44. [Google Scholar] [CrossRef]
- Pointeau, I.; Coreau, N.; Reiller, P.E. Uptake of anionic radionuclides onto degraded cement pastes and competing effect of organic ligands. Radiochim. Acta 2008, 96, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Siroux, B.; Latrille, C.; Petcut, C.; Beaucaire, C.; Tabarant, M.; Benedetti, M.F.; Reiller, P.E. On the use of a multi-site ion-exchange model to predictively simulate the adsorption behaviour of strontium and caesium onto French agricultural soils. Appl. Geochem. 2021, 132, 105052. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution Added | V/m (kgsolution/kgsolid) | [U(VI)]ini (mol/kgsolution) |
|---|---|---|
| S1-0-NaNO3 | 325 ± 19 | (9.2 ± 1.5) × 10−7 |
| S1-1.4-NaNO3 | 346 ± 20 | (8.5 ± 0.6) × 10−7 |
| S1-2.8-NaNO3 | 364 ± 12 | (7.3 ± 1.1) × 10−7 |
| Contact Time, Technique | S1-0-NaNO3 (mol/kgsolution) | S1-1.4-NaNO3 (mol/kgsolution) | S1-2.8-NaNO3 (mol/kgsolution) |
|---|---|---|---|
| 4 h, ICP-MS | (1.1 ± 0.1) × 10−4 | (9.6 ± 0.5) × 10−5 | (1.0 ± 0.1) × 10−4 |
| 48 h, ICP-MS | (8.68 ± 0.08) × 10−5 | (1.38 ± 0.03) × 10−5 | (1.67 ± 0.03) × 10−5 |
| 7 m, ICP-MS | <LOD * | <LOD * | <LOD * |
| 7 m, TRLFS | (7.2 ± 2.1) × 10−6 | (2.7 ± 0.3) × 10−6 | (2.2 ± 0.4) × 10−6 |
| S1-0-NaNO3 | S1-1.4-NaNO3 | S1-2.8-NaNO3 | |
|---|---|---|---|
| UO2(OH)3− | 1.8% | 5.8% | 11.7% |
| UO2(OH)42− | 98.2% | 94.2% | 88.3% |
| −log10[H+] | 13.3 | 13.0 | 13.2 |
| [U(VI)]solution | S1-0-NaNO3 (mol/kgsolution) | S1-1.4-NaNO3 (mol/kgsolution) | S1-2.8-NaNO3 (mol/kgsolution) |
|---|---|---|---|
| sorption | (1.3 ± 0.1) × 10−8 | (7.4 ± 1.2) × 10−8 | (6.8 ± 0.8) × 10−8 |
| desorption | (1.6 ± 0.3) × 10−8 | (5.9 ± 1.6) × 10−8 | (4.6 ± 0.9) × 10−8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macé, N.; Page, J.; Reiller, P.E. Uranium(VI) Sorption onto Hardened Cement Paste under High Saline and Alkaline Conditions. Minerals 2023, 13, 325. https://doi.org/10.3390/min13030325
Macé N, Page J, Reiller PE. Uranium(VI) Sorption onto Hardened Cement Paste under High Saline and Alkaline Conditions. Minerals. 2023; 13(3):325. https://doi.org/10.3390/min13030325
Chicago/Turabian StyleMacé, Nathalie, Jacques Page, and Pascal E. Reiller. 2023. "Uranium(VI) Sorption onto Hardened Cement Paste under High Saline and Alkaline Conditions" Minerals 13, no. 3: 325. https://doi.org/10.3390/min13030325