

The Effect of Iron- and Calcium-Rich Waste Rock’s Acid Baking Conditions on the Rare-Earth Extraction
 ,
,
Abstract
:Highlights
- Effect of low-temperature sulfation, H2SO4 to ore ratio and time on REE extraction.
- Differences in REE host minerals and their impact on these elements extraction.
- Increasing sulfuric acid availability reduces iron dissolution from Fe-rich sample.
- Inhibitory effect on the crystallization of Ca-sulfate by REE uptake during leaching.
Abstract
1. Introduction
2. Materials and Experimental Procedure
2.1. Materials
2.2. Experimental Procedures
2.3. Analytical Methods
3. Results
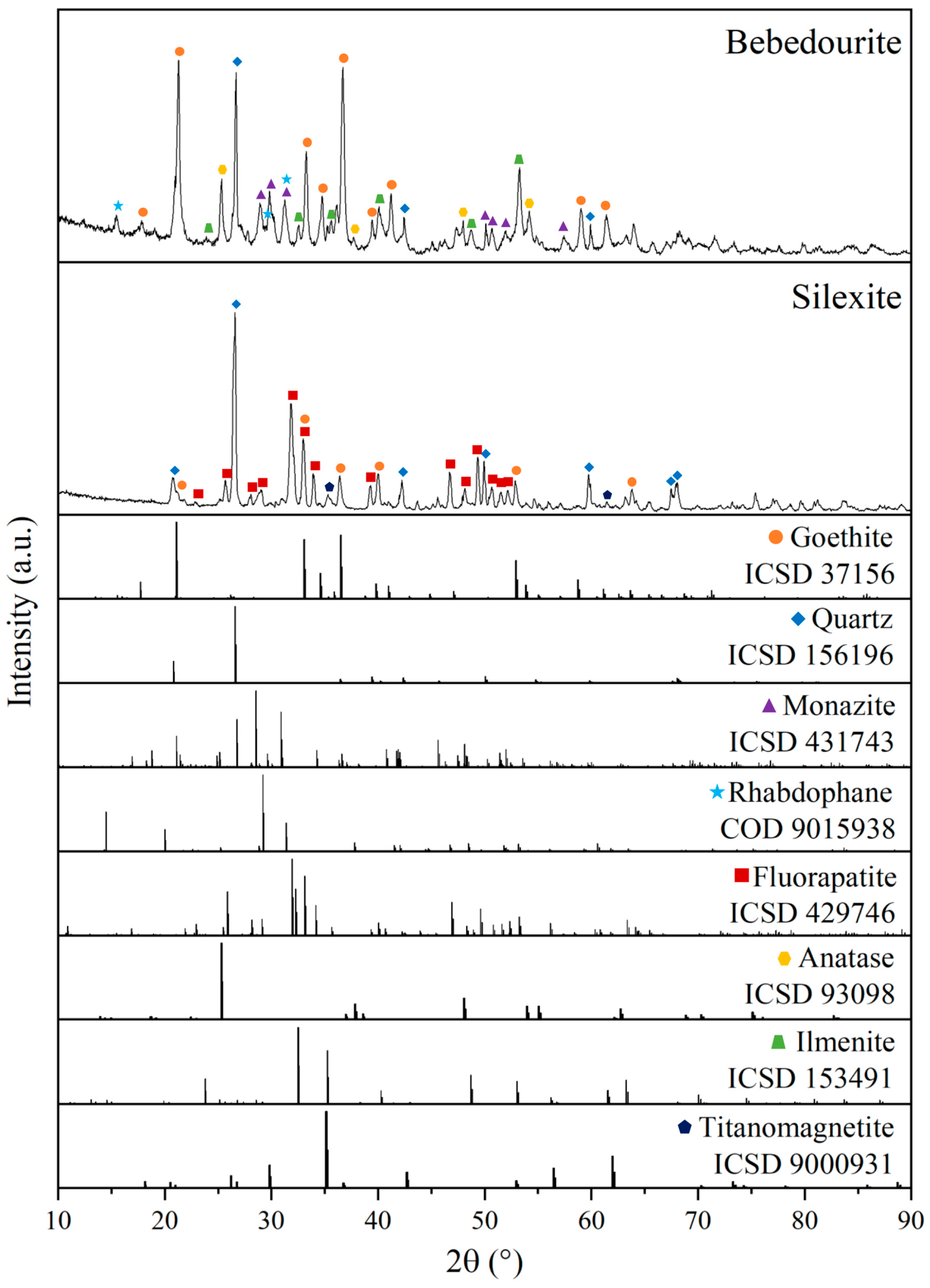
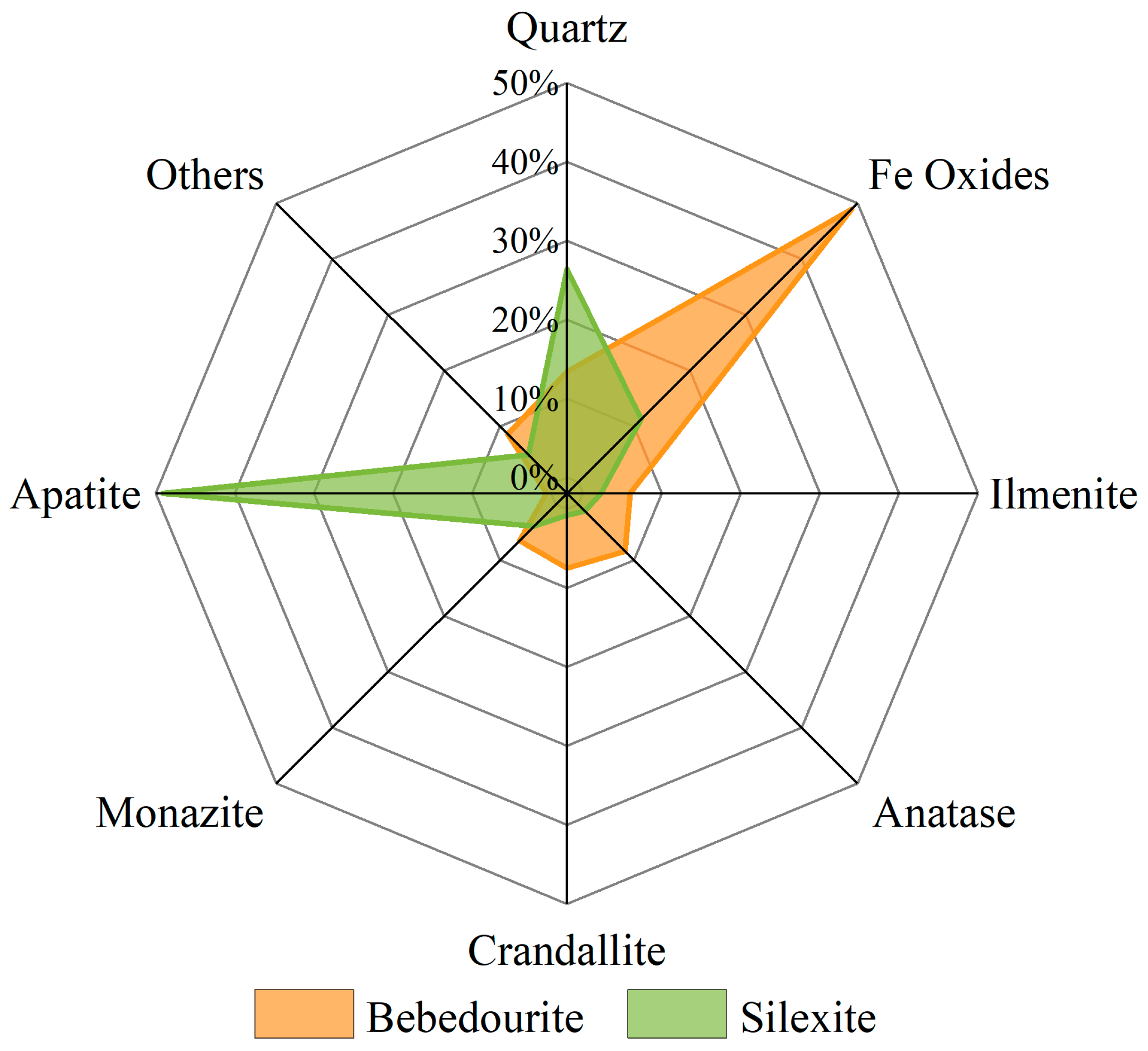
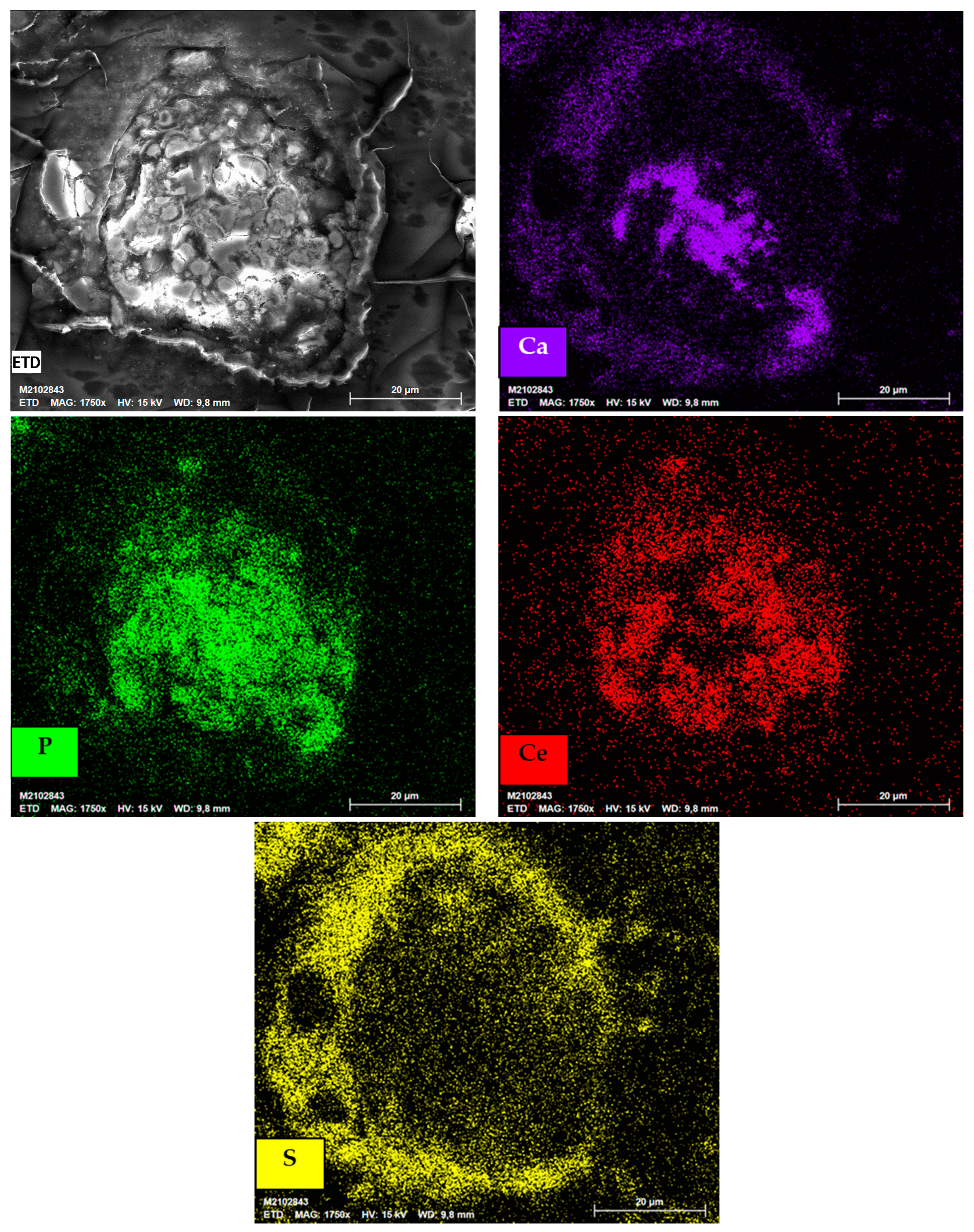
3.1. Characterization of the As-Received Samples
3.2. Implications in Rare Earth Element Extraction Associated with Impurities
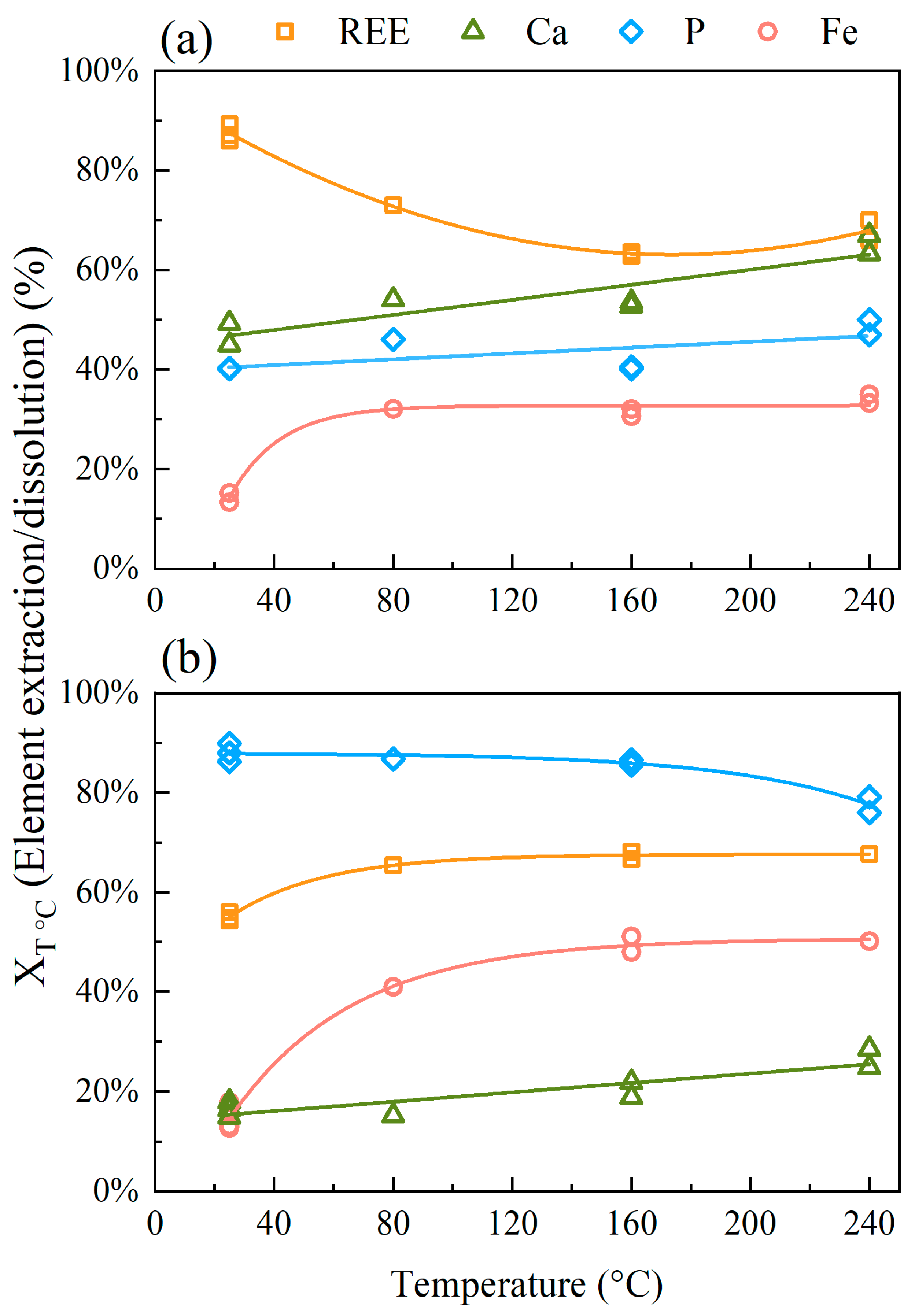
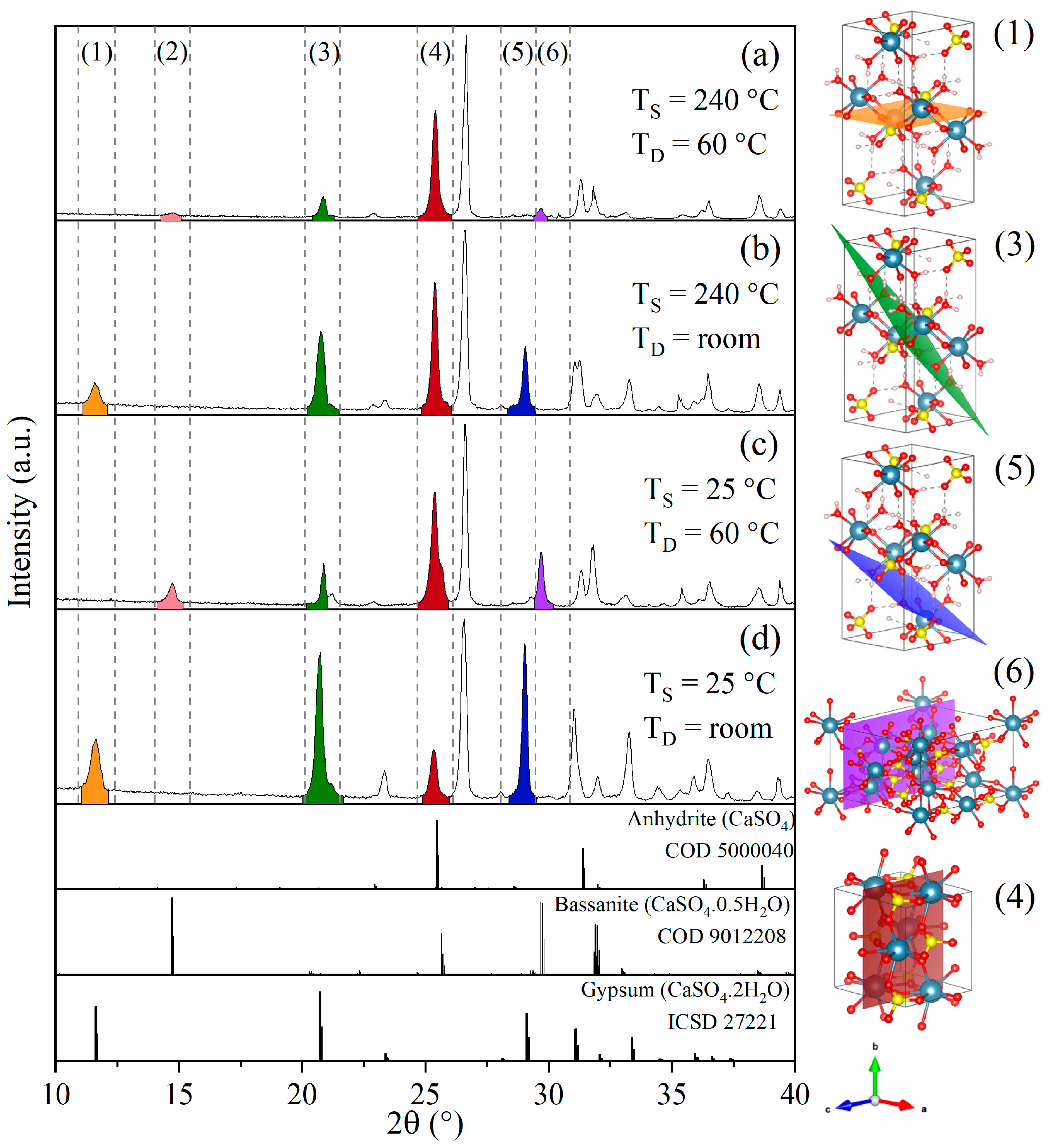
3.2.1. Effect of Temperature
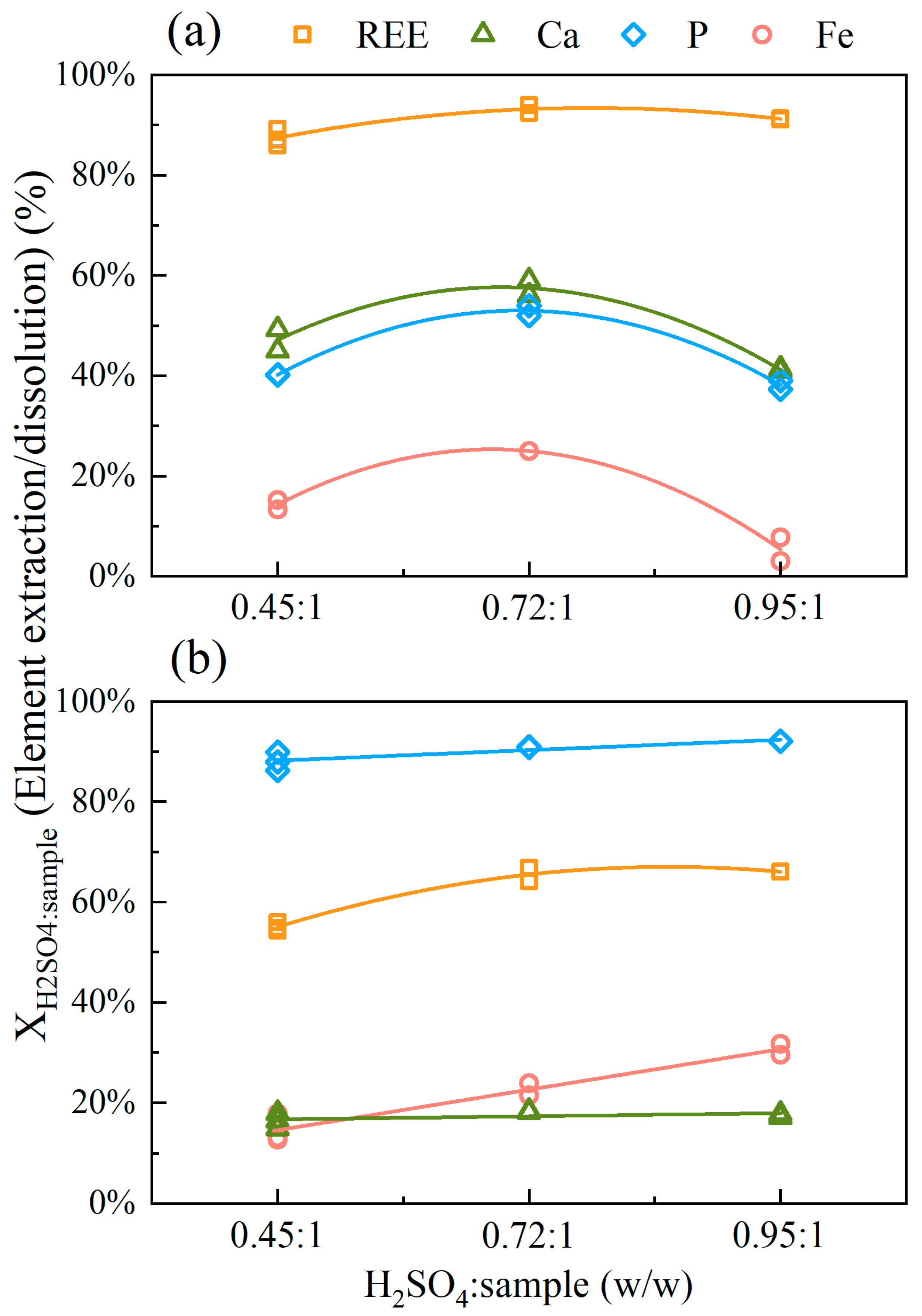
3.2.2. Effect of H2SO4:Sample Mass Ratio
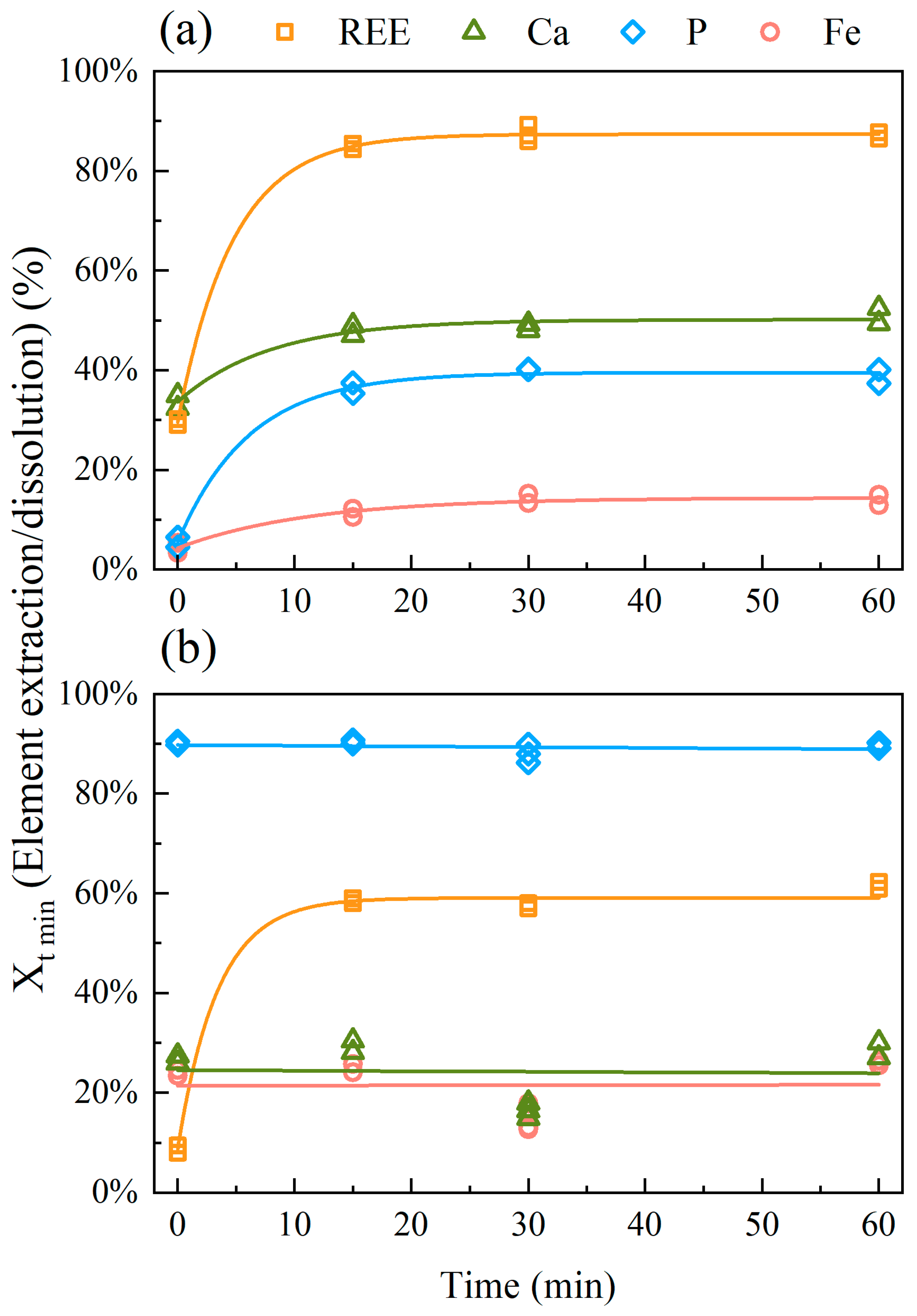
3.2.3. Effect of Time
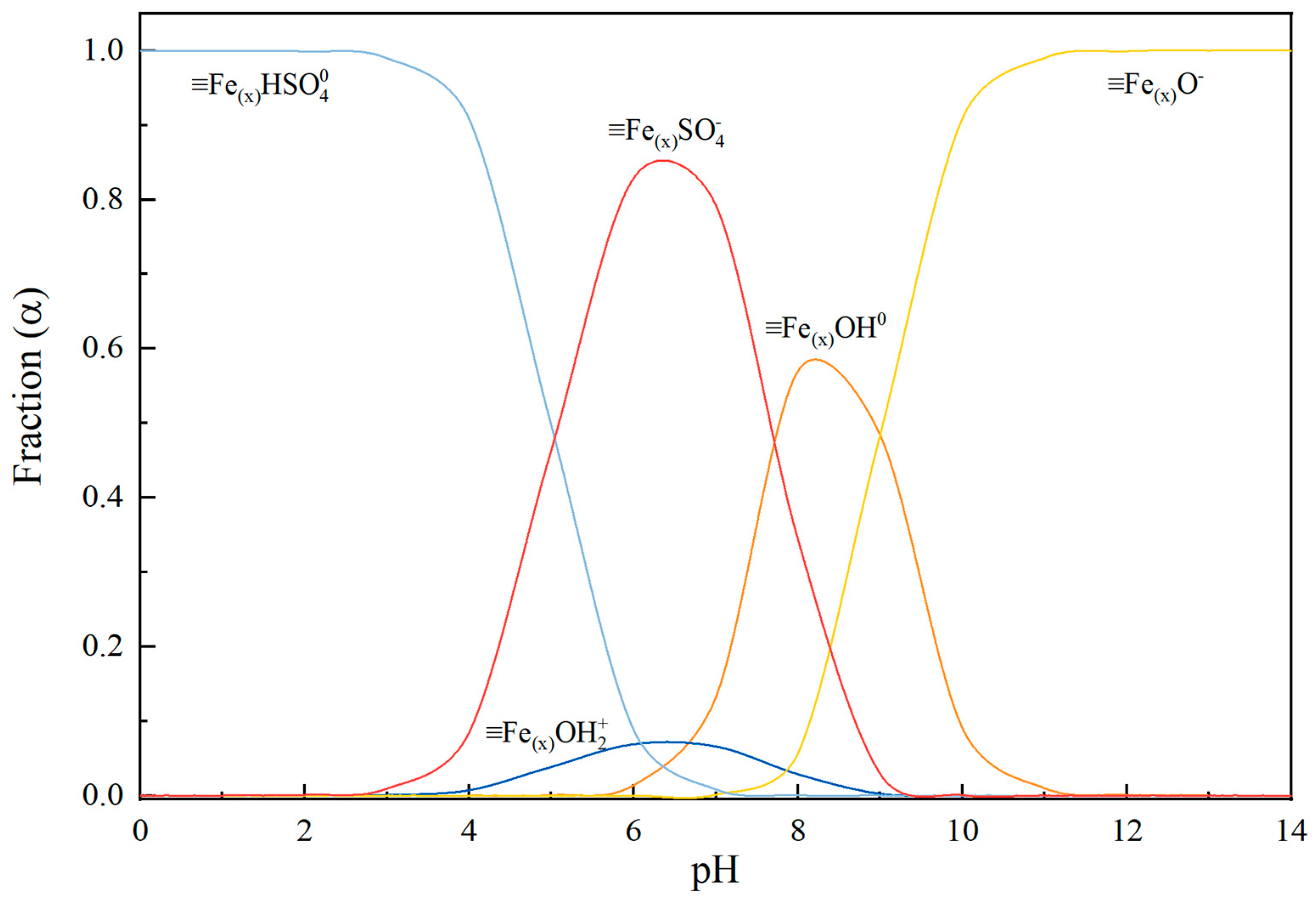
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gupta, C.K. Krishnamurthy. In Extractive Metallurgy of Rare Earths; CRC Press: Boka Radon, FL, USA, 2005. [Google Scholar]
- Lucas, J.; Lucas, P.; Le Mercier, T.; Rollat, A.; Davenport, W. Extracting Rare Earth Elements from Concentrates. In Rare Earth: Science, Technology, Production and Use; Elsevier Inc: Amsterdam, The Netherlands, 2015; pp. 50–54. [Google Scholar]
- Sousa Filho, P.C.; Serra, O.A. Rare Earths in Brazil: History, production and perspectives. Química Nova 2014, 37, 753–760. (In Portuguese) [Google Scholar] [CrossRef]
- Ribagnac, P.; Deblonde, G.J.P.; Blancher, S.B.; Lengagne, L.; Donati, L.; Malimba, C.; Courtaud, B.; Weigel, V.; Beltrami, D. Leaching of niobium- and REE-bearing iron ores: Significant reduction of H2SO4 consumption using SO2 and activated carbon. Sep. Purif. Technol. 2017, 189, 1–10. [Google Scholar] [CrossRef]
- Walawalkar, M.; Nichol, C.K.; Azimi, G. Process investigation of the acid leaching of rare earth elements from phosphogypsum using HCl, HNO3, and H2SO4. Hydrometallurgy 2016, 166, 195–204. [Google Scholar] [CrossRef]
- Beltrami, D.; Deblonde, G.J.P.; Bélair, S.; Weigel, V. Recovery of yttrium and lanthanides from sulfate solutions with high concentration of iron and low rare earth content. Hydrometallurgy 2015, 157, 356–362. [Google Scholar] [CrossRef]
- Wang, L.; Long, Z.; Huang, X.; Yu, Y.; Cui, D.; Zhang, G. Recovery of rare earths from wet-process phosphoric acid. Hydrometallurgy 2010, 101, 41–47. [Google Scholar] [CrossRef]
- Vaughan, J.; Tungpalan, K.; Fox, A.P.; Fu, W.; Gagen, E.; Nkruman, P.N.; Southam, G.; Ent, A.V.D.; Erskine, P.D.; Gow, P.; et al. Toward closing a loophole: Recovering rare earth elements from uranium metallurgical process tailings. JOM 2020, 73, 39–53. [Google Scholar] [CrossRef]
- Hamza, M.F.; El-Aassy, I.E.; Guibal, E. Integrated treatment of tailing material for the selective recovery of uranium, rare earth elements and heavy metals. Miner. Eng. 2019, 133, 138–148. [Google Scholar] [CrossRef]
- Rychkov, V.N.; Kirillov, E.V.; Kirillov, S.V.; Semenishchev, V.S.; Bunkov, G.M.; Botalov, M.S.; Smyshlyaev, D.V.; Malyshev, A.S. Recovery of rare earth elements from phosphogypsum. J. Clean. Prod. 2018, 196, 674–681. [Google Scholar] [CrossRef]
- Weshahy, A.R.; Gouda, A.A.; Atia, B.M.; Sakr, A.K.; Al-Otaibi, J.S.; Almuqrin, A.; Hanfi, M.Y.; Sayyed, M.I.; El Sheikh, R.; Radwan, H.A.; et al. Efficient recovery of rare earth elements and zinc from spent Ni-metal hydride batteries: Statistical studies. Nanomaterials 2022, 12, 2305. [Google Scholar] [CrossRef]
- Önal, M.A.R.; Borra, C.R.; Guo, M.; Blanpain, B.; Gerven, T.V. Hydrometallurgical recycling of NdFeB magnets: Complete leaching, iron removal and electrolysis. J. Rare Earths 2017, 35, 574–584. [Google Scholar] [CrossRef]
- Lee, C.H.; Chen, Y.J.; Liao, C.H.; Popuri, S.R.; Tsai, S.L.; Hung, C.E. Selective leaching process for neodymium recovery from scrap Nd-Fe-B magnet. Metall. Mater. Trans. A 2013, 44, 5825–5833. [Google Scholar] [CrossRef]
- Agência Nacional De Mineração (ANM). Sumário Mineral; Ministério de Minas e Energia: Brasil, Brazil, 2017; pp. 164–166. [Google Scholar]
- Louwerse, D. Rare Earth Element Deposits and Occurrences within Brazil and India. Bachelor’s Thesis, Delft University of Technology, Delft, The Netherlands, 2016. [Google Scholar]
- Vieira, E.V.; Lins, F.F. Processos Industriais de Concentração. In Concentração de Minérios de Terras-Raras: Uma Revisão; CETEM/CNPq: Rio de Janeiro, Brazil, 1997; Volume 7, pp. 19–32. (In Portuguese) [Google Scholar]
- Koopman, C.; Witkamp, G.J. Extraction of lanthanides from the phosphoric acid production process to gain a purified gypsum and a valuable lanthanide by-product. Hydrometallurgy 2000, 58, 51–60. [Google Scholar] [CrossRef]
- Habashi, F. Extractive metallurgy of rare earths. Can. Metall. Q. 2013, 52, 224–233. [Google Scholar] [CrossRef]
- Gontijo, V.L.; Teixeira, L.A.V.; Ciminelli, V.S.T. The reactivity of iron oxides and hydroxide during low-temperature sulfation. Hydrometallurgy 2020, 197, 105452. [Google Scholar] [CrossRef]
- Ribeiro, P.P.M.; Neumann, R.; Santos, I.D.; Rezende, M.C.; Rouse, P.R.; Dutra, A.J.B. Nickel carriers in laterite ores and their influence on the mechanism of nickel extraction by sulfation-roasting-leaching process. Miner. Eng. 2019, 131, 90–97. [Google Scholar] [CrossRef]
- Teixeira, L.A.V.; Silva, R.G.; Avelar, A.; Majuste, D.; Ciminelli, V.S.T. Selective Extraction of Rare Earth Elements from Monazite Ores with High Iron Content. Min. Metall. Explor. 2019, 36, 235–244. [Google Scholar] [CrossRef]
- Kar, B.B.; Swamy, Y.V. Some aspects of nickel extraction from chromitiferous overburden by sulphatization roasting. Miner. Eng. 2000, 13, 1635–1640. [Google Scholar] [CrossRef]
- Bainbridge, D.W. Sulfation of a Nickeliferous Laterite. Metall. Trans. 1973, 4, 1655–1658. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, B.; Schreiner, B. Separation Hydrometallurgy of Rare Earth Elements; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Soubiés, F.; Melfi, A.J.; Autefage, F. Geochemical behavior of rare earth elements in alterites from the Tapira phosphate and titanium deposit (Minas Gerais, Brazil): The importance of phosphates. Rev. Bras. Geociências 1991, 21, 3–16. (In Portuguese) [Google Scholar]
- Delvigne, J.E. Atlas of Micromorphology of Mineral Alteration and Weathering; Mineralogical Association of Canada: Ottawa, ON, Canada, 1998. [Google Scholar]
- Santos, R.L.C.; Sobral, L.G.S.; Araújo, R.V.V. Phosphate Production in Brazil: Tapira/Fosfertil Mining Complex. In Proceedings of the Encontro Nacional de Tratamento de Minérios e Metalurgia Extrativa, Recife, Brazil, 26–29 November 2002; Volume 1, pp. 439–445. (In Portuguese). [Google Scholar]
- Verbaan, N.; Bradley, K.; Brown, J.; Mackie, S. A review of hydrometallurgical flowsheets considered in current REE projects. In Symposium on Strategic and Critical Materials Proceedings; Simandl, G.J., Neetz, M., Eds.; British Columbia Ministry of Energy and Mines: Victoria, BC, Canada, 2015; Volume 3, pp. 147–162. [Google Scholar]
- Demol, J.; Ho, E.; Soldenhoff, K.; Senanayake, G. The Sulfuric Acid Bake and Leach Route for Processing of Rare Earth Ores and Concentrates: A Review. Hydrometallurgy 2019, 188, 123–139. [Google Scholar] [CrossRef]
- Thibault, Y.; McEvoy, J.G. Minimizing the impact of passivation during allanite-(Ce) decomposition in sulfuric acid media for rare earth recovery. Miner. Pet. 2020, 144, 559–571. [Google Scholar] [CrossRef]
- Lozano, A.; Ayora, C.; Martínez, A.F. Sorption of rare earth elements on schwertmannite and their mobility in acid mine drainage treatments. Appl. Geochem. 2020, 113, 104499. [Google Scholar] [CrossRef]
- Dutrizac, J.E.; Soriano, C. Behaviour of the rare earths during goethite (α-FeOOH) precipitation from sulphate-based solutions. Hydrometallurgy 2018, 176, 87–96. [Google Scholar] [CrossRef]
- Riley, E.; Dutrizac, J.E. The behavior of the rare earth elements during the precipitation of ferrihydrite from sulphate media. Hydrometallurgy 2017, 172, 69–78. [Google Scholar] [CrossRef]
- Swedlund, P.J.; Webster, J.G.; Miskelly, G.M. Goethite adsorption of Cu(II), Pb(II), Cd(II), and Zn(II) in the presence of sulfate: Properties of the ternary complex. Geochim. Cosmochim. Acta 2009, 73, 1548–1562. [Google Scholar] [CrossRef]
- Müller, B.; Sigg, L. Adsorption of Lead(II) on the goethite surface: Voltammetric Evaluation of surface complexation parameters. J. Colloid Interface Sci. 1992, 148, 517–532. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, properties, Reactions, Occurrence and Uses, 2nd ed.; VCH: Weinheim, Germany, 2003. [Google Scholar]
- Teixeira, L.A.V.; Silva, R.G.; Majuste, D.; Ciminelli, V.S.T. Stability of lanthanum in sulfate and phosphate systems and implications for selective rare earths extraction. Miner. Eng. 2020, 155, 106440. [Google Scholar] [CrossRef]
- Gontijo, V.L.; Teixeira, L.A.V.; Majuste, D.; Ciminelli, V.S.T. A review of thermodynamic data for lanthanum, iron, and thorium applied to rare earth extraction. Hydrometallurgy 2022, 213, 105951. [Google Scholar] [CrossRef]
- Li, H.; Zhao, N.; Zhang, G.; Liu, Y.; Long, Z.; Huang, X.; Grirem Advanced Materials Co., Ltd. A Process of Smelting Monazite Rare Earth Ore Rich in Fe. Patent AU 2008286599, 18 November 2010. [Google Scholar]
- Battsengel, A.; Batnasan, A.; Narankhuu, A.; Haga, K.; Watanabe, Y.; Shibayama, A. Recovery of light and heavy rare earth elements from apatite ore using sulfuric acid leaching, solvent extraction and precipitation. Hydrometallurgy 2018, 179, 100–109. [Google Scholar] [CrossRef]
- Dutrizac, J.E. The behavior of the rare earth elements during gypsum (CaSO4.2H2O) precipitation. Hydrometallurgy 2017, 174, 38–46. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Dissolution mechanism of calcium apatites in acids: A review of literature. World J. Methodol. 2012, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sadri, F.; Kim, R.; Yang, Z.; Ghahreman, A. The effect of calcium sulfate crystallization and the crystal modification on aqueous REE stability in Ca saturated REE-Ca-SO4-H2O systems. Hydrometallurgy 2018, 182, 82–96. [Google Scholar] [CrossRef]
- Vreugd, C.H.; Witkamp, G.J.; Rosmalen, G.M. Growth of gypsum III. Influence and incorporation of lanthanide and chromium ions. J. Cryst. Growth 1994, 144, 70–78. [Google Scholar] [CrossRef]
- Kurkinen, S.; Virolainen, S.; Sainio, T. Recovery of rare earth elements from phosphogypsum waste in resin-in-leach process by eluting with biodegradable complexing agents. Hydrometallurgy 2021, 201, 105569. [Google Scholar] [CrossRef]
- Silva, R.G.; Morais, C.A.; Oliveira, E.D. Evaluation of different neutralization reagents in the selective removal of impurities in rare earth sulfuric liquor. Min. Metall. Explor. 2020, 37, 65–78. [Google Scholar] [CrossRef]
- Silva, R.G.; Morais, C.A.; Oliveira, E.D. Selective precipitation of rare earth from non-purified sulfate liquors using sodium sulfate and disodium hydrogen phosphate. Miner. Eng. 2019, 134, 402–416. [Google Scholar] [CrossRef]
- Silva, R.G.; Morais, C.A.; Teixeira, L.V.; Oliveira, E.D. Selective precipitation of high-quality rare earth oxalate or carbonates from a purified sulfuric liquor containing soluble impurities. Min. Metall. Explor. 2019, 36, 967–977. [Google Scholar] [CrossRef]
- Larroumet, D.; Greenfield, D.; Akid, R.; Yarwood, J. Spectroscopic studies of the corrosion of model iron electrodes in carbonate and phosphate buffer solutions. J. Raman Spectrosc. 2008, 39, 1740–1748. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Composition (%) | Bebedourite | Silexite |
|---|---|---|
| Al2O3 | 4.0 | 0.8 |
| BaO | 2.4 | 0.4 |
| CaO | 0.9 | 24.9 |
| Fe2O3 | 45.6 | 17.2 |
| MgO | 0.3 | 0.6 |
| MnO | 1.3 | 0.6 |
| P2O5 | 7.2 | 19.5 |
| SiO2 | 10.9 | 27.0 |
| TiO2 | 11.0 | 2.7 |
| F | 0.2 | 1.4 |
| Others 1 | 1.0 | 0.3 |
| U (mg kg−1) | 99.0 | 119.5 |
| Th (mg kg−1) | 479.7 | 70.7 |
| REO 2 | 7.4 | 2.1 |
| LREE 3/REETotal | 98.9 | 98.8 |
| LOI | 7.8 | 2.5 |
| Sample | BEDE | SILX |
|---|---|---|
| d10 (µm) | 0.6 | 1.0 |
| d50 (µm) | 5.0 | 13.5 |
| d90 (µm) | 22.7 | 43.7 |
| Mean diameter (µm) | 8.8 | 18.2 |
| SSA25 °C (m2 g−1) | 21.9 | 5.7 |
| SSA240 °C (m2 g−1) | 32.3 | 7.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gontijo, V.L.; Teixeira, L.A.V.; Ciminelli, V.S.T. The Effect of Iron- and Calcium-Rich Waste Rock’s Acid Baking Conditions on the Rare-Earth Extraction. Minerals 2023, 13, 217. https://doi.org/10.3390/min13020217
Gontijo VL, Teixeira LAV, Ciminelli VST. The Effect of Iron- and Calcium-Rich Waste Rock’s Acid Baking Conditions on the Rare-Earth Extraction. Minerals. 2023; 13(2):217. https://doi.org/10.3390/min13020217
Chicago/Turabian StyleGontijo, Vitor L., Leandro Augusto Viana Teixeira, and Virgínia Sampaio Teixeira Ciminelli. 2023. "The Effect of Iron- and Calcium-Rich Waste Rock’s Acid Baking Conditions on the Rare-Earth Extraction" Minerals 13, no. 2: 217. https://doi.org/10.3390/min13020217