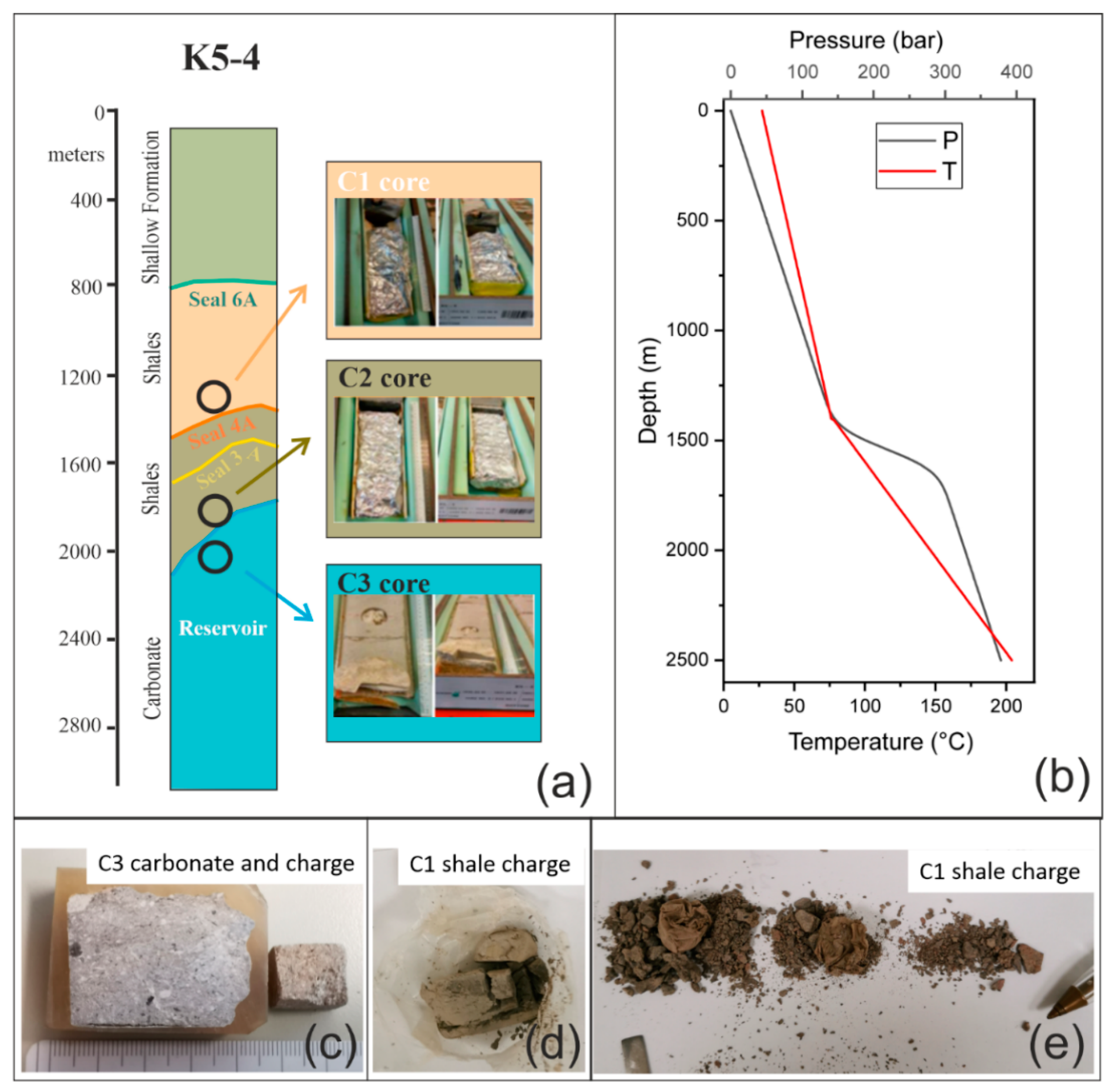
3.1. Cores Characterization
Original petrographic features of core samples were defined at various scales by XRPD, XRF, FT-IR, and SEM-EDS analyses.
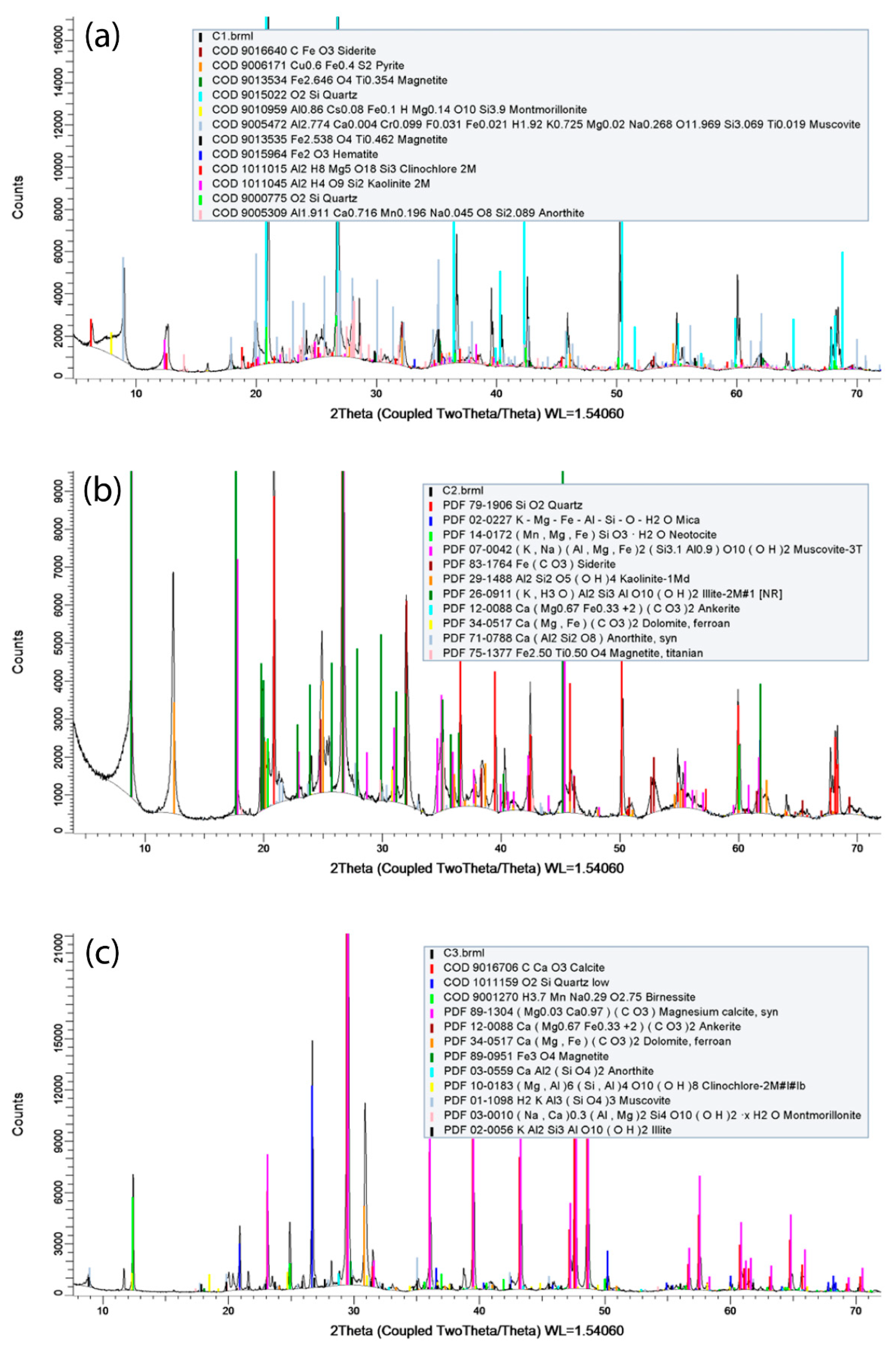
XRPD combined with Rietveld refinement (
Figure 3) allowed a semi-quantitative mineralogical composition normalized to 100 (
Table 1). Analytical results show that the shale and shaly silt overburden (C1 and C2 samples) is constituted by ca. 36 and 51% by weight of quartz and plagioclase (anorthite by Rietveld analysis), respectively. Clay minerals (illite, kaolinite and chlorite) contribute from 31 to 40%
w/w with minor mica (muscovite) content of 11%
w/w. Finally, low amounts of carbonates (calcite, dolomite, and siderite) are present (C1 sample = 7%
w/w, C2 sample = 14%
w/w). If we compare Seal 6 and Seal 4 samples, this latter shows the major contents of carbonate (14%) and clay minerals (40%
w/w) and the minor contents of quartz and anorthite (sand = 36%
w/w). The reservoir (C3 sample) is constituted by limestone (76%
w/w), mainly calcite and dolomite, subordinate quartz (13%
w/w), anorthite and mica, and a very low abundance of clay minerals (10%
w/w; illite, chlorite and kaolinite).
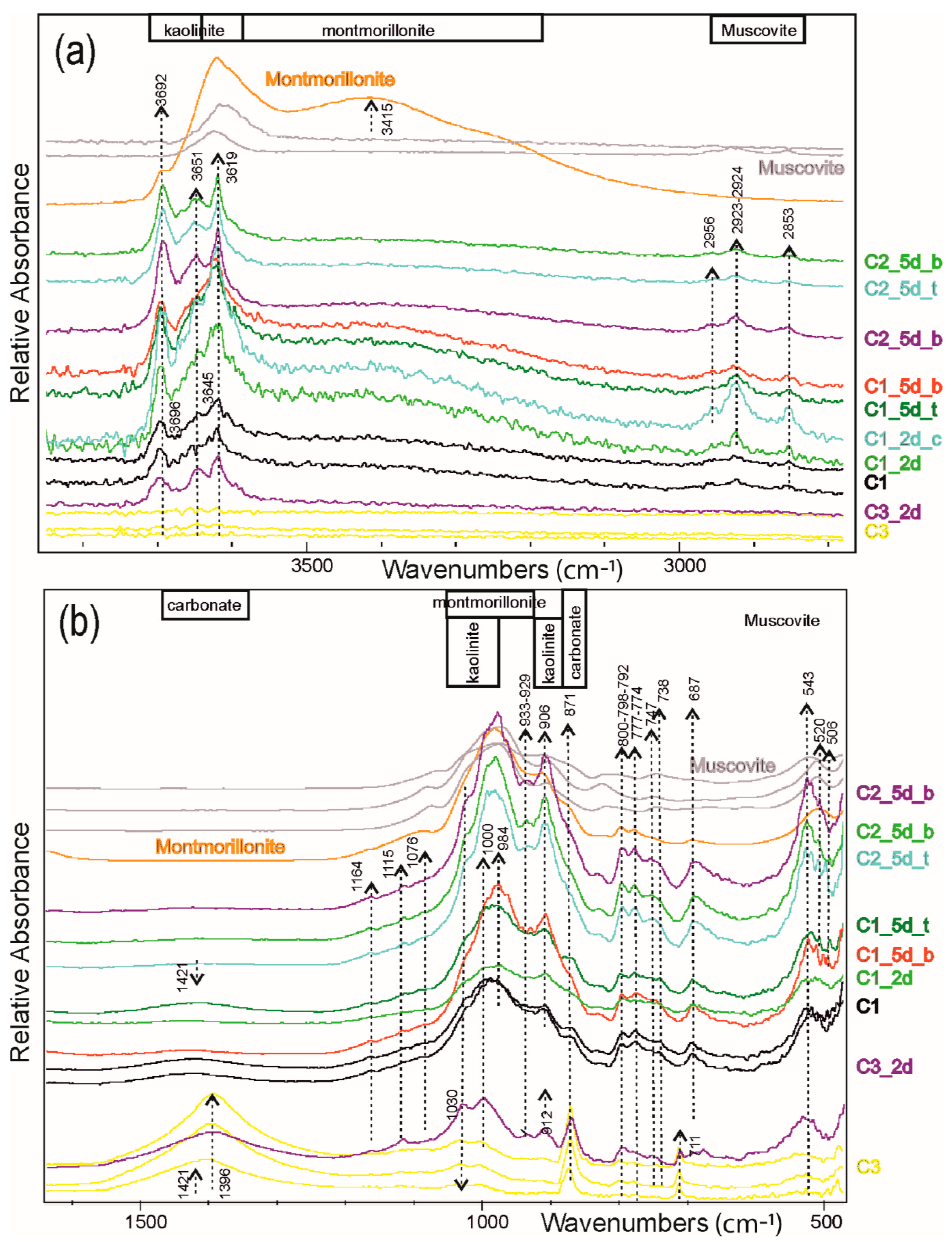
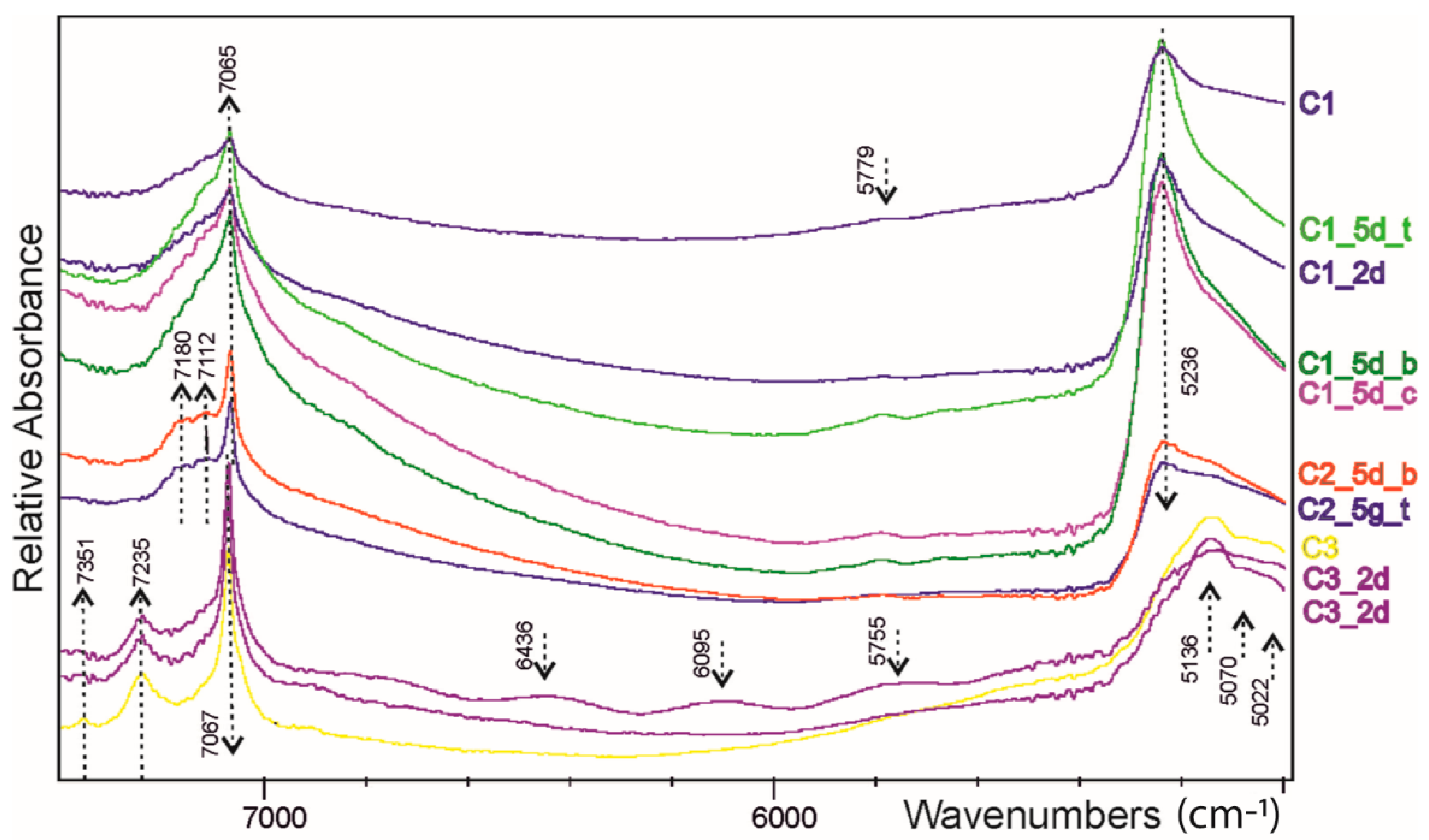
FT-IR spectra (
Figure 4 and
Figure 5) corroborate the XRPD results indicating carbonate and clay prevalence in the C3 and in the C1 samples, respectively. Actually, the C3 spectrum is dominated by the signals of calcite with typical spikes of absorption bands at 1396 (𝜈
3 CO
3−2asymmetrical stretch), 871 (𝜈
2 CO
3−2asymmetrical bend) and 711 (𝜈
4 CO
3−2aymmetrical bend) cm
−1 [
23]. A small contribution from the substitution of other (Mg, Fe, Mn) cations can be suspected by comparing the smoothed peak at 5154 cm
−1 of our sample with those of siderite (HS271.3B) in the USGS database (
https://www.usgs.gov/labs/spectroscopy-lab (accessed on 1 November 2022)). Considering the USGS database, the signals at 7092 and 7246 cm
−1 in the carbonate sample highlight the scarce presence of mica and clays, although the doublet at 7246 and 7.092 cm
−1 can be tentatively attributed to the kaolinite occurrence. Conversely, for the investigated C1 shale, the illite determines the peaks sequence in the NIR frequency (7092, 5236, 4504, 4255 cm
−1) and the shape in the OH-stretching region. In this later range, the mica contribution can be also revealed at ca. 3645 cm
−1, as well as the triplet at ca. 2656, 2923, and 2853 cm
−1. According to [
24,
25], several peaks at ca. 1030, 1000, and 984 cm
−1 (Si-O stretching), at ca. 933, 906, and 870 cm
−1 (vibration of AlAlOH and AlFeOH groups) and at ca. 543, 520, and 506 cm
−1 (Al-O and Si-O deformation) can be attributed to clays (mostly illite/montmorillonite and kaolinite). Notably, for C1, the triplet at 2957 + 2922 + 2850 cm
−1 and the smoothed signals at 1421 cm
−1 point to muscovite being richest and a significant occurrence of (ankerite, dolomite, siderite) carbonates, respectively. Based on the peak at 3619 and the hamp at 3415 cm
−1, it is demonstrated that montmorillonite/illite is the major clay in shales.
XRF results are reported in
Table 2, also considering CO
2 loss correction. The overburden shales mostly contain SiO
2 (38–41%
w/w) and Al
2O
3 (12–16%
w/w), with minor amounts of K
2O (2.6%
w/w) and Fe
2O
3 (5.2%
w/w). Boron and magnesium oxides are also present. Moreover, for XRPD, these oxides suggest the presence of quartz and clay minerals rich in potassium and iron, such as illite, chlorite, and muscovite. In the C3 sample, the most abundant oxide is CaO (40.2%
w/w) with minor amounts of SiO
2 (13%
w/w), Al
2O
3 (6%
w/w), Fe
2O
3 (1.6%
w/w), and SO
3 (1.33%
w/w). The presence of SO
3 is likely due to pyrite contribution, which was recognized in XRPD but not quantified due to the very low amount.
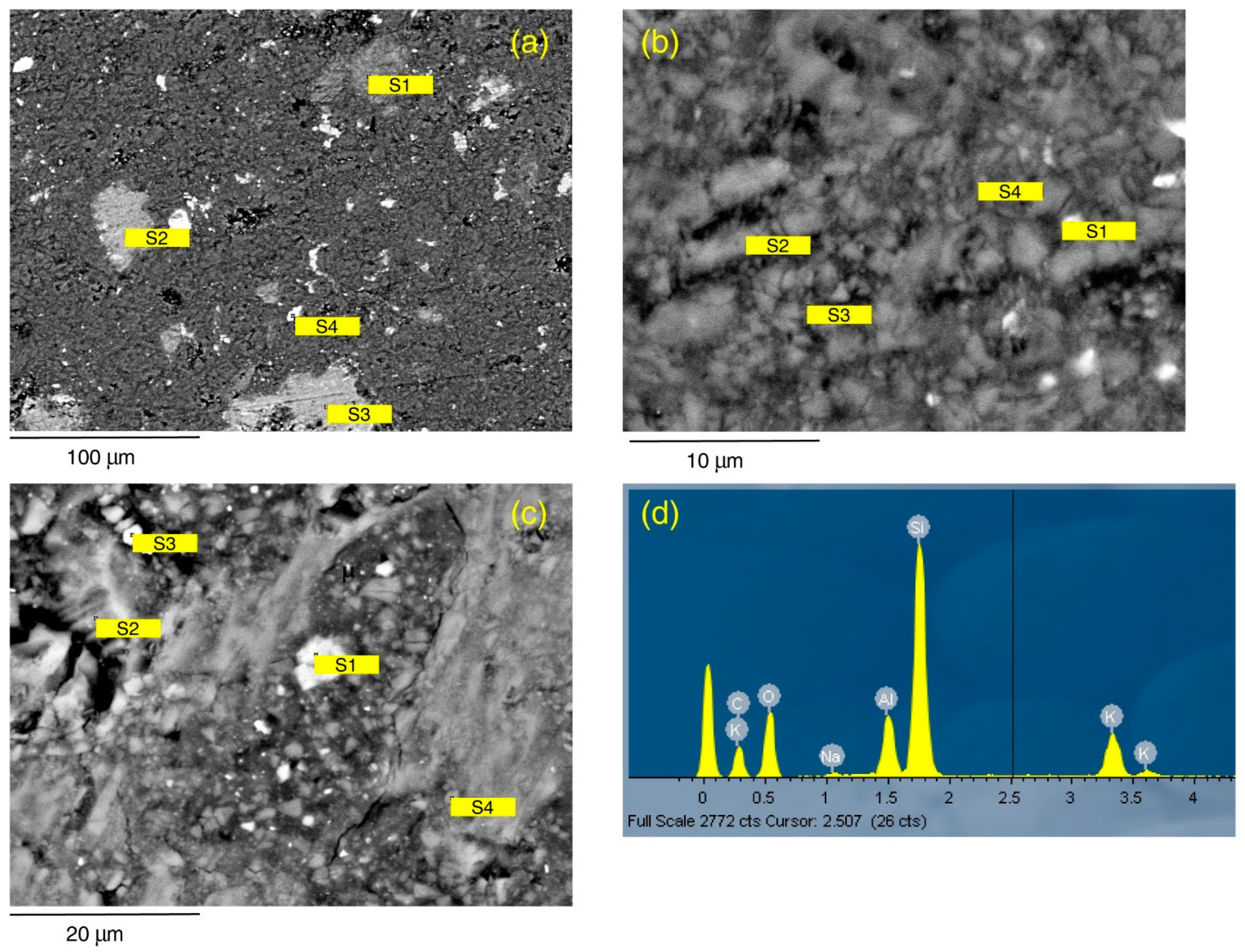
SEM-EDS investigation of Seal 6 shale (C1 sample) at a low magnification scale (
Figure 6a) shows the presence of some phenocrysts of muscovite (mean 24 × 33 µm), Fe-rich dolomite-ankerite (mean 24 × 37 µm), and anatase (mean 6 × 12 µm), which are dispersed in a dominant finer matrix. The matrix is cohesive and generally low porous. The total porosity, measured by MICP, is 8.7%, with an asymmetrical distribution of pores and mean frequency of 0.019 μm. Tortuosity is 2.174. The zoom on the matrix (
Figure 6b,c) reveals that it is constituted by microcrysts of quartz (mean 1.53 × 2.6 µm), secondary illite (mean 0.7 µm), chlorite (mean 0.93 × 1.33 µm), and kaolinite (mean 0.62 × 3.19 µm). The clay minerals exhibit both spherical and plate like shape. Illite and chlorite are generally rich in iron ion, in variable content ranging from 0.51% to 7.03%, in agreement with marine or hypersaline environments where the sedimentation rate is low. Indeed, the sources of K (for illite) and Fe ions are seawater and detrital minerals, respectively [
26]. Cubic siderite-dolomite microcrystals (mean 1.11 × 1.99 µm) are also present. Primary plagioclase (5.5 µm, spectrum 2) and quartz are visible in
Figure 6c.
Figure 6.
SEM-EDS data of C1 sample in thin section. (
a) General aspect at low magnification scale at the analytical Site 1. S1: muscovite with quartz; S2 and S3: solid solution of Fe-rich dolomite and ankerite; S4: anatase. (
b) Texture and mineralogy of the analytical Site S2. S1: Fe-rich illite; S2 and S3: quartz; S4: illite. (
c) Texture and mineralogy of the analytical Site 3. S1: solid solution of siderite-ankerite; S2: kaolinite with adsorbed Na from underlying primary plagioclase; S3: solid solution of Fe-rich chlorite and illite; S4: kaolinite. (
d) EDS spectrum of S1 in panel (a) compatible with mixture of muscovite and quartz. Mineral compositions are related to spectrum for site in
Table 3.
Figure 6.
SEM-EDS data of C1 sample in thin section. (
a) General aspect at low magnification scale at the analytical Site 1. S1: muscovite with quartz; S2 and S3: solid solution of Fe-rich dolomite and ankerite; S4: anatase. (
b) Texture and mineralogy of the analytical Site S2. S1: Fe-rich illite; S2 and S3: quartz; S4: illite. (
c) Texture and mineralogy of the analytical Site 3. S1: solid solution of siderite-ankerite; S2: kaolinite with adsorbed Na from underlying primary plagioclase; S3: solid solution of Fe-rich chlorite and illite; S4: kaolinite. (
d) EDS spectrum of S1 in panel (a) compatible with mixture of muscovite and quartz. Mineral compositions are related to spectrum for site in
Table 3.
Table 3.
Semi-quantitative analysis of C1 sample in atoms percentage.
Table 3.
Semi-quantitative analysis of C1 sample in atoms percentage.
| Spectrum | Mineral | O | Na | Mg | Al | Si | K | Ca | Ti | Mn | Fe |
|---|
| 1-Site1 | muscovite + quartz | 64.70 | 0.64 | - | 5.59 | 24.15 | 4.92 | - | - | - | - |
| 2-Site1 | Fe-dolomite + ankerite | 74.57 | - | 6.64 | - | 0.52 | - | 13.38 | - | 1.21 | 3.69 |
| 3-Site1 | Fe-dolomite + ankerite | 73.96 | - | 6.16 | - | 0.60 | - | 15.03 | - | - | 4.25 |
| 4-Site1 | anatase | 68.70 | 0.67 * | - | 0.25 * | 0.55 * | - | - | 29.83 | - | - |
| 1-Site2 | Fe-rich illite | 65.39 | 1.12 | 2.07 | 6.38 | 19.25 | 0.87 | 0.49 | - | - | 4.42 |
| 2-Site2 | quartz | 65.45 | - | - | 0.31 * | 34.24 | - | - | - | - | - |
| 3-Site2 | quartz | 66.55 | - | - | 0.38 * | 33.07 | - | - | - | - | - |
| 4-Site2 | Illite | 64.91 | 1.23 | 0.56 | 6.03 | 25.93 | 0.83 | - | - | - | 0.51 |
| 1-Site3 | siderite-dolomite | 68.06 | - | 7.62 | 1.82 * | 2.92 * | - | 1.79 | - | - | 17.78 |
| 2-Site3 | kaolinite on primary plagioclase | 68.35 | 4.65 | - | 7.79 | 17.56 | 0.17 | 1.49 | - | - | - |
| 3-Site3 | Fe-chlorite+illite | 68.84 | 2.85 | 3.44 | 4.81 | 11.50 | - | 1.55 | - | - | 7.03 |
| 4-Site3 | kaolinite | 66.30 | 2.17 | - | 5.60 | 24.52 | - | 1.23 | - | - | 0.19 |
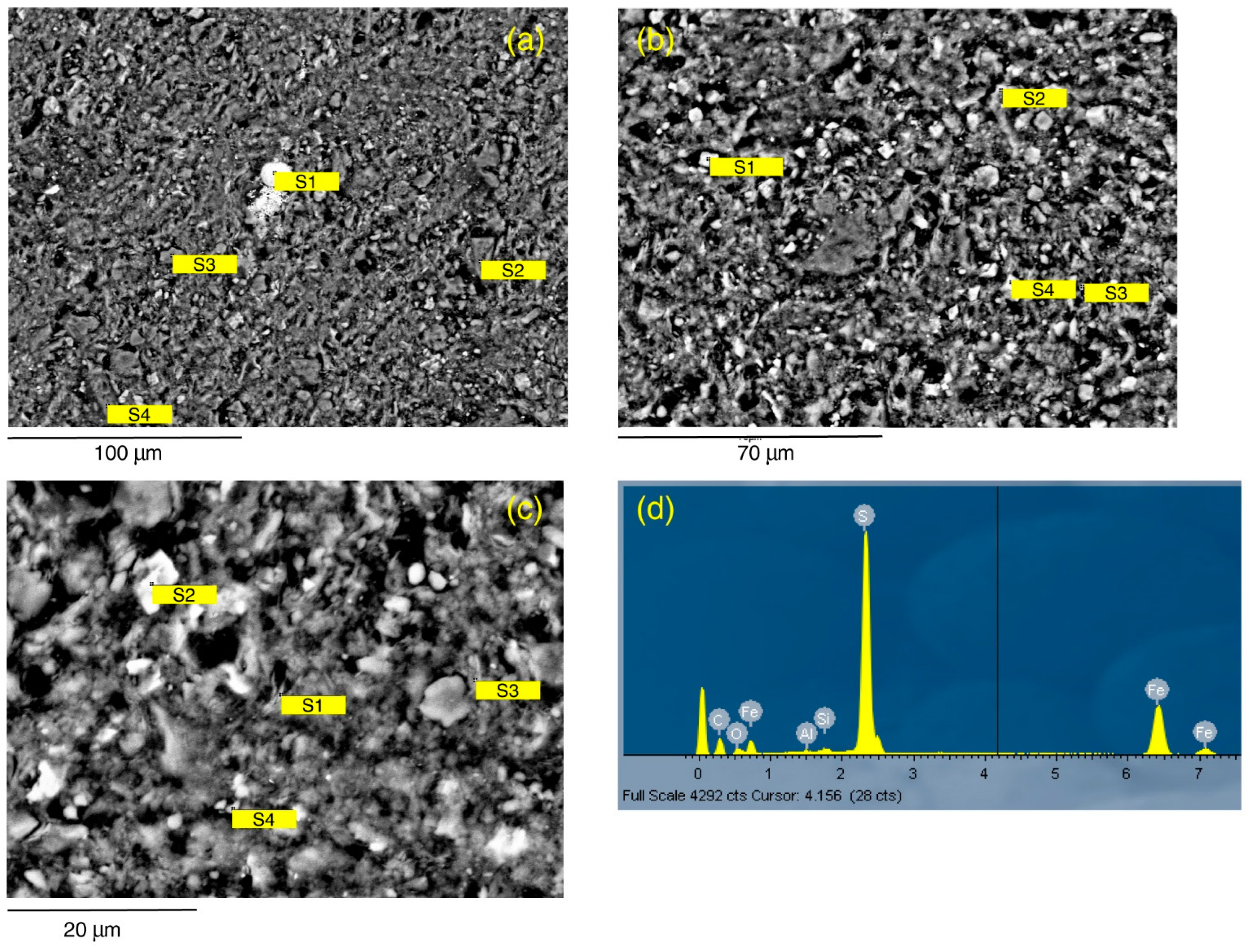
SEM analysis of Seal 4 (C2 sample) at low magnification (
Figure 7a) shows a slightly coarser and more porous structure than C1 sample. Based on MICP, this sample has a total porosity of 9.2%, with an average pore distribution of 0.15 μm. Tortuosity is 2.164. At the above spatial scale, only pyrite (mean 10 µm) and quartz macro crystals (mean 4.8 × 12.7 µm) are clearly distinguishable. An enlarged magnification (
Figure 7b) allows further distinguishing cubic crystals of dolomite-siderite (mean 2.8 µm), small crystals of anatase (mean 1.6 µm) (
Table 4) and, in the matrix, illite, both spherical (mean 2.15 µm) and plate shape (mean 1.18 × 5.05 µm), kaolinite (mean 0.71 × 4.83 µm), and quartz (mean 12 × 19 µm). The zoom on the matrix (
Figure 7c,
Table 4) shows that it is mostly constituted by secondary illite-chlorite rich in Fe, both spherical (mean 1.62 µm) and plate shape (mean 1.03 × 2.38 µm), kaolinite (mean 0.28 × 1.35 µm), muscovite (mean 0.84 × 2.3 µm), and micro crystals of dolomite-siderite (mean 2.5 µm).
SEM analysis of carbonate reservoir sample (C3) at a low magnification scale (
Figure 8a) shows the presence of primary calcite (
Table 5) and clay with a plate-like structure (mean 0.5 × 2.23 µm). The porosity, as measured by MICP, is 9.4%, close to the C2 sample but C3 has a more symmetrical distribution, with average porous dimension ranging from 0.0723 to 0.0792 μm. Tortuosity is 2.125. A fossil re-crystallized by small crystals of pyrite (mean 1 µm) is present in the center of the picture. The zoom on the matrix (
Figure 8b,c) reveals that it is constituted by secondary calcite (mean 2.19 µm), kaolinite (mean 0.8 × 6.4 µm), illite (mean 1.2 × 4.75 µm), muscovite (mean 0.52 × 5.7 µm), and quartz (mean 0.92 × 4.2 µm). Rare crystals of pyrite (3–5 µm) and sylvite are also present.
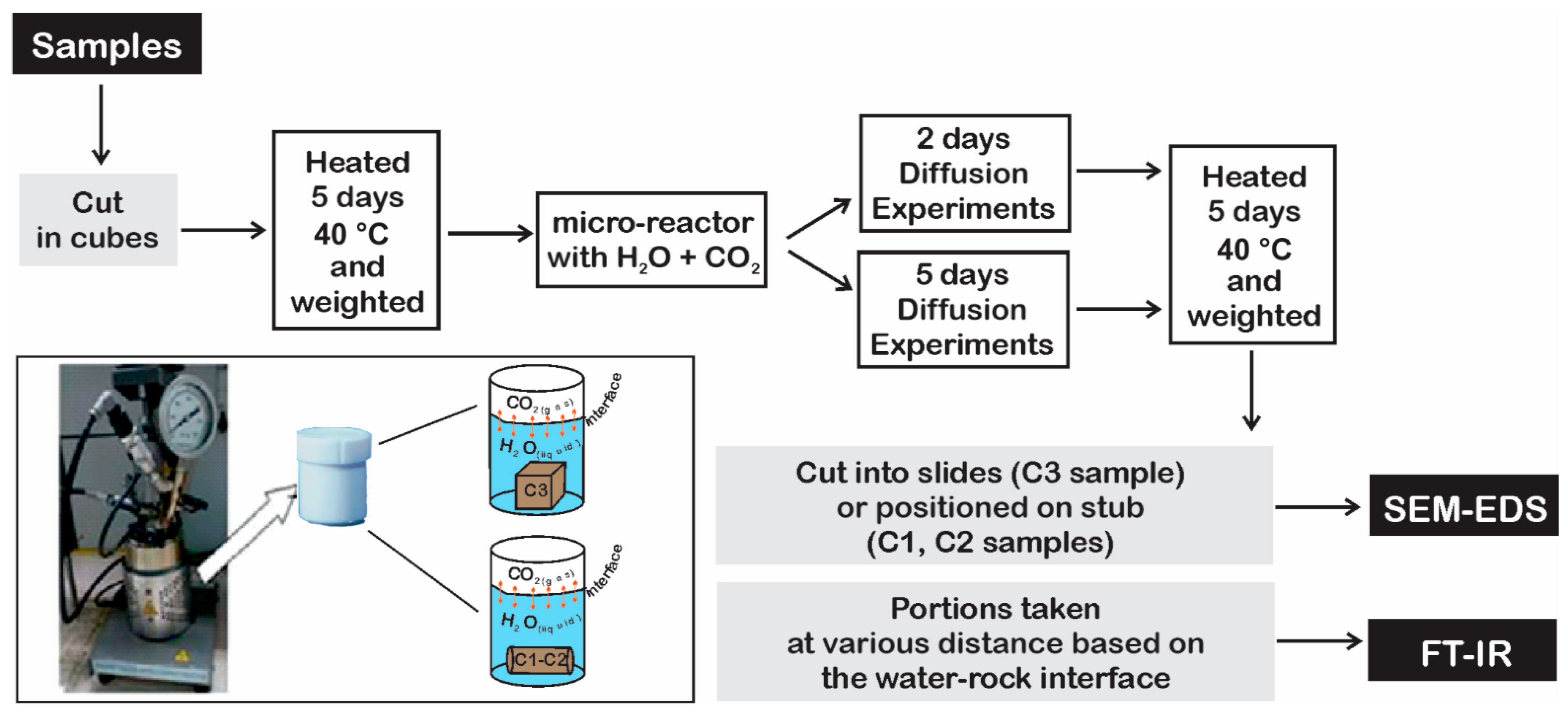
3.2. Diffusion Experiments
The reaction front due to fluid–rock interaction was evaluated by SEM-EDS analysis after two and five days of interaction inside the micro-reactor. As expected, due to the short interaction time, the results indicate limited sample changes that mostly concerned with carbonates and pyrite.
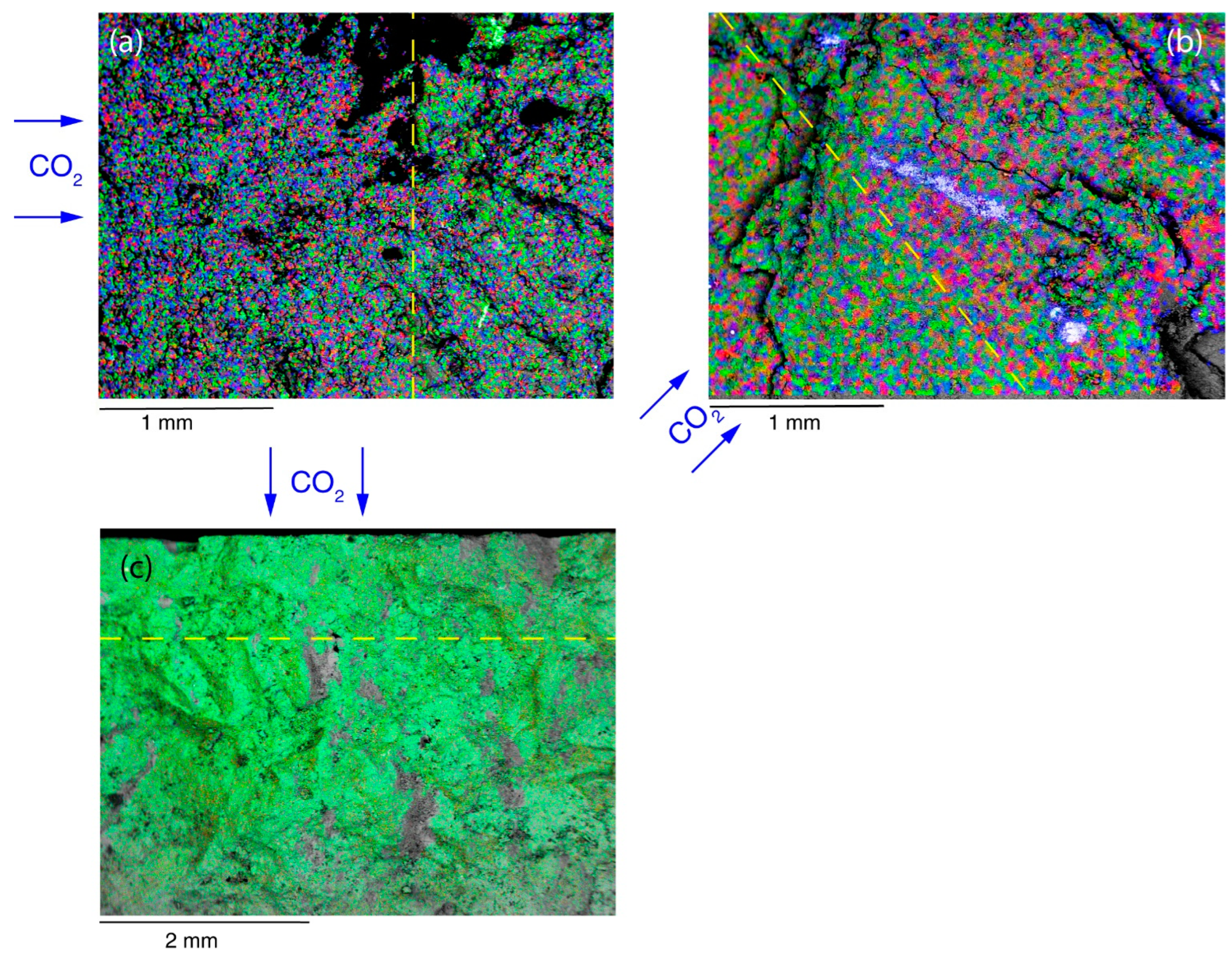
SEM-EDS investigations were conducted in two modes: (i) classical electron microscope image (e.g.,
Figure 9), and (ii) chemical map (cameo image) by chemical fake-coloring (
Figure 10). The color scale was arbitrarily changed to highlight the reaction front, thus not reflecting the real abundance of the elements. The first analysis aimed to recover chemical changes and to spot secondary minerals, whereas the cameo images were used to define the boundary between unaltered sample and the weathering due to CO
2 reaction.
A general feature observed on the SEM images after the experiments is the depletion in the pyrite microcrystals and a relative increased abundance of carbonates mostly in the form of micron sized cubes (
Figure 9). These carbonates, still pertaining to the dolomite-siderite-ankerite series, generally resulted depleted in Ca in favor of Mg and Fe at the EDS, exhibiting an evident rhombohedral habit (C3 sample,
Figure 11) and believed to represent secondary phases.
Figure 10a (C1 sample) shows this phenomenon, with a relative enrichment of Fe and Mg (red and blue colors, respectively) with respect to Ca (green color) in the portion of the specimen which reacted with CO
2. In
Figure 10b (C2 sample) a relative enrichment of Mg (green) is visible within the reaction area. The Mg relative enrichment and false scale color jeopardize the original major abondance of Fe and K (visible on the other side of reaction front) of C2 with respect to C1 samples. The reaction front of C3 sample (
Figure 10c) is slightly evident since Ca and Mg are the major elements constituting carbonates. The green color represents the iron content, which is more evident in the unaltered sample portion, thus allowing the front identification by the color intensity. The different coloring across the reaction front is due to the weathering process, which occurs as CO
2(aq) reached the minerals, and its effect on the coloring is not easily predictable, since it could be due to both matrix depletion after weathering and/or subsequent deposition of leached elements.
The distribution of minerals and elements at mm-scale allows to define two reaction fronts, the first corresponding to the area of pyrite depletion and the second coinciding with the precipitation of secondary carbonates.
The distance of these fronts from the outer part of the specimen allows defining the CO
2 penetration depth (e.g.,
Figure 9) after two and five days. These distances were measured by SEM ruler, whose instrumental accuracy is within 1 µm, but considering the difficulties in localizing the alteration front and its unevenness, we can reasonably estimate the experimental error to be approximately 10 µm. The measured penetration front and the depletion front length are reported in
Table 6.
FT-IR spectra in
Figure 4 and
Figure 5 indicate no significant variation in shales subjected to diffusion experiments, whereas after two days of hydrothermal action, the C3 carbonate significantly modified its infrared response. In particular, several peaks appeared in the OH- stretching region and between 1500 and 500 cm
−1 of the experimental C3 sample, i.e., at ca. 3695, 3648, 3633, 3618, 932–910, 540–470 cm
−1(
Figure 4), pointing to kaolinite plus illite/muscovite [
24,
25,
27]. The doublet at 3695 and 3618 cm
−1 is diagnostic for the kaolin phase and is corroborated by absorption at 933–929 and 906 cm
−1 (OH-bending/vibrations). The illite/muscovite is indicated by signals at ca. 1030, 912, and 520 cm
−1. The spectra also show a span of both 𝜈
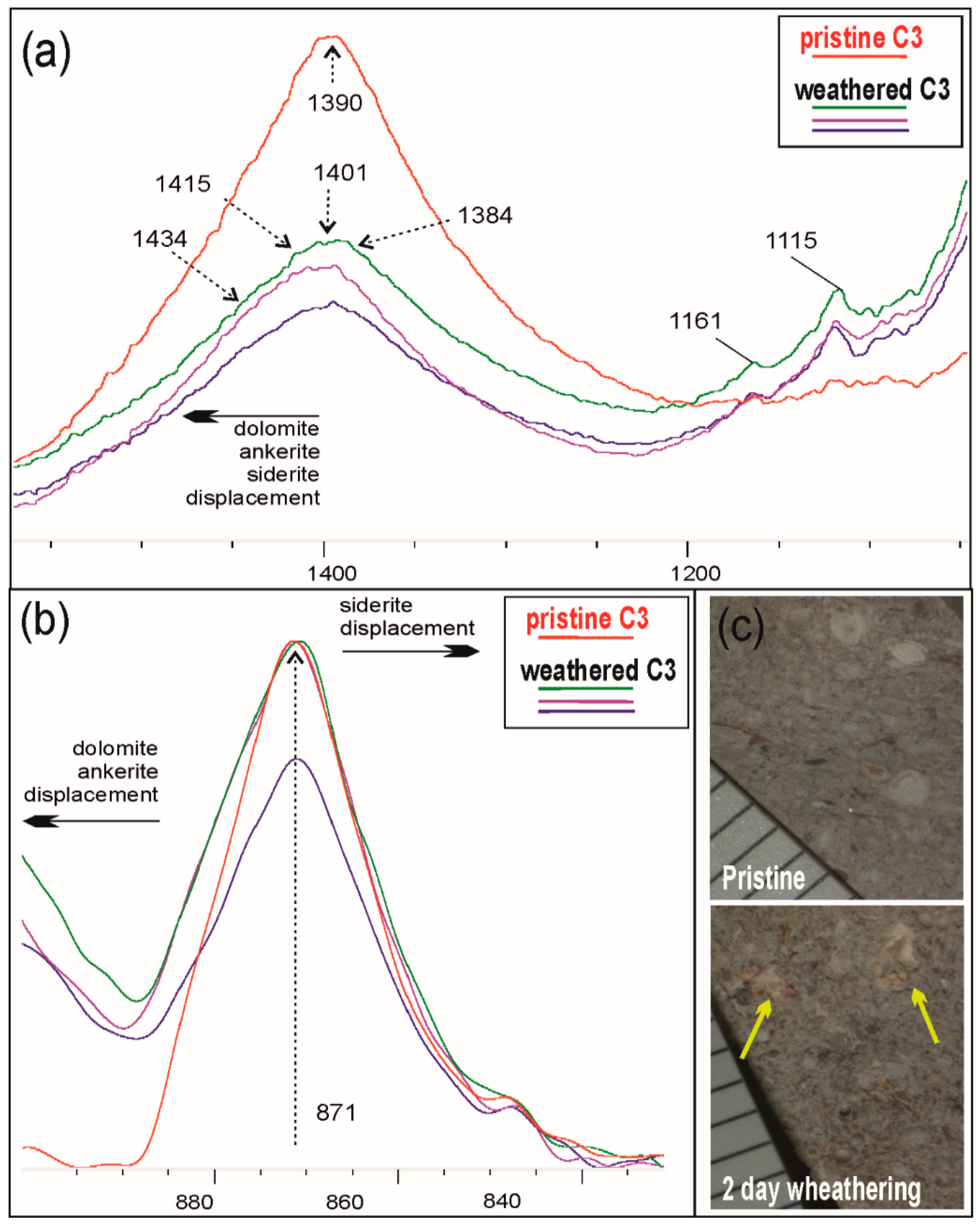
3 CO
32− asymmetrical stretch and bend bands, the first band assuming a bell-shaped between 1384 and 1434 cm
−1 and the second slightly distorting towards higher and lower wavenumbers (
Figure 12a,b). Following [
23], this indicates Mg, Fe carbonate and, in our case, may suggest calcite dissolution in agreement with the experimental sample aspect under the optical microscope (
Figure 12c). Notably, shales are slightly different, maintaining the more accentuated peaks at 3651, 3619, and 932 cm
−1 for the kaolinite abundance in sample C2, and for C1, the triplet at 2957 + 2922 + 2850 cm
−1 for muscovite being richest, and the smoothed signals at 1421 cm
−1 for the occurrence of ankerite, dolomite, and siderite carbonates, in addition to the illite (3622 and 3415 cm
−1). However, in these experimental samples, the infrared signals at 800, 777, and 747 cm
−1 (
Figure 4) show faint but sensible variations that would be interest to OH bending vibrations/interactions with cations, thus pointing to possible leeching processes. The 1000 and 900 cm
−1 bands, related to Si-O-Si stretching and Al-Al-OH bending [
28], can be tentatively attributed to structural modification of the illite clays or muscovite with possibly consequent cation losses.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










