
Calcium Carbonate Hexahydrate (Ikaite): History of Mineral Formation as Recorded by Stable Isotopes
Abstract
:1. Introduction
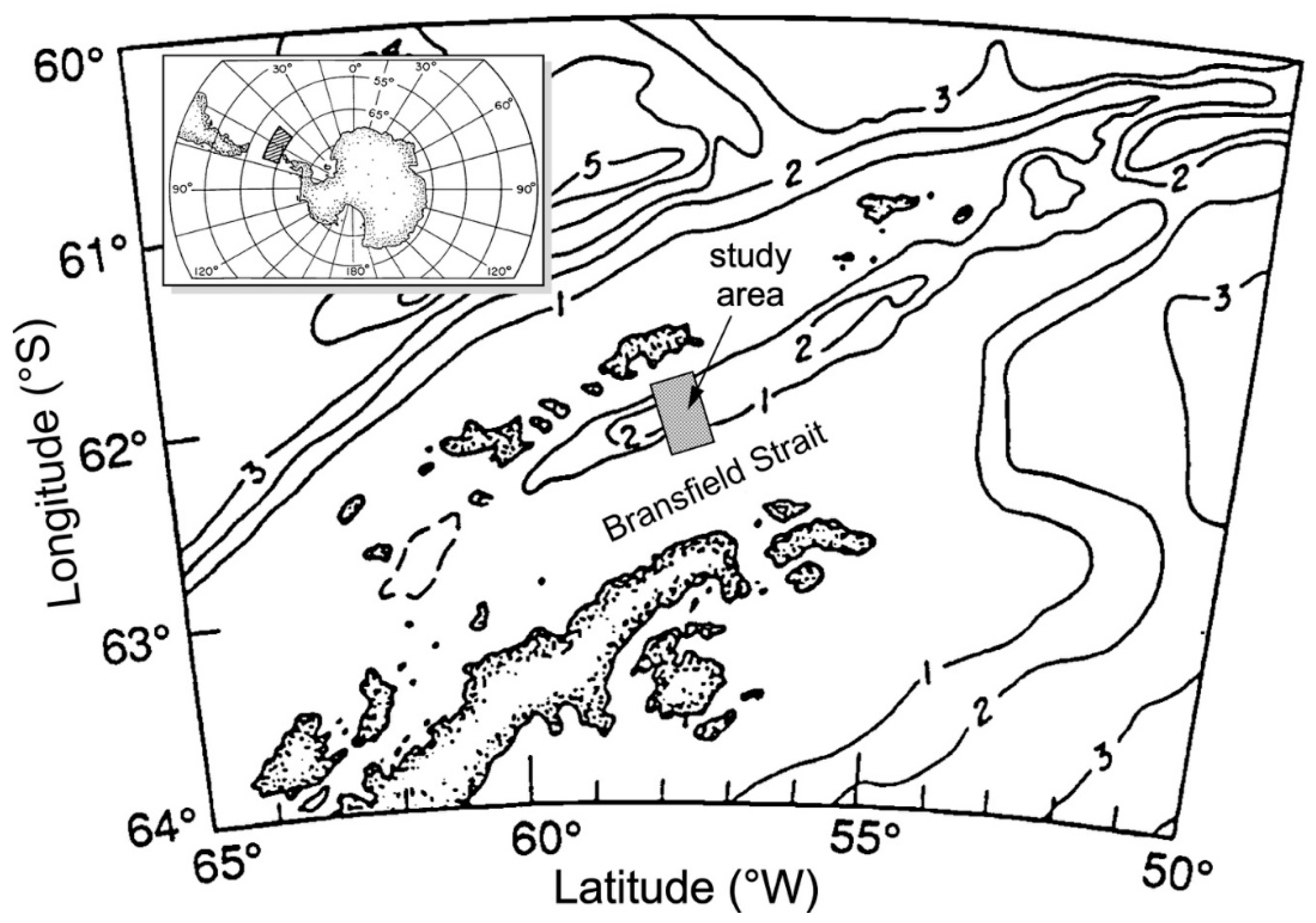
2. Methods
2.1. Shipboard Analyses
2.2. Shore-Based Analyses
2.3. Isotope Methods
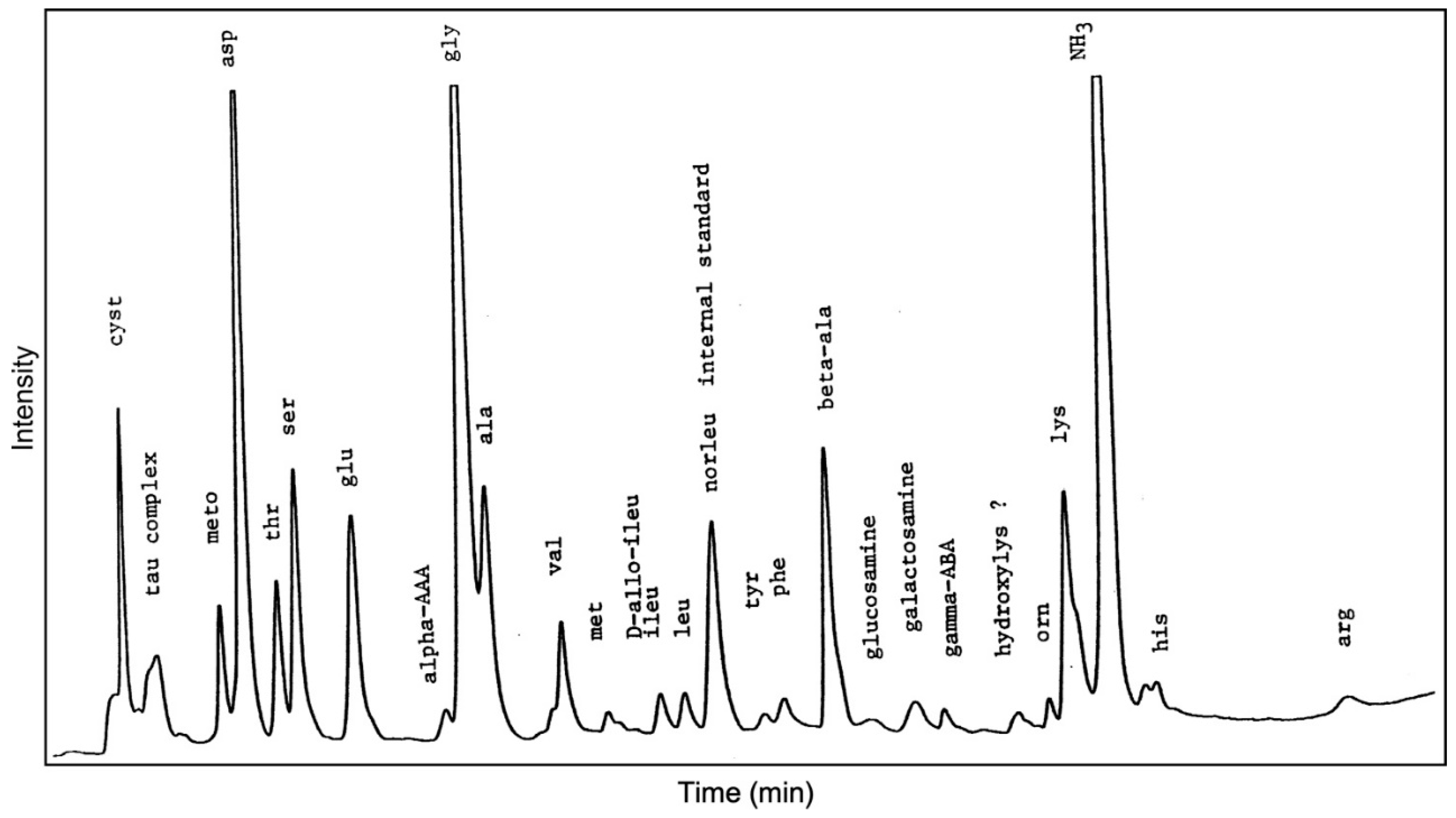
2.4. Amino Acids
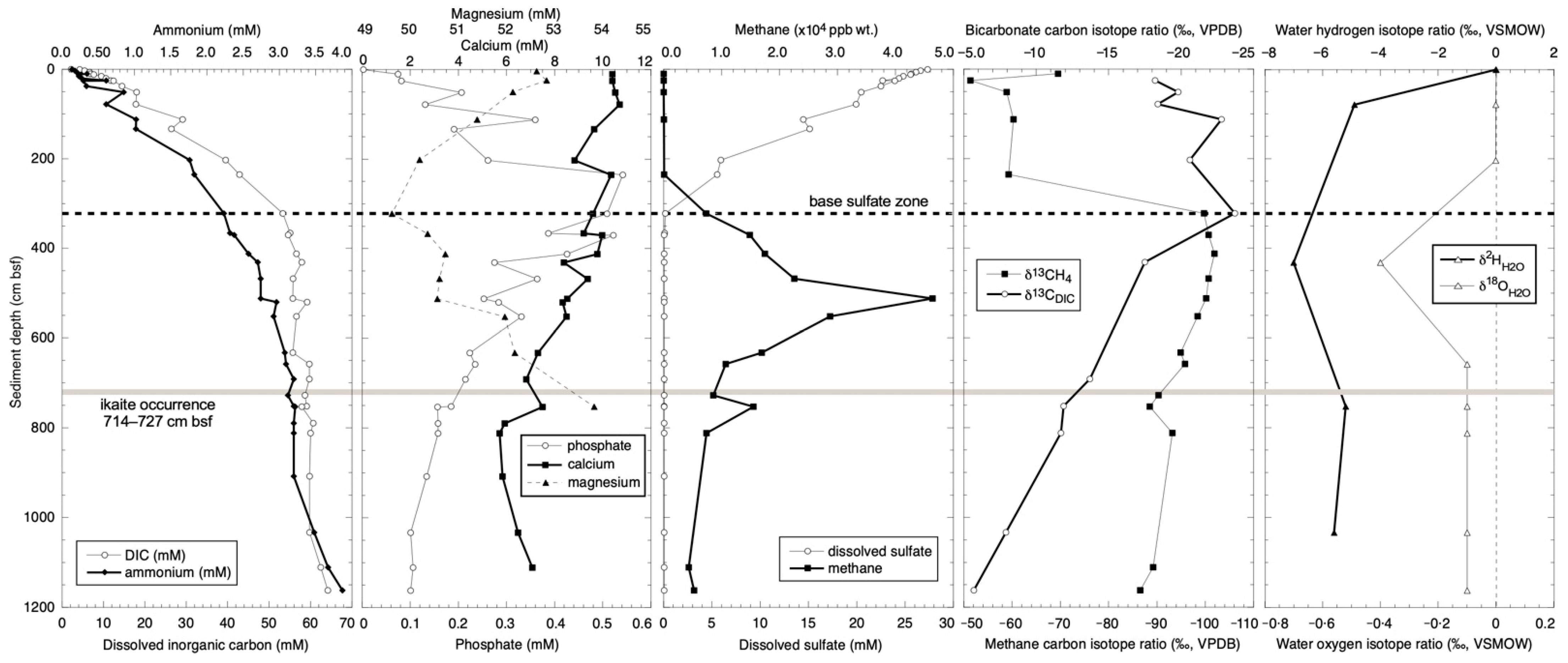
3. Diagenetic Environment
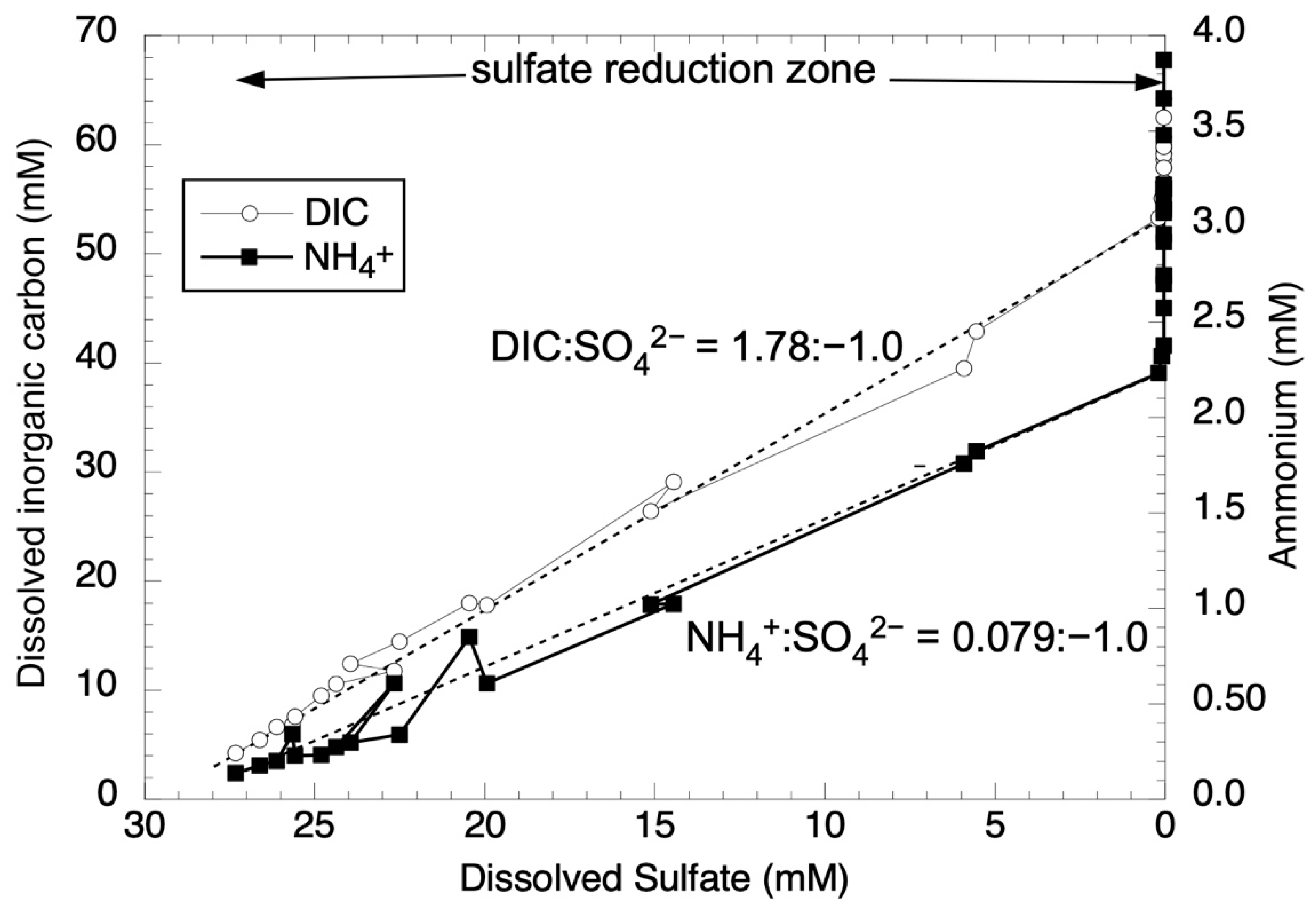
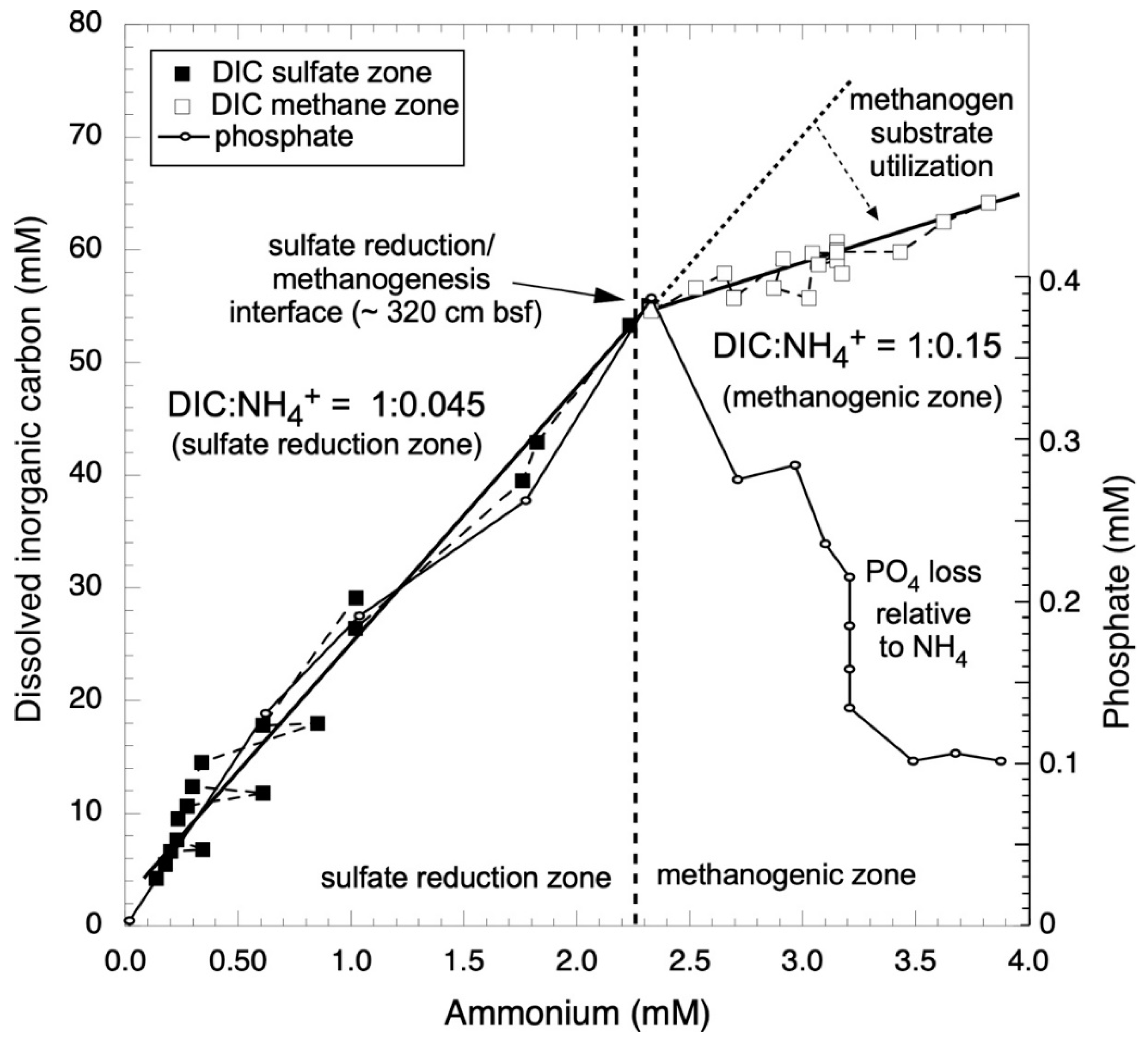
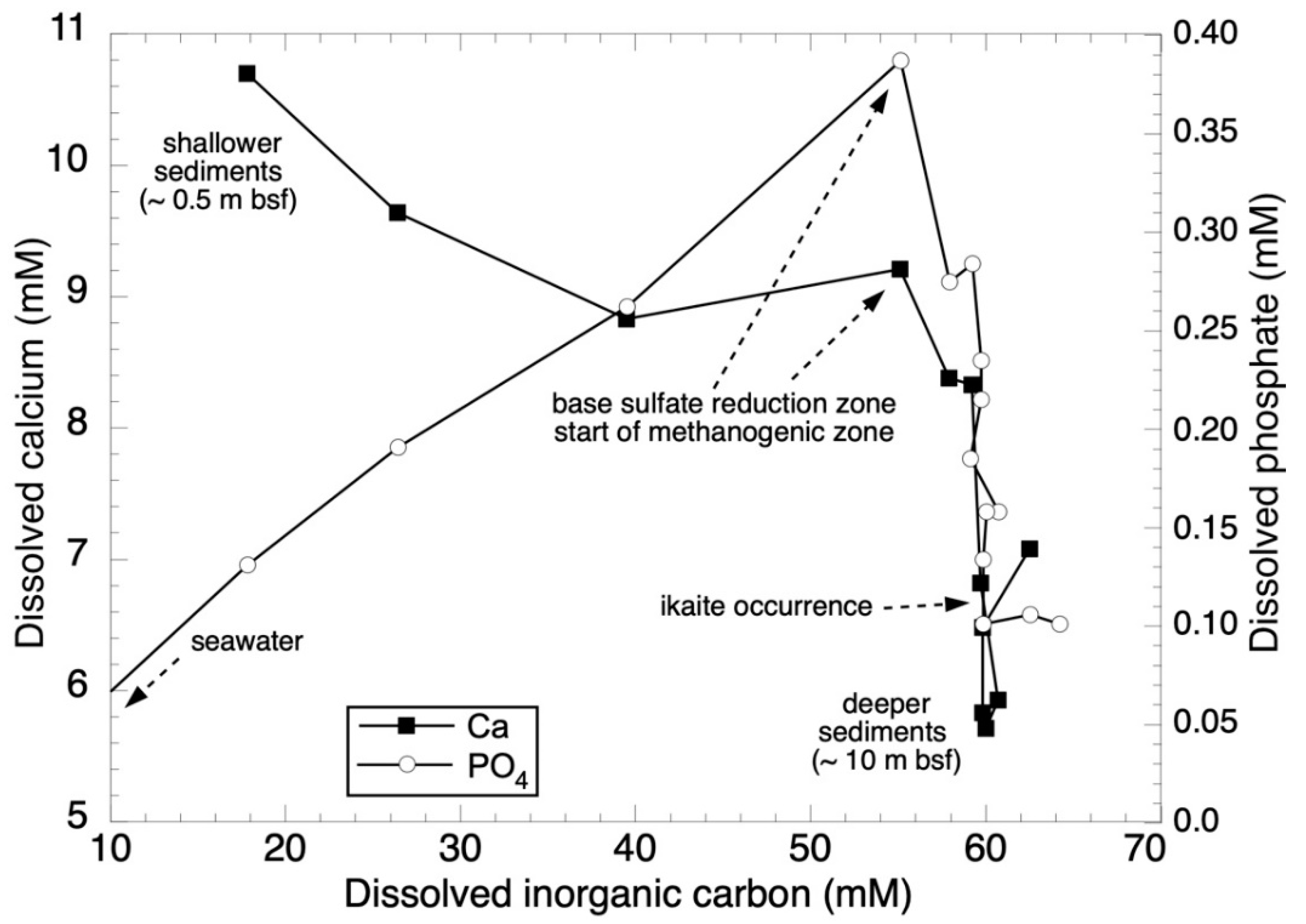
3.1. Nutrient Stoichiometries
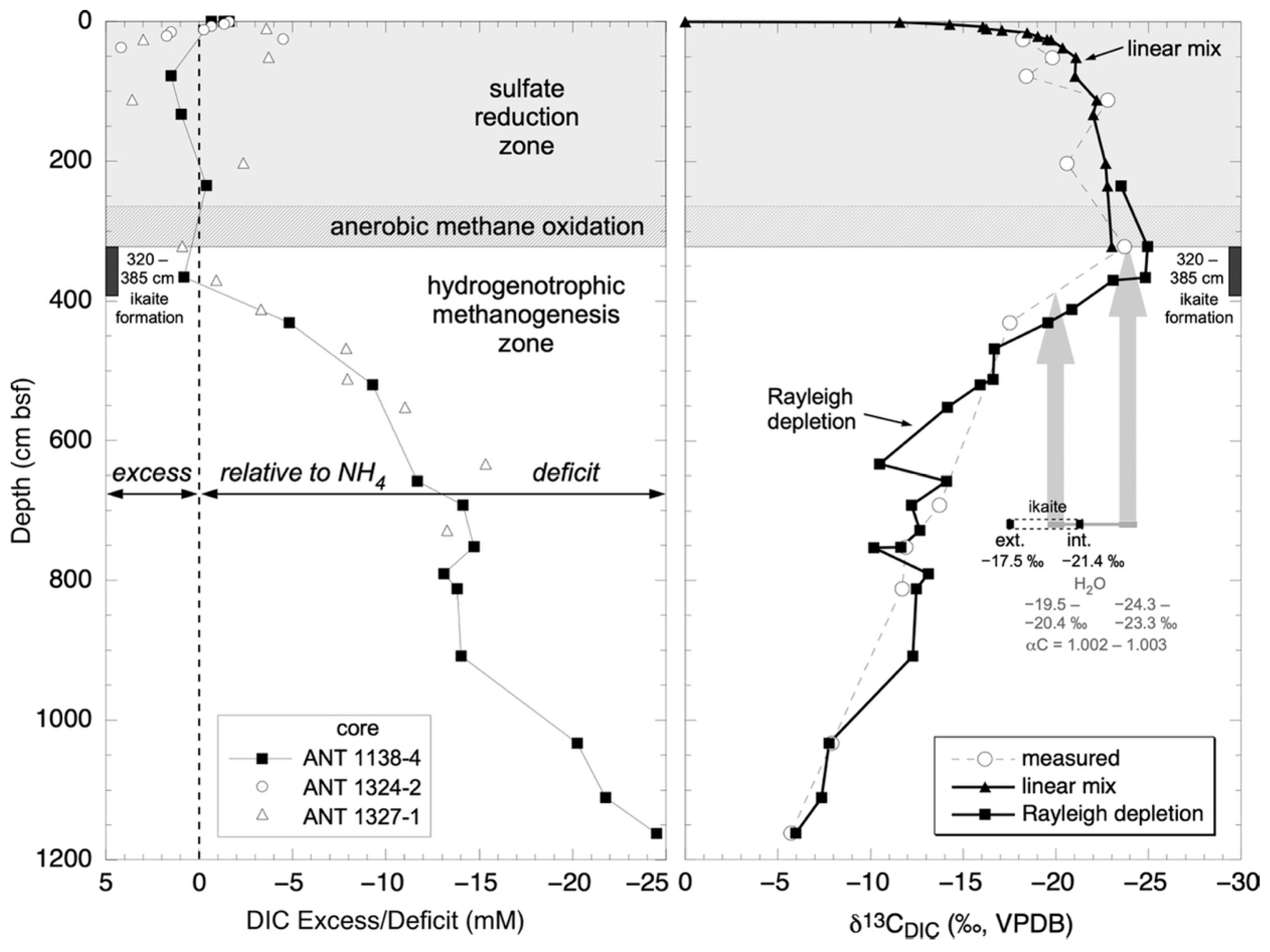
3.2. Carbon Isotope Variation in DIC
3.2.1. Linear Isotope Mixing
3.2.2. Kinetic Isotope Effects Associated with Methanogenesis
4. Calcium Carbonate Hexahydrate Precipitation
4.1. Ikaite Formation
4.2. Radiocarbon Dating
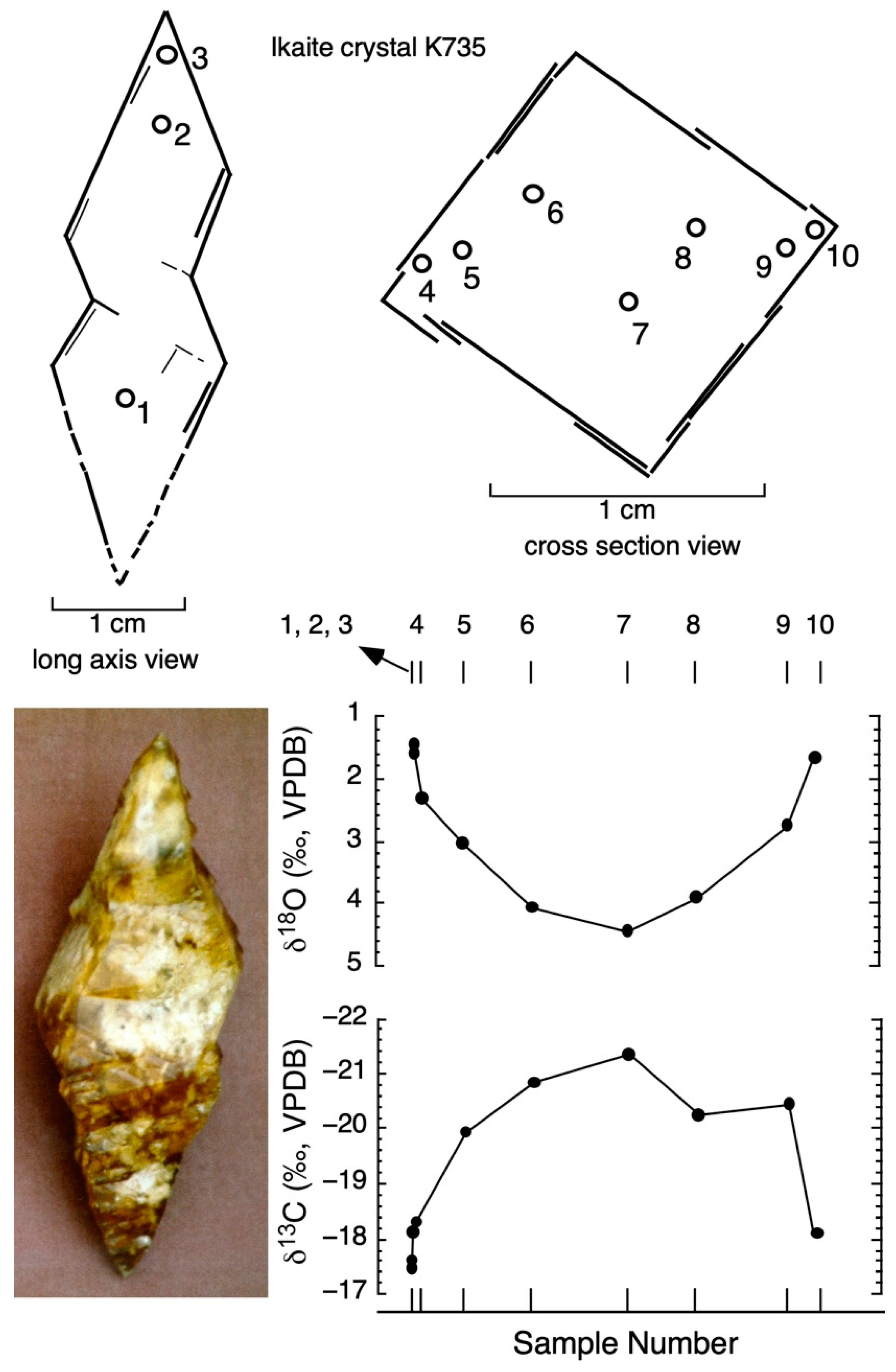
4.3. Carbon Isotope Zonation of Ikaite
4.4. Oxygen Isotope Zonation
4.5. Hydrogen Isotopes in Ikaite Hydration Water
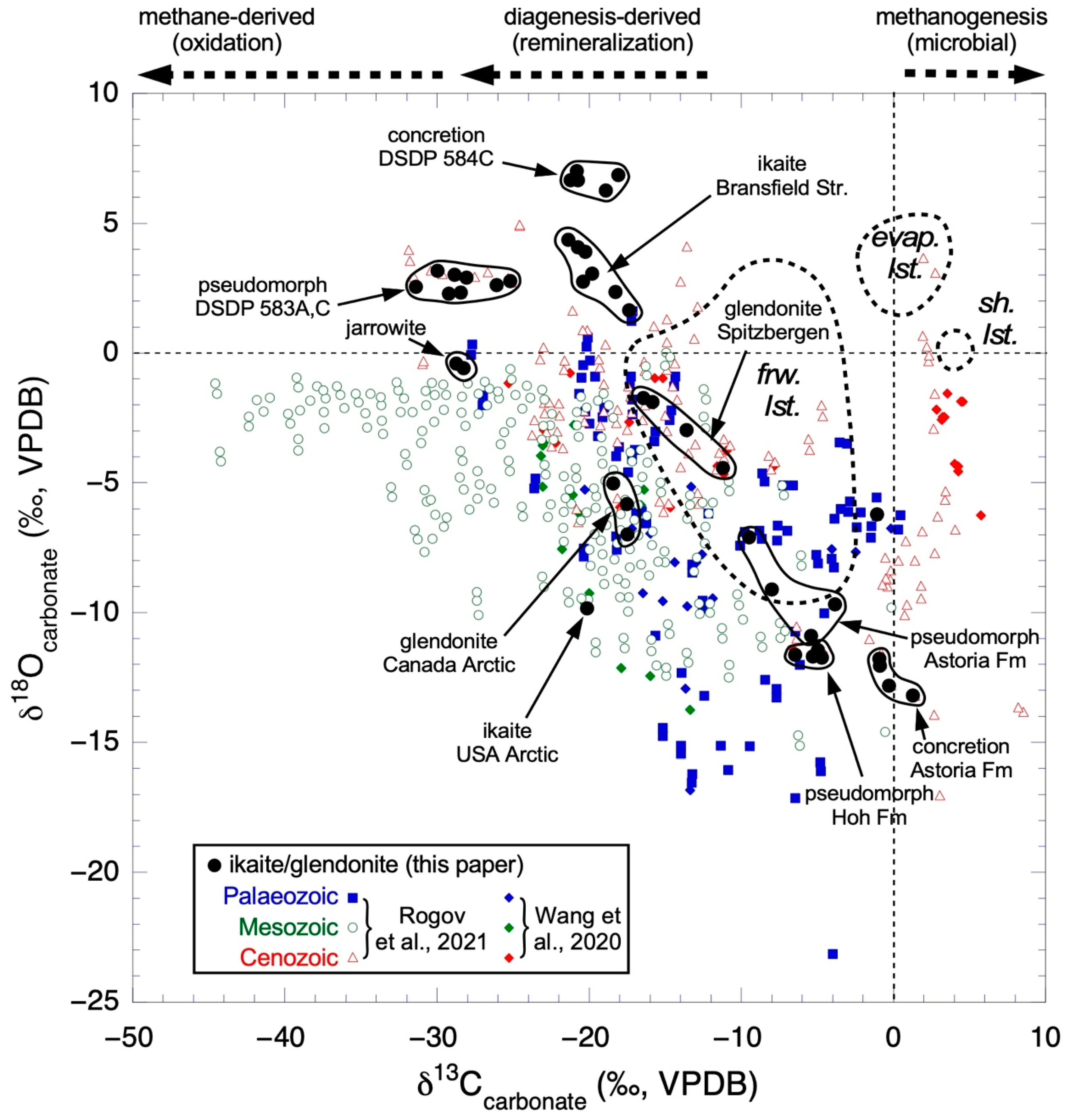
5. Implications of Ikaite Occurrences
5.1. Formation Conditions and Cold Temperature Recorders
5.2. Amino Acids
5.3. Glendonites
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hume, J.; Topley, B. CCCXC—The density of calcium carbonate hexahydrate. J. Chem. Soc. 1929, 129, 2932–2934. [Google Scholar] [CrossRef]
- Dickens, B.; Brown, W.E. The crystal structure of calcium carbonate hexahydrate at −120°. Inorg. Chem. 1970, 9, 480–486. [Google Scholar] [CrossRef]
- Daniell, J.F. Some facts relating to the formation and decomposition of sugar, and the artificial production of crystallized carbonate of lime. Annal. Chim. 1819, 10, 219–224. [Google Scholar]
- Becquerel, A.C. Du carbonate de chaux cristallise, et de l’action simultanee des matieres sucrees ou mucilagineuses sur quelques oxides metalliques, par l’intermediaire des alcalis et des terres. Annal. Chim. 1831, 47, 5–20. [Google Scholar]
- Pelouze, J. Sur la production artificielle du carbonate de chaux cristallise, et sur deux combinaisons de ce sel avec I’eau. Annal. Chim. 1831, 47, 301–307. [Google Scholar]
- Pauly, H. IKAIT Nyt mineral der danner skaer. In Naturens Verden; E. Munksgaards Forlag, 1963; pp. 168–171; 186–192. [Google Scholar]
- Stein, C.; Smith, A.J. Authigenic Carbonate Nodules in the Nankai Trough, Site 583; Init. Repts. DSDP 87; Kagami, H., Karig, D.E., Coulbourn, W.T., Eds.; U.S. Government Printing Office: Washington, DC, USA, 1986; pp. 659–668. [Google Scholar]
- Jansen, J.; Woensdregt, C.F.; Kooistra, M.J.; Van Der Gaast, S.J. Ikaite pseudomorphs in the Zaire deep-sea fan: An intermediate between calcite and porous calcite. Geology 1987, 15, 245–248. [Google Scholar] [CrossRef]
- Buchardt, B.; Seaman, P.; Stockmann, G.; Vous, M.; Wilken, U.; Düwel, L.; Kristiansen, A.; Jenner, C.; Whiticar, M.J.; Kristensen, R.M. Submarine columns of ikaite tufa. Nature 1997, 390, 129–130. [Google Scholar] [CrossRef]
- Suess, E.; Balzer, W.; Hesse, K.-F.; Müller, P.J.; Ungerer, C.A.; Wefer, G. Calcium Carbonate Hexahydrate from Organic-Rich Sediments of the Antarctic Shelf: Precursors of Glendonites. Science 1982, 216, 1128–1131. [Google Scholar] [CrossRef]
- Schubert, C.J.; Nurnberg, D.; Scheele, N.; Pauer, F.; Kriews, M. 13C depletion in ikaite crystals: Evidence for methane release from the Siberian shelves? Geo-Mar. Lett. 1997, 17, 169–175. [Google Scholar] [CrossRef]
- Zabel, M.; Schulz, H.D. Importance of submarine landslides for non-steady state conditions in pore water systems—Lower Zaire (Congo) deep-sea fan. Mar. Geol. 2001, 176, 87–99. [Google Scholar] [CrossRef]
- Hackworth, M. Mud volcanoes, gas hydrates, and carbonate formation at “warm” seeps in the northern Gulf of Mexico. Geol. Soc. Am. 2004, 36, 193. [Google Scholar]
- Greinert, J.; Derkachev, A. Glendonites and methane-derived Mg-calcites in the Sea of Okhotsk, eastern Siberia: Implications of a venting-related ikaite/glendonite formation. Mar. Geol. 2004, 204, 129–144. [Google Scholar] [CrossRef]
- Burton, E.A. Controls on marine carbonate cement mineralogy: Review and reassessment. Chem. Geol. 1993, 105, 163–179. [Google Scholar] [CrossRef]
- Bischoff, J.L.; Fitzpatrick, J.A.; Rosenbauer, R.J. The solubility and stabilization of ikaite (CaCO3·6H2O) from 0 to 25 °C: Environmental and paleoclimatic implications for thinolite tufa. J. Geol. 1993, 101, 21–33. [Google Scholar] [CrossRef]
- Johnston, J.; Merwin, H.E.; Williamson, E.D. The several forms of calcium carbonate. Am. J. Sci. 1916, 41, 473–513. [Google Scholar] [CrossRef]
- Krauss, F.; Schriever, W. Die Hydrate des Calciumcarbonats. Z. Anorg. Chem. 1930, 188, 259–273. [Google Scholar] [CrossRef]
- Brooks, R.; Clark, L.M.; Thurston, E.F. Calcium carbonate and its hydrates. Philos. Trans. R. Soc. A 1950, 243, 145–167. [Google Scholar] [CrossRef]
- Van Valkenburg, A.; Mao, H.K.; Bell, P.M. Ikaite (CaCO3·6H2O), a phase more stable than calcite and aragonite (CaCO3) at high water pressure. Geophy. Lab. Year Book 1970, 70, 237–238. [Google Scholar]
- Marland, G. Stability of calcium carbonate hexahydrate (ikaite). Geochim. Cosmochim. Acta. 1975, 39, 83–91. [Google Scholar] [CrossRef]
- Tollefsen, E.; Stockmann, G.; Skelton, A.; Mörth, C.-M.; Dupraz, C.; Sturkell, E. Chemical controls on ikaite formation. Miner. Mag. 2018, 82, 1119–1129. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.R.; Sand, K.K.; Rodriguez-Blanco, J.D.; Bovet, N.; Generosi, J.; Dalby, K.; Stipp, S.L.N. Inhibition of calcite growth: Combined effects of Mg2+ and SO42−. Cryst. Growth Des. 2016, 16, 6199–6207. [Google Scholar] [CrossRef]
- Bischoff, J.L.; Fyfe, W.S. Catalysis, inhibition, and the calcite-aragonite problem II: The vaterite-aragonite transformation. Am. Jour. Sci. 1968, 266, 80–90. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Kennedy, H.; Kattner, G.; Dieckmann, G.S.; Thomas, D.N. Experimental evidence for carbonate precipitation and CO2 degassing during sea ice formation. Geochim. Cosmochim. Acta 2004, 68, 1749–1761. [Google Scholar] [CrossRef]
- Geilfus, N.-X.; Carnat, G.; Dieckmann, G.S.; Halden, N.; Nehrke, G.; Papakyriakou, T.; Tison, J.-L.; Delille, B. First estimates of the contribution of CaCO3 precipitation to the release of CO2 to the atmosphere during young sea ice growth. J. Geophys. Res. Oceans 2013, 118, 244–255. [Google Scholar] [CrossRef] [Green Version]
- Rysgaard, S.; Wang, F.; Galley, R.J.; Grimm, R.; Notz, D.; Lemes, M.; Geilfus, N.-X.; Chaulk, A.; Hare, A.A.; Crabeck, O.; et al. Temporal dynamics of ikaite in experimental sea ice. Cryosphere 2014, 8, 1469–1478. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Dieckmann, G.S.; Wolf-Gladrow, D.A.; Nehrke, G. Laboratory study on coprecipitation of phosphate with ikaite in sea ice. J. Geophys. Res. Oceans 2014, 119, 7007–7015. [Google Scholar] [CrossRef] [Green Version]
- Dieckmann, G.S.; Nehrke, G.; Papadimitriou, S.; Göttlicher, J.; Steininger, R.; Kennedy, H.; Wolf-Gladrow, D.; Thomas, D.N. Calcium carbonate as ikaite crystals in Antarctic sea ice. Geophys. Res. Lett. 2008, 35. [Google Scholar] [CrossRef] [Green Version]
- Field, L.P.; Milodowski, A.E.; Shaw, R.P.; Stevens, L.A.; Hall, M.R.; Kilpatrick, A.; Gunn, J.; Kemp, S.J.; Ellis, M.A. Unusual morphologies and the occurrence of pseudomorphs after ikaite (CaCO3·6H2O) in fast growing, hyperalkaline speleothems. Mineral. Mag. 2017, 81, 565–589. [Google Scholar] [CrossRef] [Green Version]
- Oehlrich, M.B.; Sánchez-Pastor, N.; Mayr, C. On the study of natural and synthetic ikaite crystals. Revista Soc. Esp. Mineral. 2009, 11, 135. [Google Scholar]
- Ito, T. Ikaite from cold spring water at Shiowakka, Hokkaido, Japan. J. Miner. Pet. Econ. Geol. 1996, 91, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Boch, R.; Dietzel, M.; Reichl, P.; Leis, A.; Baldermann, A.; Mittermayr, F.; Pölt, P. Rapid ikaite (CaCO3·6H2O) crystallization in a man-made river bed: Hydrogeochemical monitoring of a rarely documented mineral formation. Appl. Geochem. 2015, 63, 366–379. [Google Scholar] [CrossRef]
- Mikkelsen, A.; Andersen, A.B.; Engelsen, S.B.; Hansen, H.C.B.; Larsen, O.; Skibsted, L.H. Presence and dehydration of ikaite, calcium carbonate hexahydrate, in frozen shrimp shell. J. Agric. Food Chem. 1999, 47, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Careche, M.; Herrero, A.M.; Carmona, P. Raman Analysis of white spots appearing in the shell of argentine red shrimp (pleoticus muelleri) during frozen storage. J. Food Sci. 2002, 67, 2892–2895. [Google Scholar] [CrossRef] [Green Version]
- Tansman, G.F.; Kindstedt, P.S.; Hughes, J.M. Crystal fingerprinting: Elucidating the crystals of Cheddar, Parmigiano-Reggiano, Gouda, and soft washed-rind cheeses using powder x-ray diffractometry. Dairy Sci. Technol. 2015, 95, 651–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohrazda, C.A. Structural study of the occurrence of Ikaite pseudomorphs in Neoproterozoic metalimestones on Islay, Scotland. Bachelor’s Thesis, Stockholm University, Stockholm, Sweden, 2017. [Google Scholar]
- Dempster, T.; Jess, S.A. Ikaite pseudomorphs in Neoproterozoic Dalradian slates record Earth’s coldest metamorphism. J. Geol. Soc. 2015, 172, 459–464. [Google Scholar] [CrossRef] [Green Version]
- Dana, E.S. A Crystallographic Study of the Thinolite of Lake Lahontan; U.S. Government Printing Office: Washington, DC, USA, 1884; Volume 6, p. 3. [Google Scholar] [CrossRef]
- David, T.W.E.; Taylor, T.G.; Woolnough, W.G.; Foxhall, H.G. Occurrence of the pseudomorph glendonite in New South Wales. Rec. Geol. Surv. NSW 1905, 8, 161–179. [Google Scholar]
- Boggs, S. Petrography and geochemistry of rhombic, calcite pseudomorphs from mid-tertiary mudstones of the pacific northwest, U.S.A. Sedimentology 1972, 19, 219–235. [Google Scholar] [CrossRef]
- Kaplan, M.E. Kal’citovye psovdomorfozy v jurskich i niznemelovych otlozenijach severa vostocnoj Sibiri (calcitic pseudomorphs in Jurassic and Lower Cretaceous deposits from the northern part of eastern Siberia). Geol. I Geofiz. 1978, 12, 62–70. [Google Scholar]
- Kemper, E.; Schmitz, H.H. Glendonite—Indikatoren des polarmarinen Ablagerungsmilieus. Geol. Rundsch. 1981, 70, 759–773. [Google Scholar] [CrossRef]
- Shearman, D.J.; Smith, A.J. Ikaite, the parent mineral of jarrowite-type pseudomorphs. Proc. Geol. Assoc. 1985, 96, 305–314. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, J.; Suess, E.; Wang, G.; Chen, C.; Xiao, S. Silicified glendonites in the Ediacaran Doushantuo Formation (South China) and their potential paleoclimatic implications. Geology 2017, 45, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, C.; Wang, J.; Suess, E.; Chen, X.; Wang, G.; Chen, C.; Xiao, S. Wide but not ubiquitous distribution of glendonites in the Doushantuo Formation, South China: Implications for Ediacaran climate. Precamb. Res. 2020, 338, 105586. [Google Scholar] [CrossRef]
- Hesse, K.; Küppers, H.; Suess, E. Refinement of the structure of Ikaite, CaCO3·6H2O. Z. Für Krist. Cryst. Mater. 1983, 163, 227–232. [Google Scholar] [CrossRef]
- Whiticar, M.J.; Suess, E. Hydrothermal hydrocarbon gases in the sediments of the King George Basin, Bransfield Strait, Antarctica. Appl. Geochem. 1990, 5, 135–147. [Google Scholar] [CrossRef]
- Holler, P. Geotechnical properties of Antarctic deep-sea sediments. Meteor Forsch. Ergebn. 1985, 39, 23–26. [Google Scholar]
- Han, M.W. Dynamics and chemistry of pore fluids in marine sediments of different tectonic settings: Oregon subduction zone and Bransfield Strait extensional basin. Ph.D. Thesis, Oregon St. University, Corvallis, OR, USA, 1988; 295p. [Google Scholar]
- Barker, P.F.; Dalziel, I.W.D. Progress in geodynamics in the Scotia Arc Region. In Geodynamics of the Eastern Pacific Region, Caribbean and Scotia Arcs; U.S. Department of Energy: Washington, DC, USA, 1983; Volume 9, pp. 137–170. [Google Scholar] [CrossRef]
- Han, M.W.; Suess, E. Lateral migration of pore fluids through sediments of an active back-arc basin, Bransfield Strait, Antarctica. Am. Geophy. Union EOS Trans. 1987, 68, 1769. [Google Scholar]
- Suess, E.; Fisk, M.; Kadko, D. Thermal interaction between back-arc volcanism and basin sediments in the Bransfield Strait. Antarc. J. US 1987, 22, 47–49. [Google Scholar]
- Hartmann, M.; Moller, P.J.; Suess, E.; Van Der Weijden, C.H. Chemistry of late Quaternary sediments and their interstitial waters from the NW-African continental margin. Meteor Forsch.-Ergebn. 1973, 24, 1–67. [Google Scholar]
- Whiticar, M.J.; Suess, E.; Wehner, H. Thermogenic hydrocarbons in surface sediments of the Bransfield Strait, Antarctic Peninsula. Nature 1985, 314, 87–90. [Google Scholar] [CrossRef]
- Swainson, I.P.; Hammond, R.P. Ikaite, CaCO3·6H2O: Cold comfort for glendonites as paleothermometers. Am. Mineral. 2001, 86, 1530–1533. [Google Scholar] [CrossRef]
- Coleman, D.D.; Risatti, J.; Schoell, M. Fractionation of carbon and hydrogen isotopes by methane-oxidizing bacteria. Geochim. Et Cosmochim. Acta 1981, 45, 1033–1037. [Google Scholar] [CrossRef]
- McCrea, J.M. On the Isotopic Chemistry of Carbonates and a Paleotemperature Scale. J. Chem. Phys. 1950, 18, 849–857. [Google Scholar] [CrossRef]
- Whiticar, M.J.; Eek, M. Challenges of 13C/12C measurements by CF-IRMS of biochemical samples at sub-nanomolar levels. In International Atomic Energy Agency, New Approaches for Stable Isotope Ratio Measurements; IAEA-TECDOC-1247; IAEA: Vienna, Austria, 2001; pp. 75–95. [Google Scholar]
- Müller, P.J.; Suess, E.; Ungerer, A.C. Amino acids and amino sugars of surface particulate and sediment trap material from waters of the Scotia sea. Deep Sea Res. Part A Oceanogr. Res. Pap. 1986, 33, 819–838. [Google Scholar] [CrossRef]
- Roth, M. Fluorescence reaction for amino acids. Anal. Chem. 1971, 43, 880–882. [Google Scholar] [CrossRef]
- Benson, J.R.; Hare, P.E. 8-phthalaldehyde: Fluorogenic detection of primary aurines in the picomole range. Proc. Natl. Acad. Sci. USA 1975, 72, 619–622. [Google Scholar] [CrossRef] [Green Version]
- Müller, P.J. Isoleucine epimarization in Quaternary planktonic foraminifera: Effects of diagenetic hydrolysis, and leaching, and Atlantic-Pacific intercore correlations. Meteor Forsch. Ergebn. 1984, 38, 25–47. [Google Scholar]
- Whiticar, M.J. The Biogeochemical Methane Cycle. In Hydrocarbons, Oils and Lipids: Diversity, Origin, Chemistry and Fate; Handbook of Hydrocarbon and Lipid Microbiology; Wilkes, H., Ed.; Springer: Cham, Switzerland, 2020; pp. 669–746. [Google Scholar] [CrossRef]
- Whiticar, M.J.; Faber, E. Methane oxidation in marine and limnic sediments and water columns. In Advances in Organic Geochemistry; Leythaeuser, D., Rullkötter, J., Eds.; Pergamon Press: New York, NY, USA, 1985; pp. 759–768. [Google Scholar]
- Hoffman, P.F.; Kaufman, A.J.; Halverson, G.P.; Schrag, D.P. A Neoproterozoic snowball earth. Science 1998, 281, 1342–1346. [Google Scholar] [CrossRef] [Green Version]
- Claypool, G.E.; Kaplan, I.R. The origin and distribution of methane in marine sediments. In Natural Gases in Marine Sediments; Kaplan, I.R., Ed.; Springer: Berlin/Heidelberg, Germany, 1974; pp. 99–139. [Google Scholar] [CrossRef]
- Whiticar, M.; Faber, E.; Schoell, M. Biogenic methane formation in marine and freshwater environments: CO2 reduction vs. acetate fermentation—Isotope evidence. Geochim. Cosmochim. Acta 1986, 50, 693–709. [Google Scholar] [CrossRef]
- Rayleigh, J.W.S. Theoretical considerations respecting the separation of gases by diffusion and similar processes. Philos. Mag. 1896, 42, 493–593. [Google Scholar] [CrossRef]
- Broecker, W.S.; Oversby, V. Chemical Equilibria in the Earth; McGraw-Hill: New York, NY, USA, 1971; 318p. [Google Scholar]
- Lennie, A.R.; Tang, C.C.; Thompson, S.P. The structure and thermal expansion behaviour of ikaite, CaCO3·6H2O, from T = 114 to T = 293 K. Miner. Mag. 2004, 68, 135–146. [Google Scholar] [CrossRef]
- Kawano, J.; Shimobayashi, N.; Miyake, A.; Kitamura, M. Precipitation diagram of calcium carbonate polymorphs: Its construction and significance. J. Phys. Condens. Matter 2009, 21, 425102. [Google Scholar] [CrossRef] [PubMed]
- Irwin, H.; Curtis, C.; Coleman, M. Isotopic evidence for source of diagenetic carbonates formed during burial of organic-rich sediments. Nature 1977, 269, 209–213. [Google Scholar] [CrossRef]
- Gautier, D.L.; Claypool, G.E. Interpretation of methanogenic biogenesis in ancient sediments by analogy with processes in modern diagenetic environments. AAPG Memoir 1984, 37, 112–123. [Google Scholar]
- Talbot, M.R.; Kelts, K. Primary and diagenetic carbonates in the anoxic sediments of Lake Bosumtwi, Ghana. Geology 1986, 14, 912. [Google Scholar] [CrossRef]
- Reeburgh, W.S.; Heggie, D.T. Microbial methane consumption reactions and their effect on methane distributions in freshwater and marine environments1. Limnol. Oceanogr. 1977, 22, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mook, W.G. Isotope geochemistry of carbonates in the weathering zone. In Handbook of Environmental Isotope Geochemistry. V. 2. The Terrestrial Environment, B; Fritz, P., Fontes, J.-C., Eds.; Elsevier: Amsterdam, The Netherlands, 1986; pp. 239–270. [Google Scholar]
- Romanek, C.S.; Grossman, E.L.; Morse, J.W. Carbon isotopic fractionation in synthetic aragonite and calcite: Effects of temperature and precipitation rate. Geochim. Cosmochim. Acta 1992, 56, 419–430. [Google Scholar] [CrossRef]
- Lu, Z.; Rickaby, R.E.; Kennedy, H.; Kennedy, P.; Pancost, R.D.; Shaw, S.; Lennie, A.; Wellner, J.; Anderson, J.B. An ikaite record of late Holocene climate at the Antarctic Peninsula. Earth Planet. Sci. Lett. 2012, 325–326, 108–115. [Google Scholar] [CrossRef]
- Matsuo, S.; Friedman, I.; I Smith, G. Studies of quaternary saline lakes—I. Hydrogen isotope fractionation in saline minerals. Geochim. Cosmochim. Acta 1972, 36, 427–435. [Google Scholar] [CrossRef]
- Emrich, K.; Ehhalt, D.H.; Vogel, J.C. Carbon isotope fractionation during the precipitation of calcium carbonate. Earth Planet. Sci. Lett. 1970, 8, 363–371. [Google Scholar] [CrossRef]
- Grossman, E. Carbon isotopic fractionation in live benthic foraminifera—Comparison with inorganic precipitate studies. Geochim. Cosmochim. Acta 1984, 48, 1505–1512. [Google Scholar] [CrossRef]
- Mees, F.; Reyes, E.; Keppens, E. Stable isotope chemistry of gaylussite and nahcolite from the deposits of the Crater Lake at Malha, northern Sudan. Chem. Geol. 1998, 146, 87–98. [Google Scholar] [CrossRef]
- Turner, J.V. Kinetic fractionation of carbon-13 during calcium carbonate precipitation. Geochim. Cosmochim. Acta 1982, 46, 1183–1191. [Google Scholar] [CrossRef]
- Sugiura, Y.; Onuma, K.; Yamazaki, A. Growth dynamics of vaterite in relation to the physico-chemical properties of its precursor, amorphous calcium carbonate, in the Ca-CO3-PO4 system. Am. Miner. 2016, 101, 289–296. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Fernández-González, A.; Prieto, M. The role of sulfate groups in controlling CaCO3 polymorphism. Geochim. Cosmochim. Acta 2010, 74, 6064–6076. [Google Scholar] [CrossRef] [Green Version]
- Clark, I.D.; Lauri, B. Kinetic enrichment of stable isotopes in cryogenic calcites. Chem. Geol. 1992, 102, 217–228. [Google Scholar]
- Grasby, S.E. Naturally precipitating vaterite (μ-CaCO3) spheres: Unusual carbonates formed in an extreme environment. Geochim. Cosmochim. Acta 2003, 67, 1659–1666. [Google Scholar] [CrossRef]
- Kodina, L.A.; Tokarev, V.G.; Vlasova, L.N.; Korobeinik, G.S. Contribution of biogenic methane to ikaite formation in the Kara Sea: Evidence from the stable carbon isotope geochemistry. Proc. Mar. Sci. 2003, 6, 349–374. [Google Scholar]
- Weiss, R.; Östlund, H.; Craig, H. Geochemical studies of the Weddell sea. Deep Sea Res. Part A. Oceanogr. Res. Pap. 1979, 26, 1093–1120. [Google Scholar] [CrossRef]
- Lacelle, D.; Lauriol, B.; Clark, I.D. Formation of seasonal ice bodies and associated cryogenic carbonates in Caverne de I’Ours, Que bec, Canada: Kinetic isotope effects and pseudo-biogenic crystal structures. J. Cave Karst Stud. 2009, 71, 49–62. [Google Scholar]
- Grossman, E.L. Applying oxygen isotope paleothermometry in deep time. Paléontol. Soc. Pap. 2012, 18, 39–68. [Google Scholar] [CrossRef]
- O’Neil, J.R.; Clayton, R.N.; Mayeda, T.K. Oxygen isotope fractionation in divalent metal carbonates. J. Chem. Phys. 1969, 51, 5547–5558. [Google Scholar] [CrossRef]
- Hays, P.; Grossman, E. Oxygen isotopes in meteoric calcite cements as indicators of continental paleoclimate. Geology 1991, 19, 441–444. [Google Scholar] [CrossRef]
- Rickaby, R.; Shaw, S.; Bennitt, G.; Kennedy, H.; Zabel, M.; Lennie, A. Potential of ikaite to record the evolution of oceanic δ18O. Geology 2006, 34, 497–500. [Google Scholar] [CrossRef]
- Bots, P.; Benning, L.G.; Rodriguez-Blanco, J.-D.; Roncal-Herrero, T.; Shaw, S. Mechanistic insights into the crystallization of amorphous calcium carbonate (ACC). Cryst. Growth Des. 2012, 12, 3806–3814. [Google Scholar] [CrossRef]
- Tarutani, T.; Clayton, R.N.; Mayeda, T.K. The effect of polymorphism and magnesium substitution on oxygen isotope fractionation between calcium carbonate and water. Geochim. Cosmochim. Acta 1969, 33, 987–996. [Google Scholar] [CrossRef]
- Kim, S.-T.; O’Neil, J.R. Equilibrium and nonequilibrium oxygen isotope effects in synthetic carbonates. Geochim. Cosmochim. Acta 1997, 61, 3461–3475. [Google Scholar] [CrossRef]
- Kluge, T.; John, C.M. A simple method for vaterite precipitation in isotopic equilibrium: Implications for bulk and clumped isotope analysis. Biogeosci. Discuss. 2014, 11, 3289–3299. [Google Scholar]
- Stewart, M.K. Hydrogen and oxygen fractionation during crystallization of mirabellite and ice. Geochim. Cosmochim. Acta 1974, 38, 167–172. [Google Scholar] [CrossRef]
- Horita, J. Stable isotope fractionation factors of water in hydrated saline mineral-brine systems. ESPL 1989, 95, 173–179. [Google Scholar] [CrossRef]
- Gonfiantini, R.; Fontes, J.R. Oxygen isotopic fractionation in the water of crystallization of gypsum. Nature 1963, 200, 644–646. [Google Scholar] [CrossRef]
- Barrer, R.M.; Denney, E. Water in hydrates I.—Fractionation of hydrogen isotopes by crystallization of salt hydrates. J. Chem. Soc. 1964, 904, 4677–4684. [Google Scholar] [CrossRef]
- Fontes, J.C.; Gonfiantini, R. Fractionnement isotopique de l’hydrogene dans l’esu de crystallisation du gypse. C.R. Acad. Sci. Ser. D 1967, 265, 4–6. [Google Scholar]
- Kemper, E. Das Klima der Kreidezeit. Geol. Jahrbuch A 1987, 96, 5–185. [Google Scholar]
- Huggett, J.M.; Schultz, B.P.; Shearman, D.J.; Smith, A.J. The petrology of ikaite pseudomorphs and their diagenesis. Proc. Geol. Assoc. 2005, 116, 3–4, 207–220. [Google Scholar] [CrossRef]
- Selleck, B.W.; Carr, P.F.; Jones, B.G. A review and synthesis of glendonites (pseudomorphs after ikaite) with new data: Assessing applicability as recorders of ancient coldwater conditions. J. Sediment. Res. 2007, 77, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Schultz, B.; Thibault, N.; Huggett, J. The minerals ikaite and its pseudomorph glendonite: Historical perspective and legacies of Douglas Shearman and Alec K. Smith. Proc. Geol. Assoc. 2022, 133, 176–192. [Google Scholar] [CrossRef]
- Butschli, O. Untersuchungen über organische Kalkgebilde, nebst Bemerkungen über organische Kieselgebilde, insbesondere über das spezifische Gewicht in Beziehung zu der Struktur, der chemischen Zusammensetzung und anderes. Göttingen. Ges. D. Wiss. Abhandl. 1908, 6, 1–177. [Google Scholar]
- Mckenzie, J.E. Calcium carbonate hexahydrate. J. Chem. Soc. Trans. 1923, 123, 2409–2417. [Google Scholar] [CrossRef]
- Kohlschutter, V.; Egg, C. Über Anderungen des Habitus und der Modifikation von Calcium-carbonat durch Lösungsgenossen. Helv. Chim. Acta 1925, 8, 479–484. [Google Scholar] [CrossRef]
- Purgstaller, B.; Dietzel, M.; Baldermann, A.; Mavromatis, V. Control of temperature and aqueous Mg2+/Ca2+ ratio on the trans formation of ikaite. Geochim. Cosmochim. Acta 2017, 217, 128–143. [Google Scholar] [CrossRef]
- Clarkson, J.R.; Price, T.J.; Adams, C.J. Role of metastable phases in the spontaneous precipitation of calcium carbonate. J. Chem. Soc. Faraday Trans. 1992, 88, 243–249. [Google Scholar] [CrossRef]
- Stockmann, G.; Tollefsen, E.; Skelton, A.; Brüchert, V.; Balic-Zunic, T.; Langhof, J.; Skogby, H.; Karlsson, A. Control of a calcite inhibitor (phosphate) and temperature on ikaite precipitation in Ikka Fjord, southwest Greenland. Appl. Geochem. 2018, 89, 11–22. [Google Scholar] [CrossRef]
- Council, T.C.; Bennett, P.C. Geochemistry of ikaite formation at Mono Lake, California: Implications for the origin of tufa mounds. Geology 1993, 21, 971–974. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Wolthers, M.; Wolf-Gladrow, D.A.; Nehrke, G. Effect of pH and phosphate on calcium carbonate polymorphs precipitated at near-freezing temperature. Cryst. Growth Des. 2015, 15, 1596–1601. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Lu, Z.; Rickaby, R.E.M.; Domack, E.W.; Wellner, J.S.; Kennedy, H.A. Ikaite abundance controlled by porewater phosphorus level: Potential links to dust and productivity. J. Geol. 2015, 123, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Ito, T. Factors controlling the transformation of natural ikaite from Shiowakka, Japan. Geochem. J. 1998, 32, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.O.; Buchardt, B.; Kühl, M.; Elberling, B. The fate of the submarine ikaite tufa columns in southwest greenland under changing climate conditions. J. Sediment. Res. 2011, 81, 553–561. [Google Scholar] [CrossRef]
- Hu, Y.B.; Wolf-Gladrow, D.A.; Dieckmann, G.S.; Völker, C.; Nehrke, G. A laboratory study of ikaite (CaCO3·6H2O) precipitation as a function of pH, salinity, temperature and phosphate concentration. Mar. Chem. 2014, 162, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ruiz, I.; Veesler, S.; Gómez-Morales, J.; Delgado-López, J.M.; Grauby, O.; Hammadi, Z.; Candoni, N.; García-Ruiz, J.M. Transient calcium carbonate hexahydrate (ikaite) nucleated and stabilized in confined nano- and picovolumes. Cryst. Growth Des. 2014, 14, 792–802. [Google Scholar] [CrossRef]
- Berner, R.A.; Morse, J.W. Dissolution kinetics of calcium carbonate in seawater. IV. Theory of calcite dissolution. Am. J. Sci. 1974, 274, 108–134. [Google Scholar] [CrossRef]
- Reddy, M.M. Crystallization of calcium carbonate in the presence of trace concentrations of phosphorus-containing anions. J. Cryst. Growth 1977, 41, 287–295. [Google Scholar] [CrossRef]
- Froelich, P.; Klinkhammer, G.; Bender, M.; Luedtke, N.; Heath, G.; Cullen, D.; Dauphin, P.; Hammond, D.; Hartman, B.; Maynard, V. Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: Suboxic diagenesis. Geochim. Cosmochim. Acta 1979, 43, 1075–1090. [Google Scholar] [CrossRef]
- Burton, E.A.; Walter, L.M. The role of pH in phosphate inhibition of calcite and aragonite precipitation rates in seawater. Geochim. Cosmochim. Acta 1990, 54, 797–808. [Google Scholar] [CrossRef]
- Lin, Y.-P.; Singer, P.C. Inhibition of calcite precipitation by orthophosphate: Speciation and thermodynamic considerations. Geochim. Cosmochim. Acta 2006, 70, 2530–2539. [Google Scholar] [CrossRef]
- Mucci, A. Growth kinetics and composition of magnesian calcite overgrowths precipitated from seawater: Quantitative influence of orthophosphate ions. Geochim. Cosmochim. Acta 1986, 50, 2255–2265. [Google Scholar] [CrossRef]
- Lebrato, M.; Garbe-Schönberg, D.; Müller, M.N.; Blanco-Ameijeiras, S.; Feely, R.A.; Lorenzoni, L.; Molinero, J.-C.; Bremer, K.; Jones, D.O.B.; Iglesias-Rodriguez, D.; et al. Global variability in seawater Mg:Ca and Sr:Ca ratios in the modern ocean. Proc. Natl. Acad. Sci. USA 2020, 117, 22281–22292. [Google Scholar] [CrossRef]
- Vickers, M.L.; Vickers, M.; Rickaby, R.E.; Wu, H.; Bernasconi, S.M.; Ullmann, C.V.; Bohrmann, G.; Spielhagen, R.F.; Kassens, H.; Schultz, B.P.; et al. The ikaite to calcite transformation: Implications for palaeoclimate studies. Geochim. Cosmochim. Acta 2022, 334, 201–216. [Google Scholar] [CrossRef]
- Broecker, W.; Yu, J. What do we know about the evolution of Mg to Ca ratios in seawater? Paleoceanography 2011, 26. [Google Scholar] [CrossRef] [Green Version]
- Vasileva, K.; Zaretskaya, N.; Ershova, V.; Rogov, M.; Stockli, L.D.; Stockli, D.; Khaitov, V.; Maximov, F.; Chernyshova, I.; Soloshenko, N. New model for seasonal ikaite precipitation: Evidence from White Sea glendonites. Mar. Geol. 2022, 449. [Google Scholar] [CrossRef]
- Mitterer, R.M.; Cunningham, R. The interaction of natural organic matter with grain surfaces: Implications for calcium carbonate precipitation. In Carbonate Cements; Society for Sedimentary Geology: Tulsa, OK, USA, 1985; pp. 17–31. [Google Scholar] [CrossRef]
- Braissant, O.; Cailleau, G.; Dupraz, C.; Verrecchia, E.P. Bacterially induced mineralization of calcium carbonate in terrestrial environments: The role of exopolysaccharides and amino acids. J. Sediment. Res. 2003, 73, 485–490. [Google Scholar] [CrossRef]
- Stougaard, P.; Jørgensen, F.; Johnsen, M.G.; Hansen, O.C. Microbial diversity in ikaite tufa columns: An alkaline, cold ecological niche in Greenland. Environ. Microbiol. 2002, 4, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Priemé, A.; Stougaard, P. Bacterial diversity in permanently cold and alkaline ikaite columns from Greenland. Extremophiles 2006, 10, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Vester, J.K.; Glaring, M.A.; Stougaard, P. Discovery of novel enzymes with industrial potential from a cold and alkaline environment by a combination of functional metagenomics and culturing. Microb. Cell Factories 2014, 13, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogov, M.A.; Ershova, V.B.; Shchepetova, E.V.; Zakharov, V.A.; Pokrovsky, B.G.; Khudoley, A.K. Earliest Cretaceous (late Berriasian) glendonites from Northeast Siberia revise the timing of initiation of transient Early Cretaceous cooling in the high latitudes. Cretac. Res. 2017, 71, 102–112. [Google Scholar] [CrossRef]
- Rogov, M.; Ershova, V.; Vereshchagin, O.; Vasileva, K.; Mikhailova, K.; Krylov, A. Database of global glendonite and ikaite records throughout the Phanerozoic. Earth Syst. Sci. Data 2021, 13, 343–356. [Google Scholar] [CrossRef]
- Morales, C.; Rogov, M.; Wierzbowski, H.; Ershova, V.; Suan, G.; Adatte, T.; Föllmi, K.B.; Tegelaar, E.; Reichart, G.-J.; De Lange, G.; et al. Glendonites track methane seepage in Mesozoic polar seas. Geology 2017, 45, 503–506. [Google Scholar] [CrossRef]
- Watkins, J.M.; Nielsen, L.C.; Ryerson, F.J.; DePaolo, D.J. The influence of kinetics on the oxygen isotope composition of calcium carbonate. Earth Planet. Sci. Lett. 2013, 375, 349–360. [Google Scholar] [CrossRef]
- Yumol, L.M.; Uchikawa, J.; Zeebe, R.E. Kinetic isotope effects during CO2 hydration: Experimental results for carbon and oxygen fractionation. Geochim. Cosmochim. Acta 2020, 279, 189–203. [Google Scholar] [CrossRef]
- Sade, Z.; Halevy, I. New constraints on kinetic isotope effects during CO2(aq) hydration and hydroxylation: Revisiting theoretical and experimental data. Geochim. Cosmochim. Acta 2017, 214, 246–265. [Google Scholar] [CrossRef]
- Galimov, E.M.; Kodina, L.A.; Stepanets, O.V.; Korobeinik, G.S. Biogeochemistry of the Russian Arctic. Kara Sea: Research results under the SIRRO project, 1995–2003. Geochem. Int. 2006, 44, 1053–1104. [Google Scholar] [CrossRef]
- Hiruta, A.; Matsumoto, R. Geochemical comparison of ikaite and methane-derived authigenic carbonates recovered from Echigo Bank in the Sea of Japan. Mar. Geol. 2021, 443, 106672. [Google Scholar] [CrossRef]
- Brandt, K. Glacioeustatic cycles in the Early Jurassic? Neues Jahrb. Geolog. Paläontol. Mon. 1986, 6, 257–274. [Google Scholar] [CrossRef]
- Frakes, L.A.; Francis, J.E. A guide to Phanerozoic cold polar climates from high-latitude ice-rafting in the Cretaceous. Nature 1988, 333, 547–549. [Google Scholar] [CrossRef]
- Sheard, M.J. Glendonites from the southern Eromanga Basin in South Australia: Palaeoclimatic indicators for Cretaceous ice. Geol. Surv. S. Australia, Quart. Geol. Notes 1990, 114, 17–23. [Google Scholar]
- Brandley, R.T.; Krause, F.F. Thinolite-type pseudomorphs after ikaite: Indicators of cold water on the subequatorial western margin of Lower Carboniferous North America. Can. Soc. Petrol. Geolog. Memoir 1994, 17, 333–344. [Google Scholar]
- Parrish, J.T.; Bradshaw, M.T.; Brakel, A.T.; Mulholland, S.M.; Totterdell, J.M.; Yeates, A.N. Palaeoclimatology of Australia during the Pangean interval. Palaeoclimates 1996, 1, 241–281. [Google Scholar]
- Johnston, J.D. Pseudomorphs after ikaite in a glaciomarine sequence in the Dalradian of Donegal, Ireland. Scott. J. Geol. 1995, 31, 3–9. [Google Scholar] [CrossRef]
- Yan, J. Ikaite pseudomorphs, cold water indicators in the Chihsia Formation (Early Permian) of South China (abstract): 30th Intern. Geol. Congr. 1996, 30, 97. [Google Scholar]
- De Lurio, J.L.; Frakes, L.A. Glendonites as a paleoenvironmental tool: Implications for early Cretaceous high latitude climates in Australia. Geochim. Cosmochim. Acta 1999, 63, 1039–1048. [Google Scholar] [CrossRef]
- Shi, G.R. Possible influence of Gondwanan glaciation on low-latitude carbonate sedimentation and trans-equatorial faunal migration: The Lower Permian of South China. Geosci. J. 2001, 5, 57–63. [Google Scholar] [CrossRef]
- Swainson, I.P.; Hammond, R.P. Hydrogen bonding in ikaite, CaCO3·6H2O. Miner. Mag. 2003, 67, 555–562. [Google Scholar] [CrossRef]
- Buchardt, B.; Israelson, C.; Seaman, P.; Stockmann, G. Ikaite tufa towers in Ikka Fjord, southwest Greenland: Their formation by mixing of seawater and alkaline spring water. J. Sed. Res. 2001, 71, 176–189. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Depth | Core | DIC | SO4 | NH4 | PO4 | Ca | Mg | Mg:Ca | CH4 | δ13CH4 | δ13CO2 | δ2H-CH4 | δ2H-H2O | δ18O-H2O |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cm | no. | mM | mM | mM | mM | mM | mM | (mol) | ppb wt. | ‰, VPDB | ‰, VPDB | ‰, VSMOW | ‰, VSMOW | ‰, VSMOW |
| seawater | 2.2 | 28.00 | 0.00 | 0.003 | 1 | −0.1 | 0.0 | 0.0 | ||||||
| 0.5 | 2 | 4.2 | 27.33 | 0.14 | ||||||||||
| 3.5 | 2 | 5.4 | 26.61 | 0.18 | ||||||||||
| 6.5 | 2 | 6.6 | 26.11 | 0.20 | ||||||||||
| 10.0 | 3 | 6.8 | 25.66 | 0.34 | 0.075 | 10.40 | 52.62 | 5.1 | 39 | −69.5 | ||||
| 12.0 | 2 | 7.6 | 25.58 | 0.23 | ||||||||||
| 15.5 | 2 | 9.5 | 24.82 | 0.23 | ||||||||||
| 20.5 | 2 | 10.6 | 24.36 | 0.27 | ||||||||||
| 25.5 | 2 | 11.8 | 22.67 | 0.61 | 0.082 | 32 | −51.4 | −18.2 | ||||||
| 26.0 | 3 | 12.4 | 23.95 | 0.30 | 10.40 | 52.83 | 5.1 | |||||||
| 37.5 | 2 | 14.5 | 22.51 | 0.34 | ||||||||||
| 51.0 | 3 | 18.0 | 20.46 | 0.85 | 0.207 | 10.53 | 52.13 | 5.0 | 39 | −58.9 | −19.8 | |||
| 78.0 | 1 | 17.8 | 19.94 | 0.61 | 0.131 | −18.4 | −4.9 | 0.0 | ||||||
| 112.0 | 3 | 29.1 | 14.45 | 1.02 | 0.360 | 10.70 | 51.39 | 4.8 | 50 | −60.3 | −22.8 | |||
| 133.0 | 1 | 26.4 | 15.12 | 1.02 | 0.191 | 9.64 | ||||||||
| 203.0 | 3 | 39.5 | 5.92 | 1.76 | 0.262 | 8.83 | 50.20 | 5.7 | −20.6 | 0.0 | ||||
| 235.0 | 1 | 42.9 | 5.54 | 1.82 | 0.542 | 10.35 | 82 | −59.3 | ||||||
| 322.0 | 3 | 53.3 | 0.20 | 2.23 | 0.509 | 9.60 | 49.62 | 5.2 | 7340 | −99.7 | −23.7 | |||
| 366.0 | 1 | 55.1 | 0.10 | 2.32 | 0.387 | 9.21 | ||||||||
| 370.0 | 3 | 54.6 | 0.05 | 2.38 | 0.522 | 9.98 | 50.36 | 5.0 | 14890 | −100.7 | ||||
| 412.0 | 3 | 56.6 | 0.05 | 2.57 | 0.426 | 9.78 | 50.73 | 5.2 | 17465 | −101.9 | ||||
| 431.0 | 1 | 57.9 | 0.05 | 2.70 | 0.275 | 8.38 | −17.5 | −7.0 | −0.4 | |||||
| 468.0 | 3 | 55.8 | 0.05 | 2.74 | 0.364 | 9.38 | 50.61 | 5.4 | 22540 | −100.7 | ||||
| 512.0 | 3 | 55.7 | 0.05 | 2.74 | 0.253 | 8.53 | 50.57 | 5.9 | 46380 | −100.2 | ||||
| 520.0 | 1 | 59.2 | 0.05 | 2.96 | 0.284 | 8.33 | ||||||||
| 552.0 | 3 | 56.6 | 0.05 | 2.92 | 0.331 | 8.50 | 51.96 | 6.1 | 28740 | −98.4 | ||||
| 633.0 | 3 | 55.7 | 0.05 | 3.07 | 0.224 | 7.30 | 52.17 | 7.1 | 16950 | −94.9 | ||||
| 658.0 | 1 | 59.7 | 0.05 | 3.09 | 0.235 | 10720 | −95.8 | −200.0 | −0.1 | |||||
| 692.0 | 1 | 59.7 | 0.05 | 3.20 | 0.215 | 6.82 | −13.7 | |||||||
| 728.0 | 3 | 58.7 | 0.05 | 3.12 | 8564 | −90.3 | ||||||||
| 752.0 | 1 | 59.1 | 0.05 | 3.20 | 0.185 | −11.9 | −5.2 | −0.1 | ||||||
| 753.0 | 3 | 57.9 | 0.05 | 3.22 | 0.157 | 7.50 | 53.82 | 7.2 | 15480 | −88.5 | ||||
| 790.0 | 1 | 60.7 | 0.05 | 3.20 | 0.158 | 5.93 | ||||||||
| 812.0 | 1 | 60.0 | 0.05 | 3.20 | 0.158 | 5.71 | 7395 | −93.2 | −11.7 | −205.0 | −0.1 | |||
| 908.0 | 1 | 59.8 | 0.05 | 3.20 | 0.134 | 5.83 | ||||||||
| 1033.0 | 1 | 59.8 | 0.05 | 3.48 | 0.101 | 6.48 | −7.9 | −5.6 | −0.1 | |||||
| 1111.0 | 1 | 62.5 | 0.05 | 3.67 | 0.106 | 7.08 | 4338 | −89.2 | −198.0 | |||||
| 1162.0 | 1 | 64.2 | 0.05 | 3.87 | 0.101 | 5247 | −86.5 | −5.7 | −194.0 | −0.1 | ||||
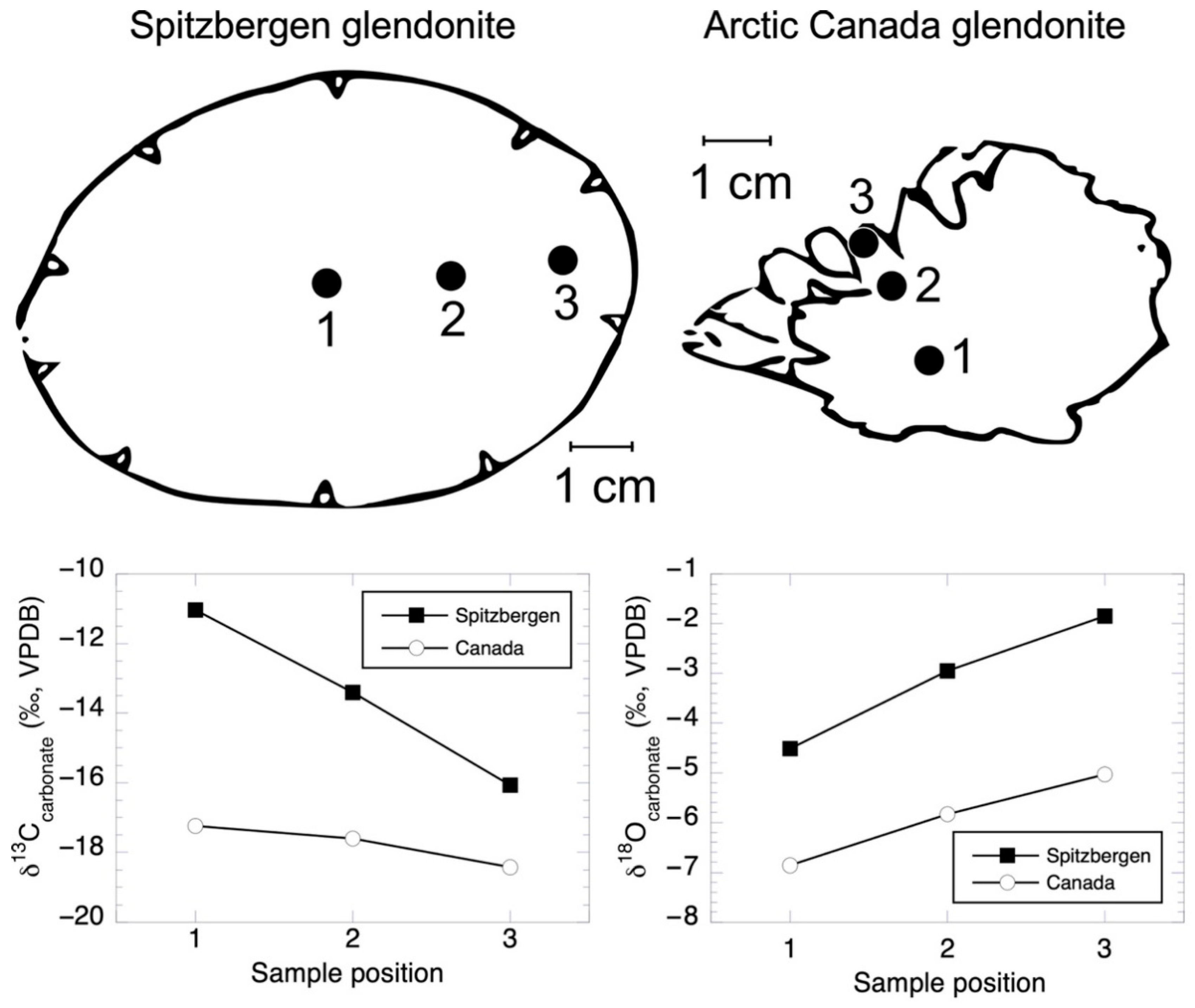
| Sample No. | Distance across Crystal (mm) | δ13Cikaite Carbonate (VPDB) | δ18Oikaite Carbonate (VPDB) | Temperature (°C) (Grossman 2012) |
|---|---|---|---|---|
| Bransfield Strait ikaite (sample K735) | ||||
| 1 | 0.0 | −17.49 | 1.58 | 8.71 |
| 2 | 0.0 | −17.60 | 1.46 | 9.19 |
| 3 | 0.0 | −18.10 | 1.60 | 8.63 |
| 4 | 1.6 | −18.33 | 2.32 | 5.85 |
| 5 | 3.0 | −19.95 | 3.04 | 3.19 |
| 6 | 5.6 | −20.85 | 4.07 | −0.39 |
| 7 | 9.1 | −21.35 | 4.45 | −1.65 |
| 8 | 11.6 | −20.25 | 3.91 | 0.15 |
| 9 | 14.9 | −20.46 | 2.75 | 4.25 |
| 10 | 15.9 | −18.10 | 1.67 | 8.36 |
| Spitzbergen glendonite | ||||
| 1 | −11.03 | −4.51 | ||
| 2 | −13.41 | −2.95 | ||
| 3 | −16.07 | −1.85 | ||
| Canada glendonite | ||||
| 1 | −17.23 | −6.86 | ||
| 2 | −17.61 | −5.83 | ||
| 3 | −18.42 | −5.04 | ||
| Amino | Plankton | Sediment | Ikaite |
|---|---|---|---|
| acid | residues/1000 | ||
| cyst | 3 | 3 | 94 ** |
| tau * | 1 | 1 | 28 ** |
| meto | 3 | 3 | 40 ** |
| asp | 108 | 142 | 209 **** |
| thr” | 57 | 62 | 42 |
| ser” | 75 | 87 | 72 |
| glu | 101 | 79 | 61 **** |
| alpha-AAA | <1 | < 1 | 8 |
| gly | 87 | 212 | 242 **** |
| ala | 106 | 93 | 44 |
| val | 71 | 52 | 29 |
| met | 13 | 9 | 6 |
| alpha-APA | <1 | < 1 | 3 *** |
| allo-ileu | <l | 1 | 1 |
| ileu | 56 | 33 | 11 |
| leu | 105 | 47 | 10 |
| tyr | 28 | 15 | 3 |
| phe | 53 | 27 | 8 |
| beta-ala | 3 | 10 | 77 *,** |
| gamma-ABA | <1 | 4 | 7 ** |
| orn | <l | 5 | 8 ** |
| lys | 70 | 60 | 64 |
| his | 15 | 13 | 9 |
| arg | 45 | 44 | 4 |
| δ13Ccarbonate (‰, VPDB) | δ18Ocarbonate (‰, VPDB) | |||
|---|---|---|---|---|
| Minimum | Maximum | Minimum | Maximum | |
| Rogov et al., 2021 [138] | ||||
| Paleozoic | −27.7 | 0.4 | −23.3 | 1.6 |
| Mesozoic | −44.6 | −0.1 | −15.1 | 0.0 |
| Cenozoic | −31.9 | 8.5 | −17.0 | 4.9 |
| Wang et al., 2021 [46] | ||||
| Paleozoic | −27.7 | 0.6 | −16.9 | 0.3 |
| Mesozoic | −45.2 | 0.0 | −14.7 | 0.0 |
| Cenozoic | −25.2 | 5.8 | −11.6 | −0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whiticar, M.J.; Suess, E.; Wefer, G.; Müller, P.J. Calcium Carbonate Hexahydrate (Ikaite): History of Mineral Formation as Recorded by Stable Isotopes. Minerals 2022, 12, 1627. https://doi.org/10.3390/min12121627
Whiticar MJ, Suess E, Wefer G, Müller PJ. Calcium Carbonate Hexahydrate (Ikaite): History of Mineral Formation as Recorded by Stable Isotopes. Minerals. 2022; 12(12):1627. https://doi.org/10.3390/min12121627
Chicago/Turabian StyleWhiticar, Michael J., Erwin Suess, Gerold Wefer, and Peter J. Müller. 2022. "Calcium Carbonate Hexahydrate (Ikaite): History of Mineral Formation as Recorded by Stable Isotopes" Minerals 12, no. 12: 1627. https://doi.org/10.3390/min12121627