
Impacts of Crystalline Host Rock on Repository Barrier Materials at 250 °C: Hydrothermal Co-Alteration of Wyoming Bentonite and Steel in the Presence of Grimsel Granodiorite
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Analytical Methods
3. Results
3.1. Aqueous Geochemistry
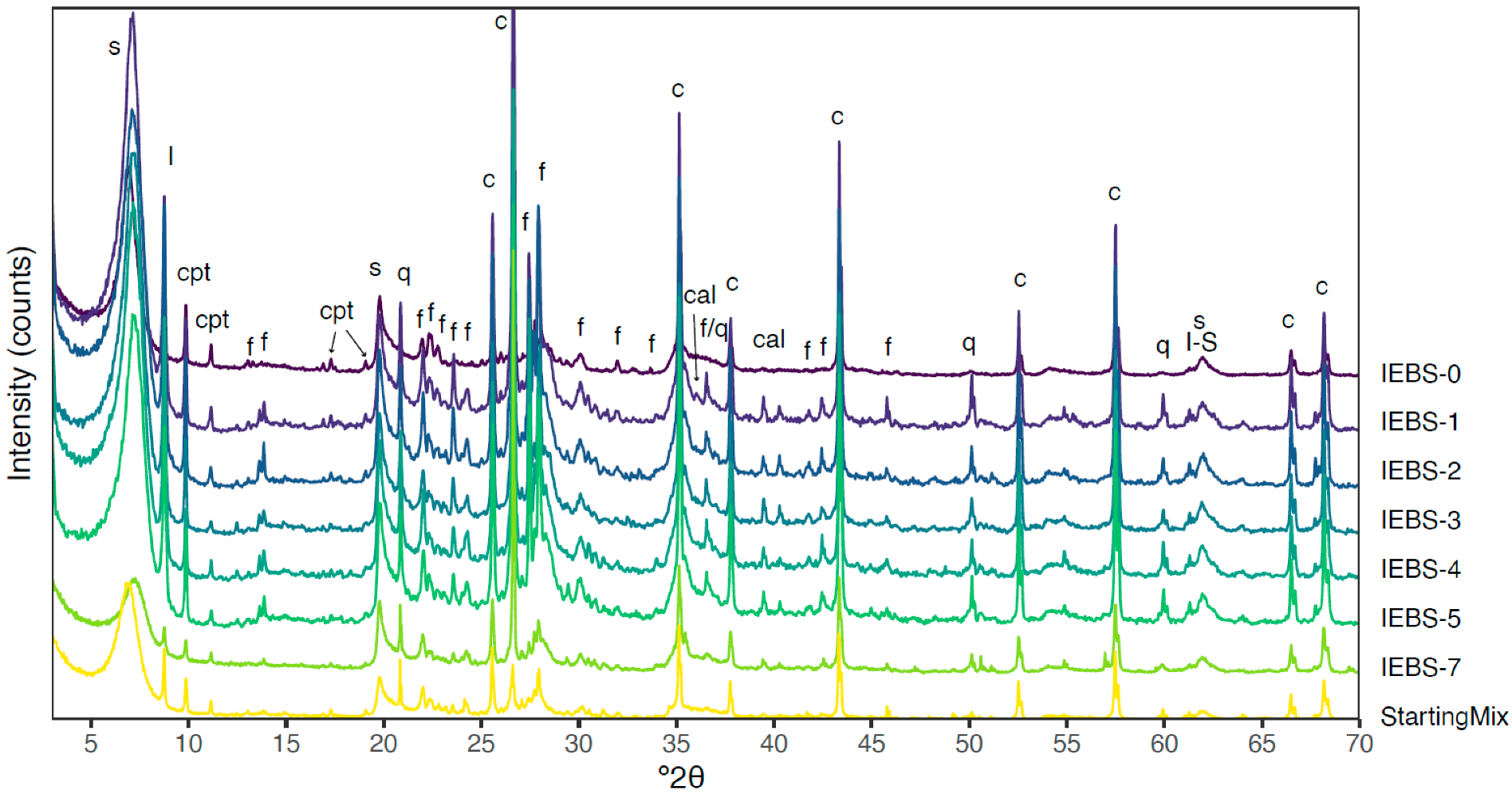
3.2. Mineralogy
3.3. Steel Coupons
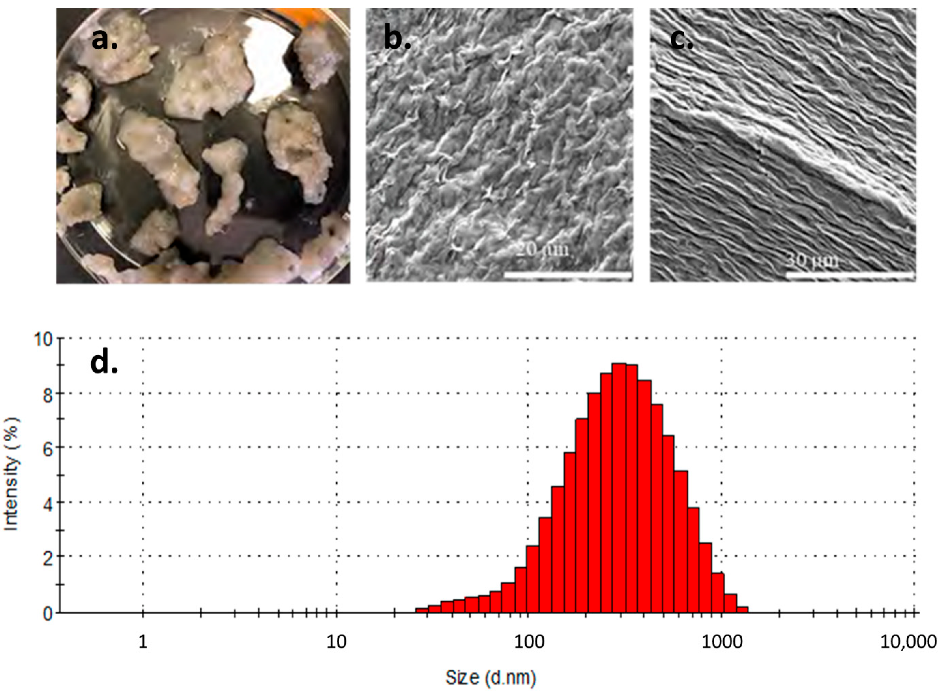
3.4. Colloid Formation
4. Discussion
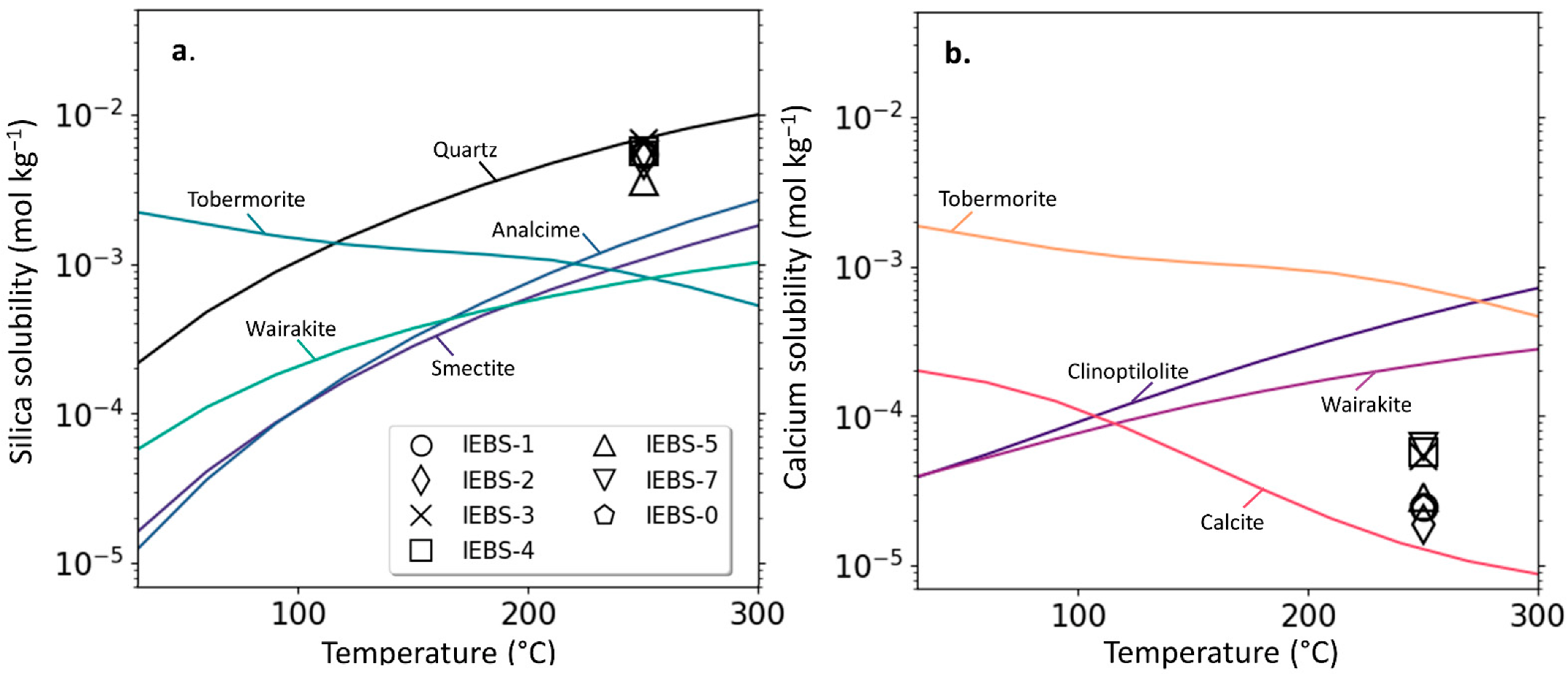
4.1. Mineral Solubilities
4.2. Clay Alteration
4.3. Secondary Calcium (Alumino-)Silicate Hydrates and Zeolite Minerals
4.4. Steel-Bentonite Interface Reactions
smectite
→ (Fe,Ni,Cr)9S8 + (Na, K, Ca)0.33Fe3(Si3.67,Al0.33)O10(OH)2
pentlandite Fe-saponite
4.5. Colloid Formation
5. Conclusions
- No montmorillonite-to-illite transition or reduction in the relative abundance of swelling clay was identified by any mineralogical or chemical analyses performed on the reacted bentonite buffer material. Dissolution of potassium-bearing mineral species in the host rock (K-feldspar and micas, Figure 3 and Table 2) was not observed to increase the potassium concentration of the solution, suggesting potential sequestration of K+ through cation exchange in clays. Illitization of montmorillonite in Wyoming bentonite in a Grimsel granodiorite wall rock environment may then be kinetically limited at the experimental conditions.
- CSH minerals were identified locally by SEM and EMP investigations but were not quantitatively identified by QXRD as a constituent of the solid reactants post-mortem. Due to the rapid decrease in calcium early in the experiments and insolubility of calcite in the carbonate-bearing GW at high temperatures, CSH formation is interpreted to have occurred early in the experiments before being retarded by low Ca concentrations.
- Zeolite formation (analcime) was observed in all experiments and was most abundant in the long term (24-week) experiment. Because zeolites were additionally identified in the experiment reacting only bentonite their formation is interpreted to result from reactions involving the alteration of montmorillonite.
- Characteristic secondary mineral assemblages formed at the steel surfaces of all steel types included in the hydrothermal experiments, dominated by an Fe-smectite with minor chlorite and CSH phases. Fe,Ni,Cr sulfides were identified at the surfaces of the reacted stainless steel coupons (316 SS in IEBS-2 and -5; 304 SS in IEBS-3); no such sulfides were identified at the surface of reacted low-carbon steel (IEBS-4). Additionally, Fe-smectite (as saponite) was not largely identified in bulk mineralogical analyses of the clay matrix, suggesting that Fe-uptake is limited to mineral precipitates at the steel surface.
- Colloid formation observed in our experiments largely formed an aggregated bentonite colloid gel. It is notable that the ionic strength at experimental conditions was relatively high (I ~0.005 to 0.04); when collected and dispersed in DI water the colloids remained dispersed and analyses showed them to be at a relatively stable size. Combined with an increase in bentonite colloid stability at higher temperatures, decreases in the ionic strength of water in contact with a bentonite buffer may increase the potential for colloid-mediated transport over time.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dohrmann, R.; Olsson, S.; Kaufhold, S.; Sellin, P. Mineralogical Investigations of the First Package of the Alternative Buffer Material Test–II. Exchangeable Cation Population Rearrangement. Clay Miner. 2013, 48, 215–233. [Google Scholar] [CrossRef]
- Dohrmann, R.; Kaufhold, S.; Lundqvist, B. The Role of Clays for Safe Storage of Nuclear Waste. In Developments in Clay Science; Elsevier: Amsterdam, The Netherlands, 2013; Volume 5, pp. 677–710. ISBN 1572-4352. [Google Scholar]
- Fernández, A.M.; Kaufhold, S.; Sánchez-Ledesma, D.; Rey, J.J.; Melón, A.; Robredo, L.; Fernández, S.; Labajo, M.A.; Clavero, M.A. Evolution of the THC Conditions in the FEBEX in Situ Test after 18 Years of Experiment: Smectite Crystallochemical Modifications after Interactions of the Bentonite with a C-Steel Heater at 100 °C. Appl. Geochem. 2018, 98, 152–171. [Google Scholar] [CrossRef]
- Hadi, J.; Wersin, P.; Serneels, V.; Greneche, J.-M. Eighteen Years of Steel–Bentonite Interaction in the FEBEX in Situ Test at the Grimsel Test Site in Switzerland. Clays Clay Miner. 2019, 67, 111–131. [Google Scholar] [CrossRef] [Green Version]
- Pusch, R. Highly Compacted Sodium Bentonite for Isolating Rock-Deposited Radioactive Waste Products. Nucl. Technol. 1979, 45, 153–157. [Google Scholar] [CrossRef]
- Sellin, P.; Leupin, O.X. The Use of Clay as an Engineered Barrier in Radioactive-Waste Management—A Review. Clays Clay Miner. 2013, 61, 477–498. [Google Scholar] [CrossRef]
- Huertas, F.; Santiago, J. The FEBEX Project. General Overview. MRS Online Proc. Libr. OPL 1997, 506, 343. [Google Scholar] [CrossRef]
- Hammarström, M.; Olsson, O. Äspö Hard Rock Laboratory—10 Years of Research; Swedish Nuclear Fuel & Waste Management Company: Stockholm, Sweden, 1996. [Google Scholar]
- Kickmaier, W.; McKinley, I. A Review of Research Carried out in European Rock Laboratories. Nucl. Eng. Des. 1997, 176, 75–81. [Google Scholar] [CrossRef]
- Greene, S.R.; Medford, J.S.; Macy, S.A. Storage and Transport Cask Data for Used Commercial Nuclear Fuel; DOE-Headquarters: Washington, DC, USA, 2013.
- Hardin, E.; Hadgu, T.; Clayton, D.J. Cavern/Vault Disposal Concepts and Thermal Calculations for Direct Disposal of 37-PWR Size DPCs; Sandia National Lab. (SNL-NM): Albuquerque, NM, USA, 2015.
- Liljenfeldt, H.; Banerjee, K.; Clarity, J.B.; Scaglione, J.; Jubin, R.T.; Sobes, V.; Howard, R.L.; Hardin, E.; Price, L.; Kalinina, E. Summary of Investigations on Technical Feasibility of Direct Disposal of Dual-Purpose Canisters; Oak Ridge National Lab. (ORNL): Oak Ridge, TN, USA, 2017.
- Wersin, P.; Johnson, L.; McKinley, I. Performance of the Bentonite Barrier at Temperatures beyond 100 °C: A Critical Review. Phys. Chem. Earth Parts ABC 2007, 32, 780–788. [Google Scholar] [CrossRef]
- Zheng, L.; Rutqvist, J.; Xu, H.; Birkholzer, J.T. Coupled THMC Models for Bentonite in an Argillite Repository for Nuclear Waste: Illitization and Its Effect on Swelling Stress under High Temperature. Eng. Geol. 2017, 230, 118–129. [Google Scholar] [CrossRef] [Green Version]
- Kaufhold, S.; Dohrmann, R. Stability of Bentonites in Salt Solutions: II. Potassium Chloride Solution—Initial Step of Illitization? Appl. Clay Sci. 2010, 49, 98–107. [Google Scholar] [CrossRef]
- Kaufhold, S.; Dohrmann, R. Stability of Bentonites in Salt Solutions|Sodium Chloride. Appl. Clay Sci. 2009, 45, 171–177. [Google Scholar] [CrossRef]
- Kaufhold, S.; Dohrmann, R.; Ufer, K.; Svensson, D.; Sellin, P. Mineralogical Analysis of Bentonite from the ABM5 Heater Experiment at Äspö Hard Rock Laboratory, Sweden. Minerals 2021, 11, 669. [Google Scholar] [CrossRef]
- Kumpulainen, S.; Kiviranta, L.; Korkeakoski, P. Long-Term Effects of an Iron Heater and Äspö Groundwater on Smectite Clays: Chemical and Hydromechanical Results from the in Situ Alternative Buffer Material (ABM) Test Package 2. Clay Miner. 2016, 51, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Sudheer Kumar, R.; Podlech, C.; Grathoff, G.; Warr, L.N.; Svensson, D. Thermally Induced Bentonite Alterations in the SKB ABM5 Hot Bentonite Experiment. Minerals 2021, 11, 1017. [Google Scholar] [CrossRef]
- Martin, P.; Barcala, J.; Huertas, F. Large-Scale and Long-Term Coupled Thermo-Hydro-Mechanic Experiments with Bentonite: The FEBEX Mock-up Test. J. Iber. Geol. 2006, 32, 259–282. [Google Scholar]
- Ouhadi, V.; Yong, R.; Goodarzi, A.; Safari-Zanjani, M. Effect of Temperature on the Re-Structuring of the Microstructure and Geo-Environmental Behaviour of Smectite. Appl. Clay Sci. 2010, 47, 2–9. [Google Scholar] [CrossRef]
- Eberl, D.; Hower, J. The Hydrothermal Transformation of Sodium and Potassium Smectite into Mixed-Layer Clay. Clays Clay Miner. 1977, 25, 215–227. [Google Scholar] [CrossRef]
- Eberl, D.; Hower, J. Kinetics of Illite Formation. Geol. Soc. Am. Bull. 1976, 87, 1326–1330. [Google Scholar] [CrossRef]
- Matteo, E.N.; Hardin, E.L.; Hadgu, T.; Park, H.D.; Rigali, M.J.; Jové-Colón, C. Status of Progress Made Toward Preliminary Design Concepts for the Inventory in Select Media for DOE-Managed HLW/SNF; Sandia National Lab. (SNL-NM): Albuquerque, NM, USA, 2016.
- Rodríguez, M.A. Anticipated Degradation Modes of Metallic Engineered Barriers for High-Level Nuclear Waste Repositories. JOM 2014, 66, 503–525. [Google Scholar] [CrossRef]
- Shabalin, B.; Lavrynenko, O.; Pavlenko, O.Y. Development of the Nano-Mineral Phases at the Steel-Bentonite Interface in Time of the Evolution of Geological Repository for Radioactive Waste; Springer: Berlin/Heidelberg, Germany, 2018; pp. 29–44. [Google Scholar]
- Meunier, A.; Velde, B.; Velde, B. Illite: Origins, Evolution and Metamorphism; Springer: Berlin/Heidelberg, Germany, 2004; ISBN 3-540-20486-5. [Google Scholar]
- Alvarez, M.; Lapitz, P.; Ruzzante, J. AE Response of Type 304 Stainless Steel during Stress Corrosion Crack Propagation. Corros. Sci. 2008, 50, 3382–3388. [Google Scholar] [CrossRef]
- Bourdelle, F.; Truche, L.; Pignatelli, I.; Mosser-Ruck, R.; Lorgeoux, C.; Roszypal, C.; Michau, N. Iron–Clay Interactions under Hydrothermal Conditions: Impact of Specific Surface Area of Metallic Iron on Reaction Pathway. Chem. Geol. 2014, 381, 194–205. [Google Scholar] [CrossRef]
- Cheshire, M.C.; Caporuscio, F.A.; Jové-Colón, C.; Norskog, K.E. Fe-Saponite Growth on Low-Carbon and Stainless Steel in Hydrothermal-Bentonite Experiments. J. Nucl. Mater. 2018, 511, 353–366. [Google Scholar] [CrossRef]
- Kursten, B.; Van Iseghem, P. In Situ Corrosion Studies on Candidate Container Materials for the Underground Disposal of High Level Radioactive Waste in Boom Clay. In Proceedings of the 54th Annual Conference and Exposition, San Antonio, TX, USA, 25–30 April 1999. [Google Scholar]
- Pignatelli, I.; Bourdelle, F.; Bartier, D.; Mosser-Ruck, R.; Truche, L.; Mugnaioli, E.; Michau, N. Iron–Clay Interactions: Detailed Study of the Mineralogical Transformation of Claystone with Emphasis on the Formation of Iron-Rich T–O Phyllosilicates in a Step-by-Step Cooling Experiment from 90 °C to 40 °C. Chem. Geol. 2014, 387, 1–11. [Google Scholar] [CrossRef]
- Smailos, E.; Azkarate, I.; Kursten, B.; Marx, G.; Cunado, M. Long-Term Performance of Candidate Materials for HLW/Spent Fuel Disposal Containers; FZKA: Karlsruhe, Germany, 2003. [Google Scholar]
- Kober, F.; Giroud, N.; Uyama, M.; Hitomi, T.; Hayagane, S.; Kadota, N.; Saito, H.; Okamoto, S.; Aoshima, K.; Osawa, M. FEBEX-DP. In Metal Corrosion and Iron-Bentonite Interaction Studies; Nagra Arbeitsbericht (NAGRA): Wettingen, Switzerland, 2017. [Google Scholar]
- Pusch, R.; Kasbohm, J.; Knutsson, S.; Yang, T.; Nguyen-Thanh, L. The Role of Smectite Clay Barriers for Isolating High-Level Radioactive Waste (HLW) in Shallow and Deep Repositories. Procedia Earth Planet. Sci. 2015, 15, 680–687. [Google Scholar] [CrossRef] [Green Version]
- Cathelineau, M.; Guillaume, D.; Mosser-Ruck, R.; Dubessy, J.; Charpentier, D.; Villiéras, F.; Michau, N. Dissolution-Crystallization Processes Affecting Di-Octahedral Smectite in Presence of Iron Metal: Implication on Mineral Distribution in Clay Barriers. In Proceedings of the International Meeting, Clays in Natural & Engineered Barriers for Radioactive Waste Confinement, Tours, France, 14–18 March 2005. [Google Scholar]
- Jodin-Caumon, M.-C.; Mosser-Ruck, R.; Rousset, D.; Randi, A.; Cathelineau, M.; Michau, N. Effect of a Thermal Gradient on Iron-Clay Interactions. Clays Clay Miner. 2010, 58, 667–681. [Google Scholar] [CrossRef]
- Savage, D.; Benbow, S.; Watson, C.; Takase, H.; Ono, K.; Oda, C.; Honda, A. Natural Systems Evidence for the Alteration of Clay under Alkaline Conditions: An Example from Searles Lake, California. Appl. Clay Sci. 2010, 47, 72–81. [Google Scholar] [CrossRef]
- Möri, A.; Alexander, W.; Geckeis, H.; Hauser, W.; Schäfer, T.; Eikenberg, J.; Fierz, T.; Degueldre, C.; Missana, T. The Colloid and Radionuclide Retardation Experiment at the Grimsel Test Site: Influence of Bentonite Colloids on Radionuclide Migration in a Fractured Rock. Colloids Surf. Physicochem. Eng. Asp. 2003, 217, 33–47. [Google Scholar] [CrossRef]
- Telfeyan, K.; Reimus, P.W.; Boukhalfa, H.; Ware, S.D. Aging Effects on Cesium-137 (137 Cs) Sorption and Transport in Association with Clay Colloids. J. Colloid Interface Sci. 2020, 566, 316–326. [Google Scholar] [CrossRef]
- Reimus, P.W.; Zavarin, M.; Wang, Y. Colloid-Facilitated Radionuclide Transport: Current State of Knowledge from a Nuclear Waste Repository Risk Assessment Perspective; Los Alamos National Lab. (LANL): Los Alamos, NM, USA, 2017.
- Andersson, J.; Skagius, K.; Winberg, A.; Lindborg, T.; Ström, A. Site-Descriptive Modelling for a Final Repository for Spent Nuclear Fuel in Sweden. Environ. Earth Sci. 2013, 69, 1045–1060. [Google Scholar] [CrossRef]
- Mejías, M.; Renard, P.; Glenz, D. Hydraulic Testing of Low-Permeability Formations: A Case Study in the Granite of Cadalso de Los Vidrios, Spain. Eng. Geol. 2009, 107, 88–97. [Google Scholar] [CrossRef]
- Stober, I. Researchers Study Conductivity of Crystalline Rock in Proposed Radioactive Waste Site. Eos Trans. Am. Geophys. Union 1996, 77, 93–94. [Google Scholar] [CrossRef]
- Missana, T.; Geckeis, H. The CRR Final Project Report Series II: Supporting Laboratory Experiments with Radionuclides and Bentonite Colloids; National Cooperative for the Disposal of Radioactive Waste (NAGRA): Wettingen, Switzerland, 2006. [Google Scholar]
- Bennett, D.; Gens, R. Overview of European Concepts for High-Level Waste and Spent Fuel Disposal with Special Reference Waste Container Corrosion. J. Nucl. Mater. 2008, 379, 1–8. [Google Scholar] [CrossRef]
- Sevougian, S.D.; Mariner, P.; Connolly, L.A.; MacKinnon, R.J.; Rogers, R.; Dobson, D.; Prouty, J.L. DOE SFWST Campaign R&D Roadmap Update Rev. 1; Sandia National Lab. (SNL-NM): Albuquerque, NM, USA, 2019.
- Birkholzer, J.T.; Tsang, C.-F.; Bond, A.E.; Hudson, J.A.; Jing, L.; Stephansson, O. 25 Years of DECOVALEX-Scientific Advances and Lessons Learned from an International Research Collaboration in Coupled Subsurface Processes. Int. J. Rock Mech. Min. Sci. 2019, 122, 103995. [Google Scholar] [CrossRef]
- Hansen, J.; Doudou, S.; Palmu, M.; White, M. DOPAS Work Package 6 Deliverable D6. 4: DOPAS Project Final Summary Report; DOPAS: Eurajoki, Finland, 2016. [Google Scholar]
- Matteo, E.N. Prioritization of Cross-Cutting Research and Development Activities: Engineered Barrier System; Sandia National Lab. (SNL-NM): Albuquerque, NM, USA, 2020.
- Seyfried, W.; Janecky, D.R.; Berndt, M.E.; Ulmer, G.; Barnes, H. Rocking Autoclaves for Hydrothermal Experiments II. The Flexible Reaction-Cell System. Hydrothermal Exp. Technol. 1987, 23, 216–239. [Google Scholar]
- Sauer, K.; Caporuscio, F.; Rock, M.; Cheshire, M.; Jové-Colón, C. Hydrothermal Interaction of Wyoming Bentonite and Opalinus Clay. Clays Clay Miner. 2020, 68, 144–160. [Google Scholar] [CrossRef]
- Kersting, A.; Zavarin, M.; Zhao, P.; Dai, Z.; Carroll, S.; Wang, Y.; Miller, A.; James, S.; Reimus, P.; Zheng, L. Radionuclide Interaction and Transport in Representative Geologic Media. In Used Fuel Disposition Campaign Milestone Report FCRD-UFD-2012-000154; Prepared for U.S. Department of Energy: Used Fuel Disposition Program; Fuel Cycle Research & Development: Washington, DC, USA, 2012. [Google Scholar]
- Cheshire, M.C.; Caporuscio, F.A.; Rearick, M.S.; Jové-Colón, C.; McCarney, M.K. Bentonite Evolution at Elevated Pressures and Temperatures: An Experimental Study for Generic Nuclear Repository Designs. Am. Mineral. 2014, 99, 1662–1675. [Google Scholar] [CrossRef] [Green Version]
- Parkhurst, D.L.; Appelo, C. Description of Input and Examples for PHREEQC Version 3: A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; US Geological Survey: Reston, VA, USA, 2013.
- Blanc, P.; Lassin, A.; Piantone, P. Thermoddem, Version V1: 10: A Database Devoted to Waste Minerals; Thermoddem: Orléans, France, 2007. [Google Scholar]
- Chung, F.H. Quantitative Interpretation of X-Ray Diffraction Patterns of Mixtures. I. Matrix-Flushing Method for Quantitative Multicomponent Analysis. J. Appl. Crystallogr. 1974, 7, 519–525. [Google Scholar] [CrossRef]
- Pouchou, J.-L.; Pichoir, F. Quantitative Analysis of Homogeneous or Stratified Microvolumes Applying the Model “PAP.” In Electron Probe Quantitation; Springer: Berlin/Heidelberg, Germany, 1991; pp. 31–75. [Google Scholar]
- Chermak, J. Low Temperature Experimental Investigation of the Effect of High PH NaOH Solutions on the Opalinus Shale, Switzerland. Clays Clay Miner. 1992, 40, 650–658. [Google Scholar] [CrossRef]
- Cuadros, J.; Linares, J. Experimental Kinetic Study of the Smectite-to-Illite Transformation. Geochim. Cosmochim. Acta 1996, 60, 439–453. [Google Scholar] [CrossRef]
- Howard, J.J.; Roy, D.M. Development of Layer Charge and Kinetics of Experimental Smectite Alteration. Clays Clay Miner. 1985, 33, 81–88. [Google Scholar] [CrossRef]
- Jakobsson, S.P.; Moore, J.G. Hydrothermal Minerals and Alteration Rates at Surtsey Volcano, Iceland. Geol. Soc. Am. Bull. 1986, 97, 648–659. [Google Scholar] [CrossRef]
- Mosser-Ruck, R.; Cathelineau, M. Experimental Transformation of Na, Ca-Smectite under Basic Conditions at 150 °C. Appl. Clay Sci. 2004, 26, 259–273. [Google Scholar] [CrossRef]
- McCaleb, S.B. Hydrothermal Products Formed from Montmorillonite Clay Systems. In Clays and Clay Minerals; Elsevier: Amsterdam, The Netherlands, 1962; pp. 276–294. [Google Scholar]
- Savage, D.; Walker, C.; Arthur, R.; Rochelle, C.; Oda, C.; Takase, H. Alteration of Bentonite by Hyperalkaline Fluids: A Review of the Role of Secondary Minerals. Phys. Chem. Earth Parts ABC 2007, 32, 287–297. [Google Scholar] [CrossRef]
- Sánchez, L.; Cuevas, J.; Ramírez, S.; De León, D.R.; Fernández, R.; Villa, R.V.D.; Leguey, S. Reaction Kinetics of FEBEX Bentonite in Hyperalkaline Conditions Resembling the Cement–Bentonite Interface. Appl. Clay Sci. 2006, 33, 125–141. [Google Scholar] [CrossRef]
- Fernández, R.; Cuevas, J.; Sánchez, L.; de la Villa, R.V.; Leguey, S. Reactivity of the Cement–Bentonite Interface with Alkaline Solutions Using Transport Cells. Appl. Geochem. 2006, 21, 977–992. [Google Scholar] [CrossRef]
- Savage, D.; Noy, D.; Mihara, M. Modelling the Interaction of Bentonite with Hyperalkaline Fluids. Appl. Geochem. 2002, 17, 207–223. [Google Scholar] [CrossRef]
- Savage, D. Review of the Potential Effects of Alkaline Plume Migration from a Cementitious Repository for Radioactive Waste; UK Environment Agency: Bristol, UK, 1997.
- Savage, D.; Benbow, S. Low PH Cements; Swedish Nuclear Power Inspectorate: Stockholm, Sweden, 2007. [Google Scholar]
- Jové-Colón, C.; Caporuscio, F.A.; Sauer, K.; Cheshire, M. Engineered Barrier Material Interactions at Elevated Temperatures: Bentonite-Metal Interactions under Elevated Temperature Conditions; Sandia National Laboratories (SNL-NM): Albuquerque, NM, USA, 2019. [Google Scholar]
- Kloprogge, J.T.; Komarneni, S.; Amonette, J.E. Synthesis of Smectite Clay Minerals: A Critical Review. Clays Clay Miner. 1999, 47, 529–554. [Google Scholar] [CrossRef]
- Kursten, B.; Smailos, E.; Azkarate, I.; Werme, L.; Smart, N.; Marx, G.; Cuñado, M.; Santarini, G. Corrosion Evaluation of Metallic HLW/Spent Fuel Disposal Containers-Review; Forschungszentrum Karlsruhe GmbH: Karlsruhe, Germany, 2004. [Google Scholar]
- Mosser-Ruck, R.; Cathelineau, M.; Guillaume, D.; Charpentier, D.; Rousset, D.; Barres, O.; Michau, N. Effects of Temperature, PH, and Iron/Clay and Liquid/Clay Ratios on Experimental Conversion of Dioctahedral Smectite to Berthierine, Chlorite, Vermiculite, or Saponite. Clays Clay Miner. 2010, 58, 280–291. [Google Scholar] [CrossRef]
- Mosser-Ruck, R.; Pignatelli, I.; Bourdelle, F.; Abdelmoula, M.; Barres, O.; Guillaume, D.; Charpentier, D.; Rousset, D.; Cathelineau, M.; Michau, N. Contribution of Long-Term Hydrothermal Experiments for Understanding the Smectite-to-Chlorite Conversion in Geological Environments. Contrib. Miner. Pet. 2016, 171, 1–21. [Google Scholar] [CrossRef]
- García-García, S.; Wold, S.; Jonsson, M. Effects of Temperature on the Stability of Colloidal Montmorillonite Particles at Different PH and Ionic Strength. Appl. Clay Sci. 2009, 43, 21–26. [Google Scholar] [CrossRef]
- García-García, S.; Jonsson, M.; Wold, S. Temperature Effect on the Stability of Bentonite Colloids in Water. J. Colloid Interface Sci. 2006, 298, 694–705. [Google Scholar] [CrossRef]
- Lu, N.; Reimus, P.W.; Parker, G.R.; Conca, J.L.; Triay, I.R. Sorption Kinetics and Impact of Temperature, Ionic Strength and Colloid Concentration on the Adsorption of Plutonium-239 by Inorganic Colloids. Radiochim. Acta 2003, 91, 713–720. [Google Scholar] [CrossRef]
- Tari, G.; Olhero, S.; Ferreira, J. Influence of Temperature on Stability of Electrostatically Stabilized Alumina Suspensions. J. Colloid Interface Sci. 2000, 231, 221–227. [Google Scholar] [CrossRef]
- Derjaguin, B.V.; Landau, L. Theory of the Stability of Strongly Charged Lyophobic Sols and of the Adhesion of Strongly Charged Particles in Solutions of Electrolytes. Prog. Surf. Sci. 1941, 43, 30–59. [Google Scholar] [CrossRef]
- Verwey, E.J.W. Theory of the Stability of Lyophobic Colloids. J. Phys. Chem. 1947, 51, 631–636. [Google Scholar] [CrossRef] [Green Version]
- Derjaguin, B.V. Some Results from 50 Years’ Research on Surface Forces. In Surface Forces and Surfactant Systems; Springer: Berlin/Heidelberg, Germany, 1987; pp. 17–30. [Google Scholar]
- Hiemenz, P.C.; Rajagopalan, R. Principles of Colloid and Surface Chemistry, Revised and Expanded; CRC Press: Boca Raton, FL, USA, 2016; ISBN 1-315-27428-0. [Google Scholar]
- Boulton, G.; Caban, P. Groundwater Flow beneath Ice Sheets: Part II—Its Impact on Glacier Tectonic Structures and Moraine Formation. Quat. Sci. Rev. 1995, 14, 563–587. [Google Scholar] [CrossRef]
- Boulton, G.; Caban, P.; Van Gijssel, K. Groundwater Flow beneath Ice Sheets: Part I—Large Scale Patterns. Quat. Sci. Rev. 1995, 14, 545–562. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title | Duration | GW (g) | GG (g) | WB (g) | EBS Type | EBS (g) | Fe (g) | Fe3O4 (g) | WRR (by Mass) |
|---|---|---|---|---|---|---|---|---|---|
| IEBS-0 | 8 weeks | 160 | - | 16.78 | - | - | 0.5 | 0.5 | 9:1 |
| IEBS-1 | 6 weeks | 144 | 3.47 | 10.91 | - | - | 0.49 | 0.5 | 9:1 |
| IEBS-2 | 6 weeks | 182 | 3.19 | 11.02 | 316 SS | nm | 0.49 | 0.5 | 12:1 |
| IEBS-3 | 6 weeks | 110 | 3.41 | 11.05 | 304 SS | 2.74 | 0.5 | 0.59 | 7:1 |
| IEBS-4 | 6 weeks | 185 | 3.28 | 11.00 | LCS | 5.06 | 0.5 | 0.51 | 12:1 |
| IEBS-5 | 8 weeks | 150 | 3.29 | 11.01 | 316 SS | 5.07 | 0.5 | 0.5 | 9:1 |
| IEBS-7 | 24 weeks | 270 | 6.51 | 21.5 | 316 SS | 5.07 | 0.97 | 0.97 | 9:1 |
| Parameter | Value |
|---|---|
| pH | 8.4 |
| Ionic strength | 0.005 |
| Constituent | Concentration (mol L−1) |
| Na+ | 2 × 10−2 |
| K+ | 8 × 10−5 |
| Ca2+ | 2 × 10−4 |
| Mg2+ | 5 × 10−4 |
| Cl− | 4 × 10−4 |
| CO32− | 2 × 10−2 |
| Si | 6 × 10−4 |
| SO42− | 3 × 10−3 |
| Phase | WB | GG | 80 WB:20 GG | IEBS-0 | IEBS-1 | IEBS-2 | IEBS-3 | IEBS-4 | IEBS-5 | IEBS-7 |
|---|---|---|---|---|---|---|---|---|---|---|
| Quartz | 1.5 | 24.1 | 6.9 | 1.3 | 11.4 | 8.2 | 8.2 | 8.2 | 9 | 8.6 |
| K-Feldspar | 0.7 | 10.3 | 3 | 2.1 | 3.5 | 3.7 | 4.2 | 4.4 | 4.4 | 2.3 |
| Plagioclase | 6.2 | 39.3 | 14.1 | 2.3 | 13.6 | 14.8 | 12.7 | 13.9 | 13.4 | 6.1 |
| Apatite | 0.5 | 0.1 | 0.4 | 0.4 | 0.3 | 0.2 | 0.4 | |||
| Pyrite | 0.2 | 0.3 | 0.2 | 0.1 | 0.1 | 0.1 | 0.1 | 0.2 | ||
| Calcite | 0.8 | 0.2 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | |||
| Dolomite | 2.3 | 0.6 | 0.4 | 0.4 | 0.1 | 0.2 | 0.1 | |||
| Amphibole | 0.1 | 0.9 | 0.3 | 0 | 0 | 0 | 0 | 0 | ||
| Gypsum | 0 | 0 | 0 | 0 | 0 | |||||
| Clinoptilolite | 13 | 9.9 | 5.2 | 8 | 6.4 | 5.1 | 3.8 | 5.5 | 3 | |
| Cristobalite | 1.5 | 1.1 | 1 | 0.6 | ||||||
| Buffer | 0.4 | 1 | ||||||||
| Analcime | 2.9 | |||||||||
| Amorphous + Other | 2.9 | 4.2 | ||||||||
| Smectite + Illite + I/S | 71 | 5.5 | 55.3 | 84.8 | 58.7 | 64.3 | 66.4 | 63.2 | 62.4 | 72 |
| Mica | 3.8 | 14.3 | 6.3 | 3.4 | 1.5 | 2.5 | 5.7 | 4.3 | 1.5 | |
| Chlorite | 2 | 1.8 | 2 | 0.4 | 0.2 | 0.3 | 0.2 | 0.2 | ||
| TOTAL | 100 | 100.1 | 100 | 100 | 100 | 100.1 | 100 | 100 | 100 | 102.2 |
| Expt. | Expt. Duration | Steel Type | Average Precipitation Thickness (μm) | Precipitation Rate (μm Day−1) |
|---|---|---|---|---|
| IEBS-3 | 6 weeks | 304 SS | 31.60 (±27.01) | 0.88 |
| IEBS-2 | 6 weeks | 316 SS | 2.27 (±1.40) | 0.06 |
| IEBS-5 | 8 weeks | 316 SS | 38.72 (±27.76) | 0.69 |
| IEBS-7 | 24 weeks | 316 SS | 45.94 (±23.58) | 0.25 |
| IEBS-4 | 6 weeks | LCS | 40.17 (±30.17) | 1.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zandanel, A.; Sauer, K.B.; Rock, M.; Caporuscio, F.A.; Telfeyan, K.; Matteo, E.N. Impacts of Crystalline Host Rock on Repository Barrier Materials at 250 °C: Hydrothermal Co-Alteration of Wyoming Bentonite and Steel in the Presence of Grimsel Granodiorite. Minerals 2022, 12, 1556. https://doi.org/10.3390/min12121556
Zandanel A, Sauer KB, Rock M, Caporuscio FA, Telfeyan K, Matteo EN. Impacts of Crystalline Host Rock on Repository Barrier Materials at 250 °C: Hydrothermal Co-Alteration of Wyoming Bentonite and Steel in the Presence of Grimsel Granodiorite. Minerals. 2022; 12(12):1556. https://doi.org/10.3390/min12121556
Chicago/Turabian StyleZandanel, Amber, Kirsten B. Sauer, Marlena Rock, Florie A. Caporuscio, Katherine Telfeyan, and Edward N. Matteo. 2022. "Impacts of Crystalline Host Rock on Repository Barrier Materials at 250 °C: Hydrothermal Co-Alteration of Wyoming Bentonite and Steel in the Presence of Grimsel Granodiorite" Minerals 12, no. 12: 1556. https://doi.org/10.3390/min12121556