
On the Sorption Mode of U(IV) at Calcium Silicate Hydrate: A Comparison of Adsorption, Absorption in the Interlayer, and Incorporation by Means of Density Functional Calculations

Abstract
:1. Introduction
2. Computational Details
3. Models
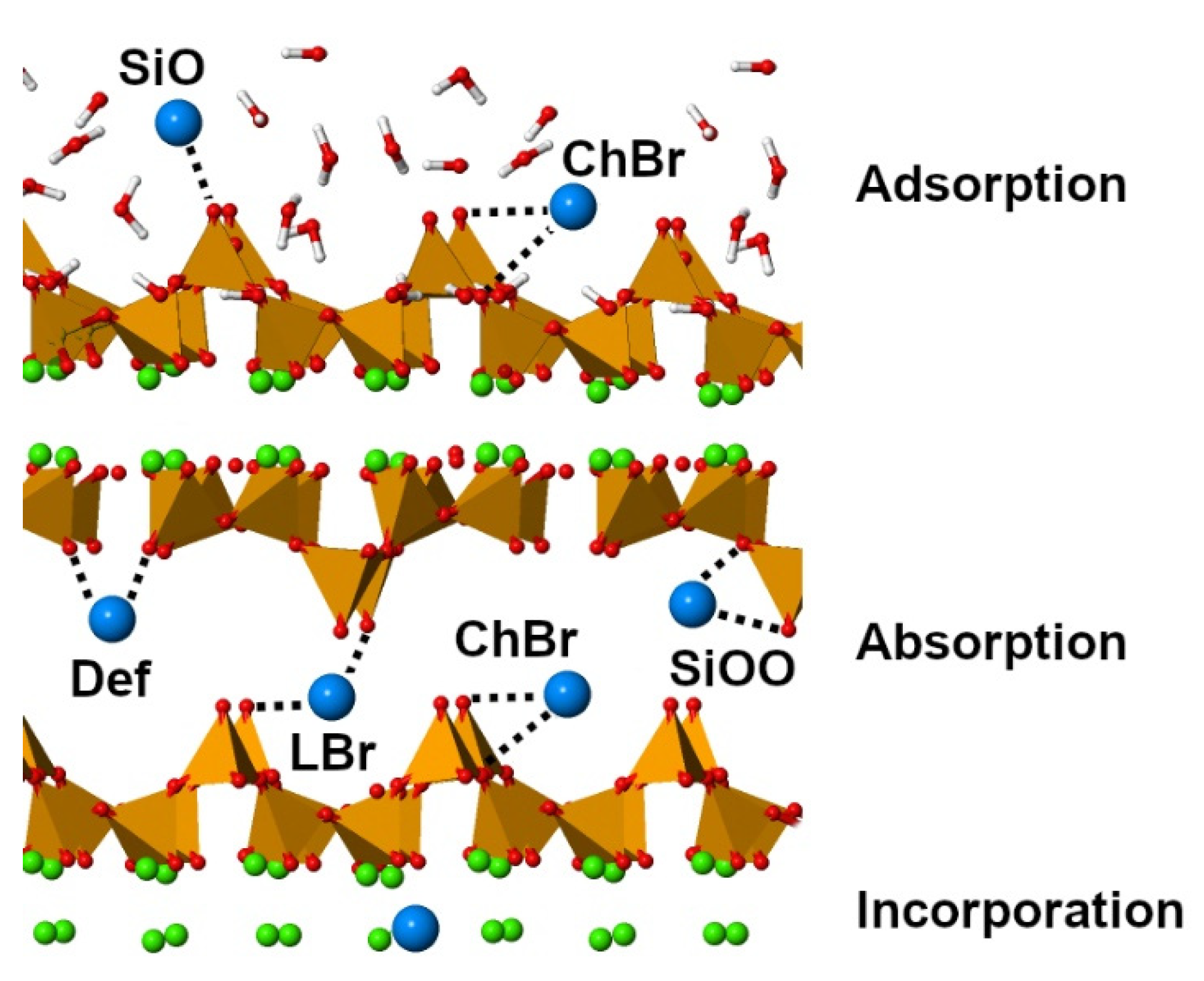
4. Sorption Sites
5. Results
5.1. Geometries
5.2. Energies
6. Comparison with Experiment
7. Summary
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geckeis, H.; Lützenkirchen, J.; Polly, R.; Rabung, T.; Schmidt, M. Mineral–Water Interface Reactions of Actinides. Chem. Rev. 2013, 113, 1016–1062. [Google Scholar] [CrossRef]
- Lothenbach, B.; Nonat, A. Calcium silicate hydrates: Solid and liquid phase composition. Cem. Concr. Res. 2015, 78, 57–70. [Google Scholar] [CrossRef]
- Nonat, A. The structure and stoichiometry of C-S-H. Cem. Concr. Res. 2004, 34, 1521–1528. [Google Scholar] [CrossRef]
- Richardson, I.G. The calcium silicate hydrates. Cem. Concr. Res. 2008, 38, 137–158. [Google Scholar] [CrossRef]
- Richardson, I.G. Model structures for C-(A)-S-H (i). Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 903–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grangeon, S.; Claret, F.; Linard, Y.; Chiaberge, C. X-ray diffraction: A powerful tool to probe and understand the structure of nanocrystalline calcium silicate hydrates. Acta Crystallogr. Sect. B Struct. Sci. 2013, 69, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Gaona, X.; Dähn, R.; Tits, J.; Scheinost, A.C.; Wieland, E. Uptake of Np(IV) by C–S–H phases and cement paste: An EXAFS study. Environ. Sci. Technol. 2011, 45, 8765–8771. [Google Scholar] [CrossRef]
- Macé, N.; Wieland, E.; Dähn, R.; Tits, J.; Scheinost, A.C. EXAFS investigation on U(VI) immobilization in hardened cement paste: Influence of experimental conditions on speciation. Radiochim. Acta 2013, 101, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Wieland, E.; Mace, N.; Dähn, R.; Kunz, D.; Tits, J. Macro- and micro-scale studies on U(VI) immobilization in hardened cement paste. J. Radioanal. Nucl. Chem. 2010, 286, 793–800. [Google Scholar] [CrossRef]
- Tits, J.; Wieland, E. Actinide Sorption by Cementitious Materials; Report 18-02; Paul Scherrer Institut (PSI): Villigen, Switzerland, 2018. [Google Scholar]
- Grangeon, S.; Claret, F.; Roosz, C.; Sato, T.; Gaboreau, S.; Linard, Y. Structure of nanocrystalline calcium silicate hydrates: Insights from X-ray diffraction, synchrotron X-ray absorption and nuclear magnetic resonance. J. Appl. Crystallogr. 2016, 49, 771–783. [Google Scholar] [CrossRef]
- Kremleva, A.; Krüger, S.; Rösch, N. Uranyl(VI) sorption in calcium silicate hydrate phases. A quantum chemical study of tobermorite models. Appl. Geochem. 2020, 113, 104463. [Google Scholar] [CrossRef]
- Stucki, J.W.; Komadel, P.; Wilkinson, H.T. Microbial Reduction of Structural Iron(III) in Smectites. Soil Sci. Soc. Am. J. 1987, 51, 1663–1665. [Google Scholar] [CrossRef]
- Häußler, V.; Amayri, S.; Beck, A.; Platte, T.; Stern, T.A.; Vitova, T.; Reich, T. Uptake of actinides by calcium silicate hydrate (C-S-H) phases. Appl. Geochem. 2018, 98, 426–434. [Google Scholar] [CrossRef]
- Ochs, M.; Mallants, D.; Wang, L. Radionuclide and Metal Sorption on Cement and Concrete; Springer International Publishing: Cham, Switzerland, 2016; Volume 2. [Google Scholar]
- Tits, J.; Gaona, X.; Laube, A.; Wieland, E. Influence of the redox state on the neptunium sorption under alkaline conditions: Batch sorption studies on titanium dioxide and calcium silicate hydrates. Radiochim. Acta 2014, 102, 385–400. [Google Scholar] [CrossRef] [Green Version]
- Reich, T. (Johannes Gutenberg Universität Mainz, Mainz, Germany). Private communication, 2022.
- Stietz, J.; Amayri, S.; Häußler, V.; Prieur, D.; Reich, T. Uptake and speciation of plutonium by hardened cement in the presence of gluconate at high and low ionic strengths. Front. Nucl. Eng. 2022; in preparation. [Google Scholar]
- Shannon, R. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- MacDonald, A.H.; Vosko, S. A relativistic density functional formalism. J. Phys. C Solid State Phys. 1979, 12, 2977. [Google Scholar] [CrossRef]
- Kremleva, A.; Krüger, S.; Rösch, N. Toward a reliable energetics of adsorption at solvated mineral surfaces: A computational study of uranyl(VI) on 2: 1 clay minerals. J. Phys. Chem. C 2016, 120, 324–335. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 5, 799–805. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide. J. Chem. Phys. 1994, 100, 7535–7542. [Google Scholar] [CrossRef]
- Pearson, R.G. Ionization potentials and electron affinities in aqueous solution. J. Am. Chem. Soc. 1986, 108, 6109–6114. [Google Scholar] [CrossRef]
- Bryantsev, V.S.; Diallo, M.S.; Goddard III, W.A. Calculation of solvation free energies of charged solutes using mixed cluster/continuum models. J. Phys. Chem. B 2008, 112, 9709–9719. [Google Scholar] [CrossRef]
- Richardson, I.G. Tobermorite/jennite- and tobermorite/calcium hydroxide-based models for the structure of C-S-H: Applicability to hardened pastes of tricalcium silicate, beta-dicalcium silicate, Portland cement, and blends of Portland cement with blast-fumace slag, metakaolin, or silica fume. Cem. Concr. Res. 2004, 34, 1733–1777. [Google Scholar] [CrossRef]
- Renaudin, G.; Russias, J.; Leroux, F.; Frizon, F.; Cau-Dit-Coumes, C. Structural characterization of C-S-H and C-A-S-H samples-Part I: Long-range order investigated by Rietveld analyses. J. Solid State Chem. 2009, 182, 3312–3319. [Google Scholar] [CrossRef]
- Bonaccorsi, E.; Merlino, S.; Kampf, A.R. The crystal structure of tobermorite 14 Å (Plombierite), a C-S-H phase. J. Am. Ceram. Soc. 2005, 88, 505–512. [Google Scholar] [CrossRef]
- Grangeon, S.; Fernandez-Martinez, A.; Baronnet, A.; Marty, N.; Poulain, A.; Elkaim, E.; Roosz, C.; Gaboreau, S.; Henocq, P.; Claret, F. Quantitative X-ray pair distribution function analysis of nanocrystalline calcium silicate hydrates: A contribution to the understanding of cement chemistry. J. Appl. Crystallogr. 2017, 50, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.; Nonat, A. From C–S–H to C–A–S–H: Experimental study and thermodynamic modelling. Cem. Concr. Res. 2015, 68, 124–138. [Google Scholar] [CrossRef]
- Neck, V.; Kim, J.I. Solubility and hydrolysis of tetravalent actinides. Radiochim. Acta 2001, 89, 1–16. [Google Scholar] [CrossRef]


{kind=link}
| Site | Species | U-OSi | U-OH2 | U-OH | U-Oav | U-Si | U-Ca | ΔE | ΔErel |
|---|---|---|---|---|---|---|---|---|---|
| Surface | |||||||||
| SiO | U(OH)5− | 218 | 215, 227, 229, 239, 245 | 229 (6) | 378 | 416, 466 | −200 | 42 | |
| SiOO | U(OH)40 | 229, 236 | 225, 227, 229, 231 | 229 (6) | 293 | 388, 452, 485 | −183 | 59 | |
| ChBr2 | U(OH)3+ | 229, 241 | 221, 224, 230 | 229 (5) | 333, 334, 343 | 383, 402, 416, 423, 436, 468 | −128 | 114 | |
| ChBr3 | U(OH)3+ | 222, 251, 262 | 223, 229, 232 | 236 (6) | 307, 355 | 391, 414, 429 | −128 | 114 | |
| Def2 | U(OH)3+ | 215, 219 | 262 | 218, 221, 240 | 229 (6) | 358, 370 | 403, 464 | −102 | 140 |
| Interlayer | |||||||||
| LBr2 | U(OH)3+ | 227, 237 | 245 | 216, 226, 231 | 230 (6) | 350, 358 | 404, 407, 446 | −125 | 117 |
| SiO | U(OH)3+ | 238 | 231, 242, 251 | 217, 221, 243 | 235 (7) | 349 | 365, 415 | −177 | 65 |
| SiOO | U(OH)40 | 236, 238 | 218, 223, 231, 232 | 230 (6) | 299 | 349, 417 | −104 | 137 | |
| ChBr1 | U(OH)22+ | 225, 272, 276 | 243, 264 | 203, 234 | 245 (7) | 306, 357, 400 | 374, 431,447, 489, 491 | −81 | 161 |
| ChBr3 | U(OH)3+ | 216, 230, 257 | 224, 225, 231 | 231 (6) | 301, 360, 420 | 342, 450, 454, 479 | −215 | 27 | |
| ChBr3’ | U(OH)3+ | 215, 228, 254 | 221, 232, 239 | 232 (6) | 333, 340, 341, 444 | 407, 412, 426 | −179 | 63 | |
| Def2 | U(OH)40 | 214, 221 | 219, 233, 238, 242 | 228 (6) | 360, 362 | 406, 411 | −76 | 166 | |
| CaO layer | |||||||||
| IncO7 | U4+ | 225, 228, 229, 231, 231, 252, 259 | 236 (7) | 319, 343, 344, 355, 363, 368 | 376, 379, 398, 406, 409, 415 | −208 | 33 | ||
| IncW7 | U(OH)3+ | 227, 227, 227, 232, 253, 260 | 221 | 235 (7) | 318, 341, 345, 364, 367 | 372, 382, 403, 403, 404, 425 | −242 | 0 | |
| Experimentc | |||||||||
| Np(IV) d | 231 (7.7) | 363 (4.3) | 419 (8.0) | ||||||
| Pu(IV) e | 225 (7) | 315 (2), 354 (5) | 412 (6) | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiorescu, I.; Kremleva, A.; Krüger, S. On the Sorption Mode of U(IV) at Calcium Silicate Hydrate: A Comparison of Adsorption, Absorption in the Interlayer, and Incorporation by Means of Density Functional Calculations. Minerals 2022, 12, 1541. https://doi.org/10.3390/min12121541
Chiorescu I, Kremleva A, Krüger S. On the Sorption Mode of U(IV) at Calcium Silicate Hydrate: A Comparison of Adsorption, Absorption in the Interlayer, and Incorporation by Means of Density Functional Calculations. Minerals. 2022; 12(12):1541. https://doi.org/10.3390/min12121541
Chicago/Turabian StyleChiorescu, Ion, Alena Kremleva, and Sven Krüger. 2022. "On the Sorption Mode of U(IV) at Calcium Silicate Hydrate: A Comparison of Adsorption, Absorption in the Interlayer, and Incorporation by Means of Density Functional Calculations" Minerals 12, no. 12: 1541. https://doi.org/10.3390/min12121541