
A Combined Extended X-ray Absorption Fine Structure Spectroscopy and Density Functional Theory Study of Americium vs. Yttrium Adsorption on Corundum (α–Al2O3)
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Density Functional Theory Calculations
2.2. Eu3+ Sorption Isotherms in the Absence and Presence of Y3+
2.3. Extended X-ray Absorption Fine Structure Spectroscopy (EXAFS)
3. Results and Discussion
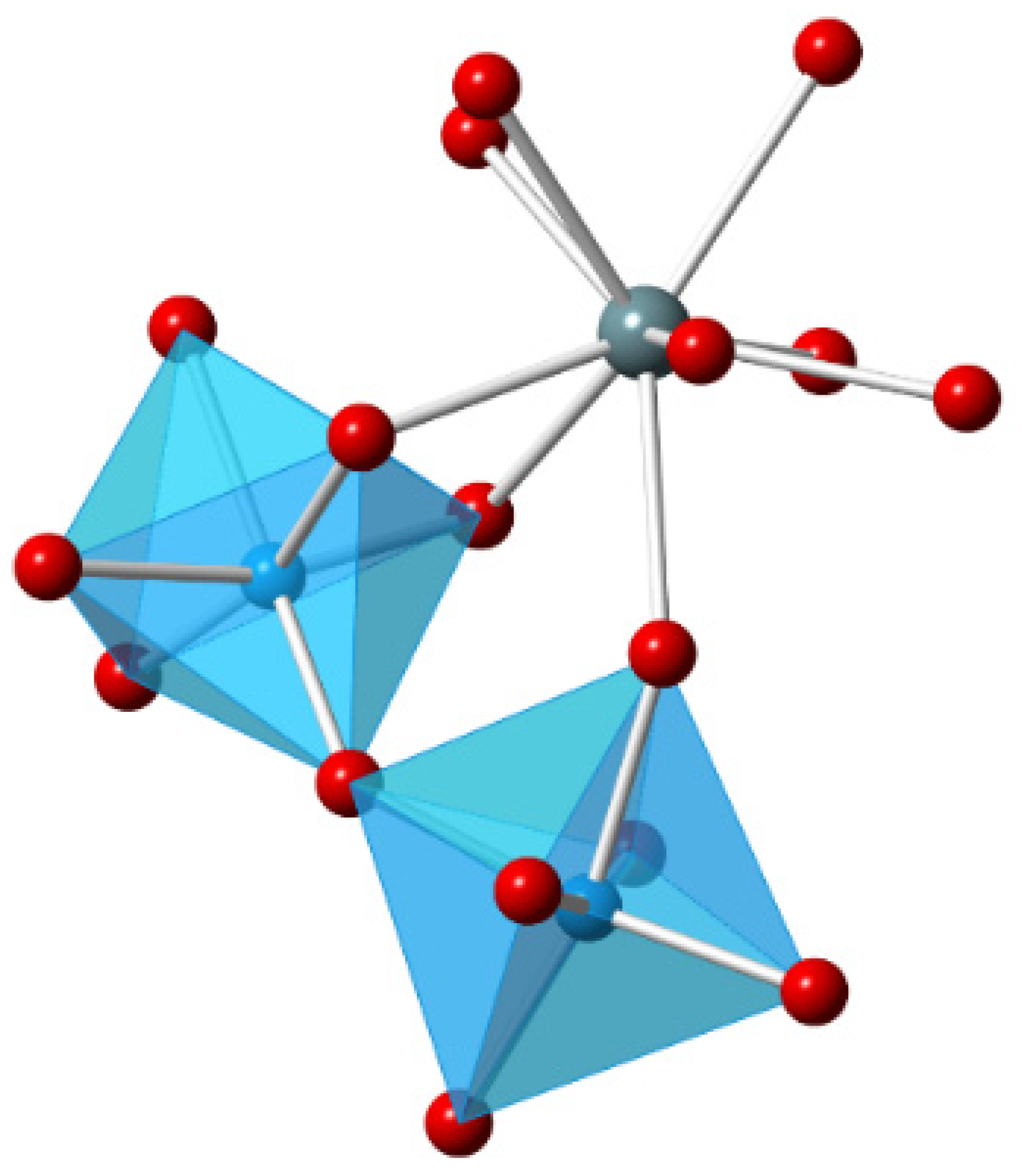
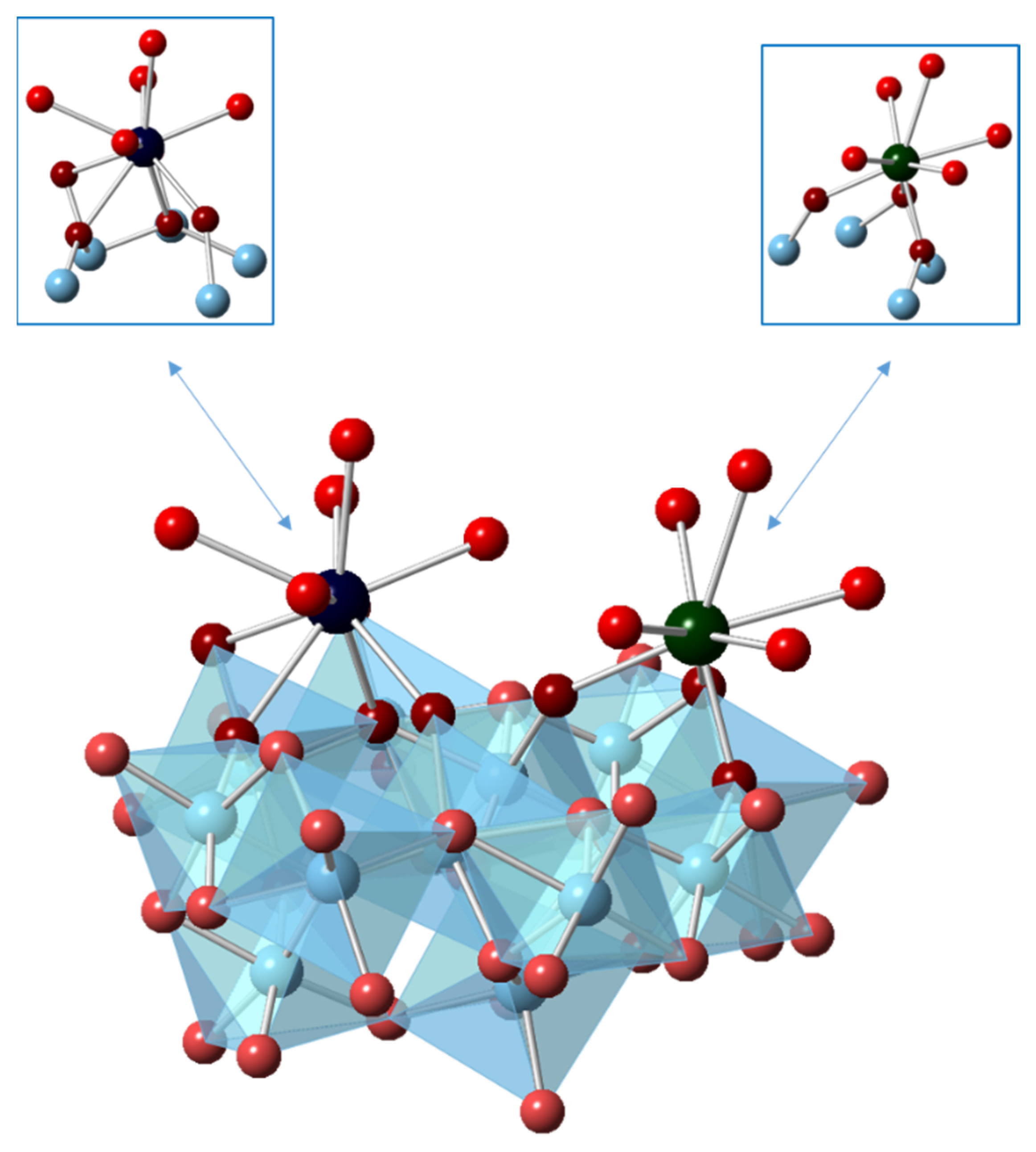
3.1. Density Functional Theory Calculations
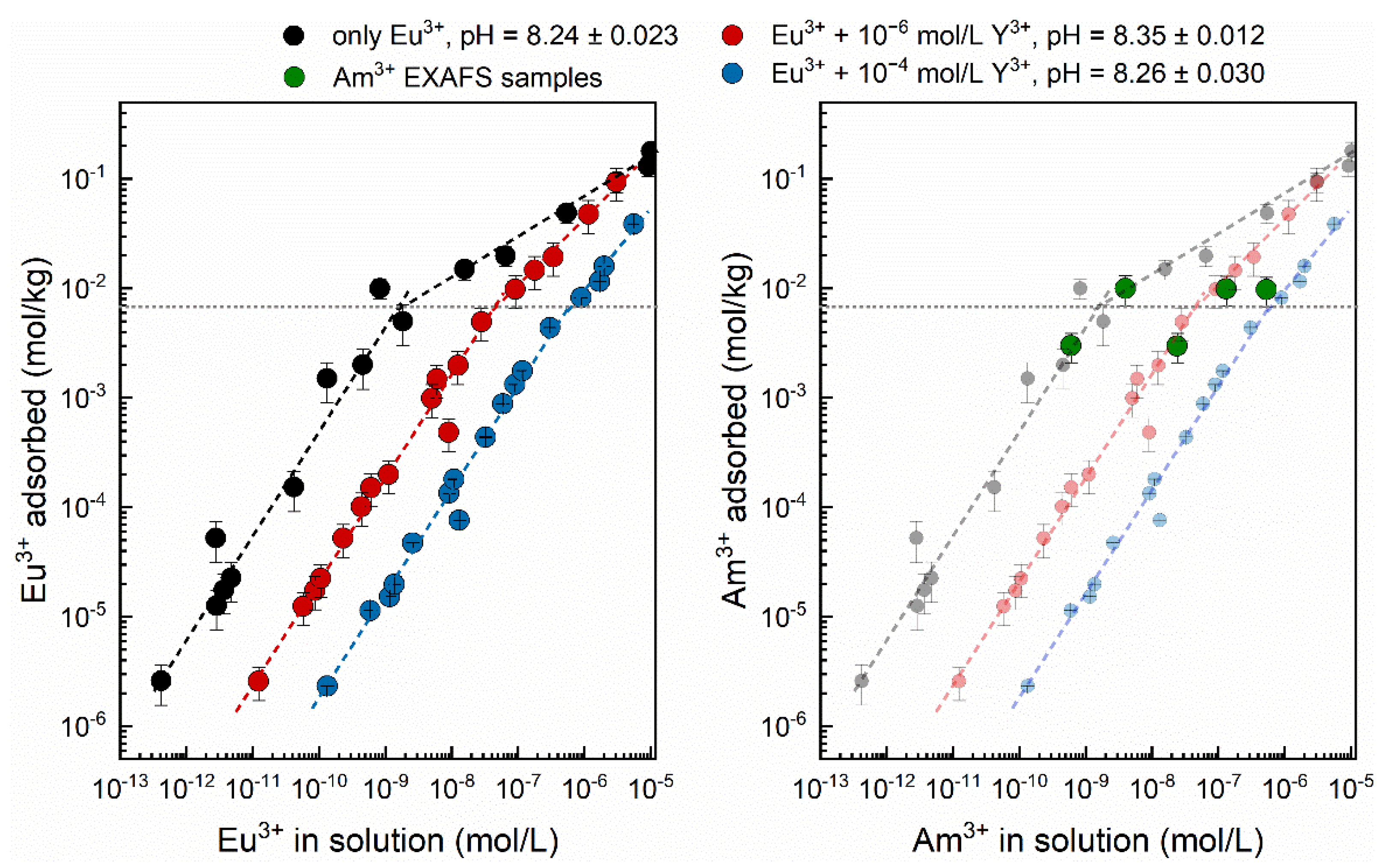
3.2. Eu3+ Sorption Isotherms
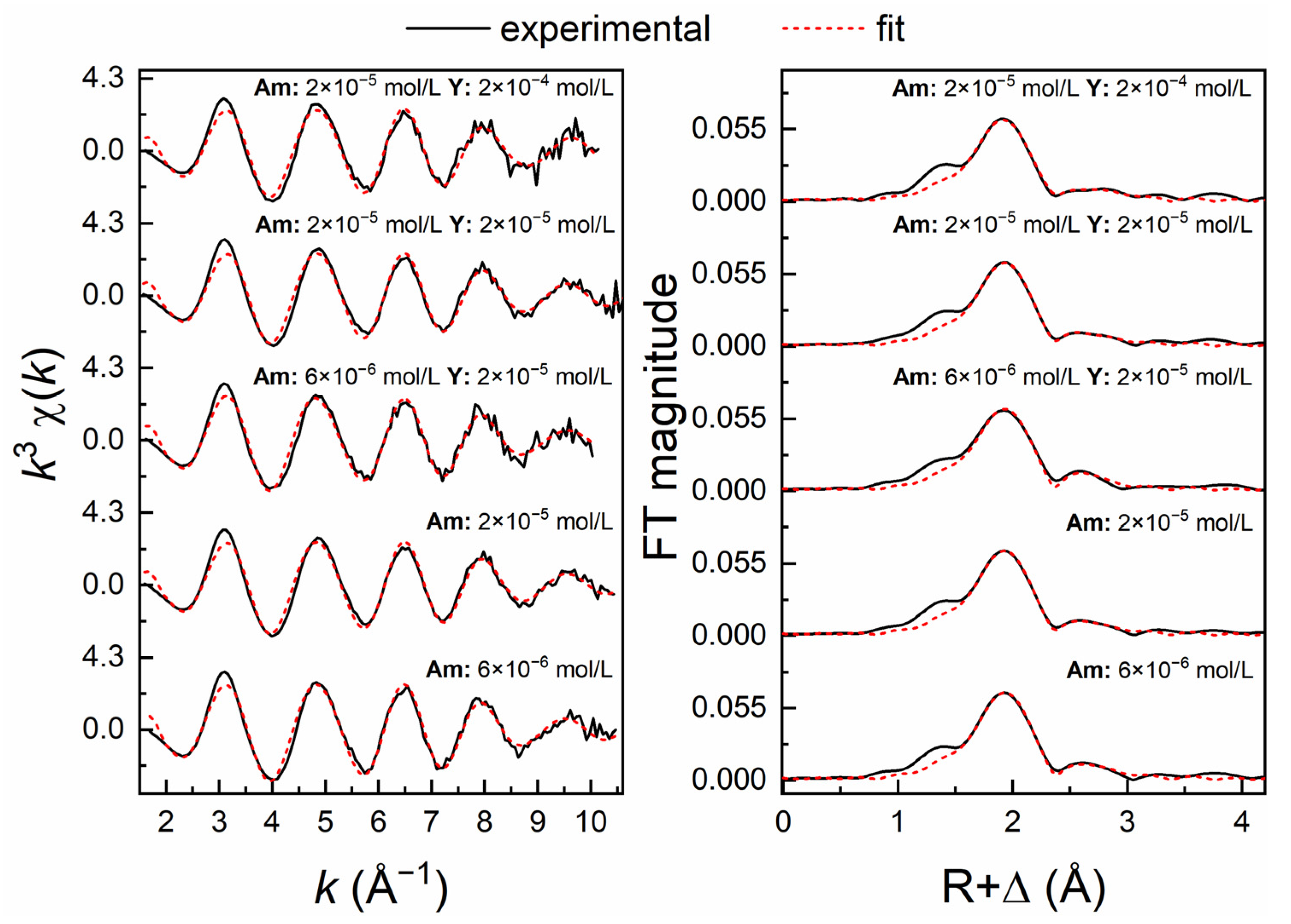
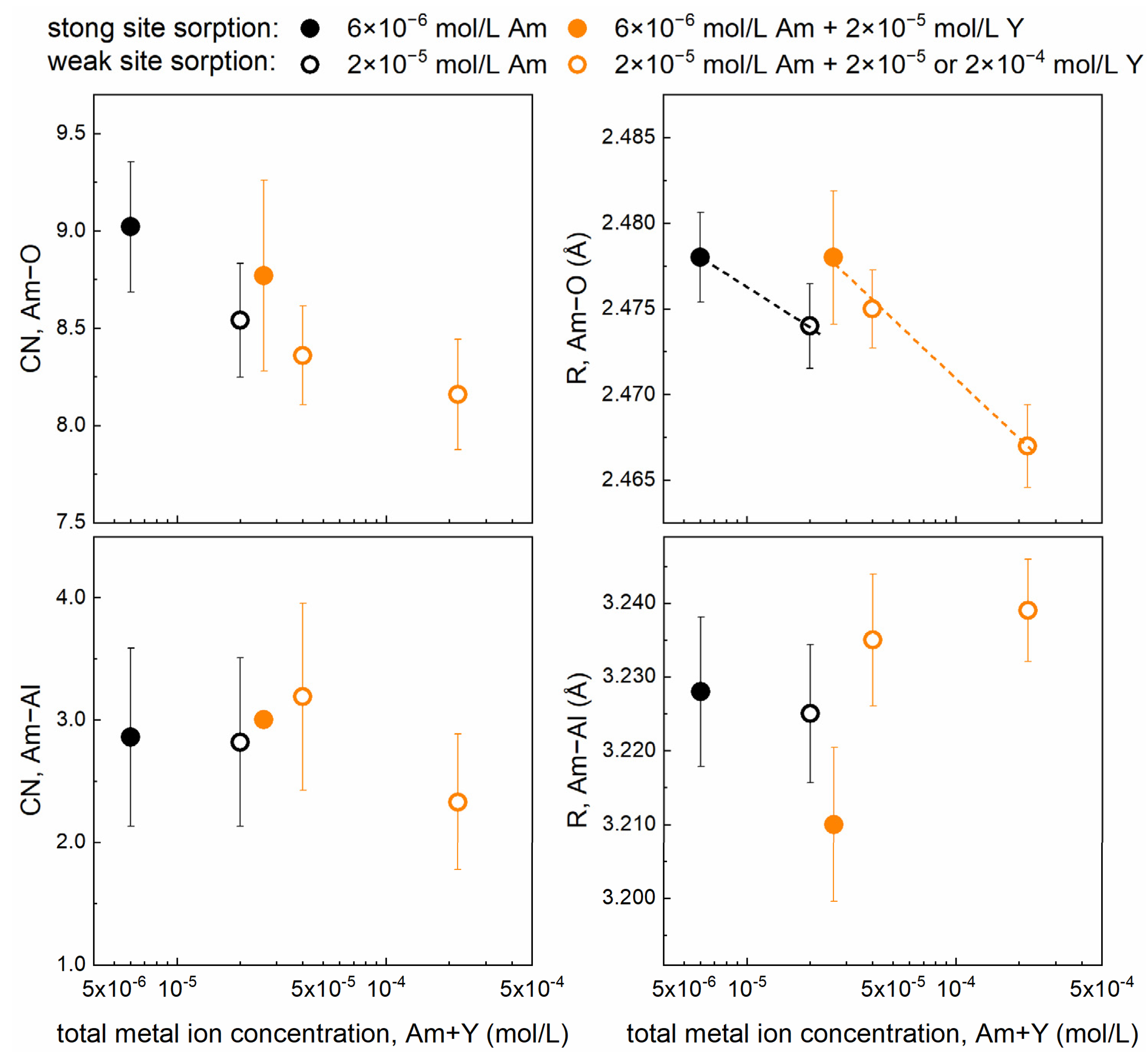
3.3. Am3+ EXAFS Investigations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lokshin, E.P.; Ivanenko, V.I.; Tareeva, O.A.; Korneikov, R.I. Sorption extraction of lanthanides from phosphoric acid solutions. Russ. J. Appl. Chem. 2009, 82, 537–544. [Google Scholar] [CrossRef]
- Tu, Y.J.; Lo, S.C.; You, C.F. Selective and fast recovery of neodymium from seawater by magnetic iron oxide Fe3O4. Chem. Eng. J. 2015, 262, 966–972. [Google Scholar] [CrossRef]
- Moldoveanu, G.A.; Papangelakis, V.G. An overview of rare-earth recovery by ion-exchange leaching from ion-adsorption clays of various origins. Mineral. Mag. 2016, 80, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Lozano, A.; Ayora, C.; Fernández-Martínez, A. Sorption of rare earth elements onto basaluminite: The role of sulfate and pH. Geochim. Cosmochim. Acta 2019, 258, 50–62. [Google Scholar] [CrossRef]
- Kammerlander, K.K.K.; Köhler, L.K.; Huittinen, N.; Bok, F.; Steudtner, R.; Oschatz, C.; Vogel, M.; Stumpf, T.; Brunner, E. Sorption of europium on diatom biosilica as model of a “green” sorbent for f-elements. Appl. Geochem. 2021, 126, 9. [Google Scholar] [CrossRef]
- Rabung, T.; Schild, D.; Geckeis, H.; Klenze, R.; Fanghänel, T. Cm(III) Sorption onto Sapphire (α-Al2O3) Single Crystals. J. Phys. Chem. B 2004, 108, 17160–17165. [Google Scholar] [CrossRef]
- Rabung, T.; Pierret, M.C.; Bauer, A.; Geckeis, H.; Bradbury, M.H.; Baeyens, B. Sorption of Eu(III)/Cm(III) on Ca-montmorillonite and Na-illite. Part 1: Batch sorption and time-resolved laser fluorescence spectroscopy experiments. Geochim. Cosmochim. Acta 2005, 69, 5393–5402. [Google Scholar] [CrossRef]
- Hartmann, E.; Baeyens, B.; Bradbury, M.H.; Geckeis, H.; Stumpf, T. A Spectroscopic Characterization and Quantification of M(III)/Clay Mineral Outer-Sphere Complexes. Environ. Sci. Technol. 2008, 42, 7601–7606. [Google Scholar] [CrossRef]
- Huittinen, N.; Rabung, T.; Lützenkirchen, J.; Mitchell, S.C.; Bickmore, B.R.; Lehto, J.; Geckeis, H. Sorption of Cm(III) and Gd(III) onto gibbsite, α-Al(OH)3: A batch and TRLFS study. J. Colloid Interface Sci. 2009, 332, 158–164. [Google Scholar] [CrossRef]
- Tan, X.; Fang, M.; Wang, X. Sorption speciation of lanthanides/actinides on minerals by TRLFS, EXAFS and DFT studies: A review. Molecules 2010, 15, 8431–8468. [Google Scholar] [CrossRef]
- Huittinen, N.; Rabung, T.; Andrieux, P.; Lehto, J.; Geckeis, H. A comparative batch sorption and time-resolved laser fluorescence spectroscopy study on the sorption of Eu(III) and Cm(III) on synthetic and natural kaolinite. Radiochim. Acta 2010, 98, 613–620. [Google Scholar] [CrossRef]
- Janot, N.; Benedetti, M.F.; Reiller, P.E. Colloidal α-Al2O3, Europium(III) and Humic Substances Interactions: A Macroscopic and Spectroscopic Study. Environ. Sci. Technol. 2011, 45, 3224–3230. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, A.; Marsac, R.; Rabung, T.; Lützenkirchen, J.; Geckeis, H. Sorption of Cm(III) and Eu(III) onto clay minerals under saline conditions: Batch adsorption, laser-fluorescence spectroscopy and modeling. Geochim. Cosmochim. Acta 2015, 151, 192–202. [Google Scholar] [CrossRef]
- Virtanen, S.; Meriläinen, S.; Eibl, M.; Rabung, T.; Lehto, J.; Huittinen, N. Sorption competition and kinetics of trivalent cations (Eu, Y and Cm) on corundum (α-Al2O3): A batch sorption and TRLFS study. Appl. Geochem. 2018, 92, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Eibl, M.; Virtanen, S.; Pischel, F.; Bok, F.; Lönnrot, S.; Shaw, S.; Huittinen, N. A spectroscopic study of trivalent cation (Cm3+ and Eu3+) sorption on monoclinic zirconia (ZrO2). Appl. Surf. Sci. 2019, 487, 1316–1328. [Google Scholar] [CrossRef]
- Neumann, J.; Brinkmann, H.; Britz, S.; Lutzenkirchen, J.; Bok, F.; Stockmann, M.; Brendler, V.; Stumpf, T.; Schmidt, M. A comprehensive study of the sorption mechanism and thermodynamics of f-element sorption onto K-feldspar. J. Colloid Interface Sci. 2021, 591, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.A.; Kent, D.B. Surface Complexation Modeling in Aqueous Geochemistry. Rev. Mineral. 1990, 23, 177–260. [Google Scholar]
- Ochs, M.; Davis, J.A.; Olin, M.; Payne, T.E.; Tweed, C.J.; Askarieh, M.M.; Altmann, S. Use of thermodynamic sorption models to derive radionuclide Kd values for performance assessment: Selected results and recommendations of the NEA sorption project. Radiochim. Acta 2006, 94, 779–785. [Google Scholar] [CrossRef]
- Fedoroff, M.; Lefevre, G.; Duc, M.; Milonjić, S.; Nešković, C. Sorption mechanisms and sorption models. Mater. Sci. Forum 2004, 453–454, 305. [Google Scholar] [CrossRef]
- Payne, T.E.; Brendler, V.; Comarmond, M.J.; Nebelung, C. Assessment of surface area normalisation for interpreting distribution coefficients (Kd) for uranium sorption. J. Environ. Radioact. 2011, 102, 888–895. [Google Scholar] [CrossRef]
- Stockmann, M.; Schikora, J.; Becker, D.A.; Flugge, J.; Noseck, U.; Brendler, V. Smart Kd-values, their uncertainties and sensitivities—Applying a new approach for realistic distribution coefficients in geochemical modeling of complex systems. Chemosphere 2017, 187, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Kulik, D.; Berner, U.; Curti, E. Modelling Chemical Equilibrium Partitioning with the GEMS-PSI Code; Paul Scherrer Institute: Villigen, Switzerland, 2004; pp. 109–122. [Google Scholar]
- Rabung, T.; Stumpf, T.; Geckeis, H.; Klenze, R.; Kim, J.I. Sorption of Am(III) and Eu(III) onto γ-alumina: Experiment and modelling. Radiochim. Acta 2000, 88, 711–716. [Google Scholar] [CrossRef]
- Rabung, T.; Geckeis, H.; Wang, X.K.; Rothe, J.; Denecke, M.A.; Klenze, R.; Fanghänel, T. Cm(III) sorption onto γ-Al2O3: New insight into sorption mechanisms by time-resolved laser fluorescence spectroscopy and extended X-ray absorption fine structure. Radiochim. Acta 2006, 94, 609–618. [Google Scholar] [CrossRef]
- Rabung, T.; Geckeis, H.; Kim, J.I.; Beck, H.P. Sorption of Eu(III) on a Natural Hematite: Application of a Surface Complexation Model. J. Colloid Interface Sci. 1998, 208, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Pourret, O.; Guo, H.; Bonhoure, J. Rare earth elements sorption to iron oxyhydroxide: Model development and application to groundwater. Appl. Geochem. 2017, 87, 158–166. [Google Scholar] [CrossRef]
- Pourret, O.; Davranche, M. Rare earth element sorption onto hydrous manganese oxide: A modeling study. J. Colloid Interface Sci. 2013, 395, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouby, M.; Lützenkirchen, J.; Dardenne, K.; Preocanin, T.; Denecke, M.A.; Klenze, R.; Geckeis, H. Sorption of Eu(III) onto titanium dioxide: Measurements and modeling. J. Colloid Interface Sci. 2010, 350, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Ridley, M.K.; Hiemstra, T.; Machesky, M.L.; Wesolowski, D.J.; van Riemsdijk, W.H. Surface speciation of yttrium and neodymium sorbed on rutile: Interpretations using the charge distribution model. Geochim. Cosmochim. Acta 2012, 95, 227–240. [Google Scholar] [CrossRef]
- Kasar, S.; Kumar, S.; Kar, A.S.; Godbole, S.V.; Tomar, B.S. Sorption of Eu(III) by amorphous titania, anatase and rutile: Denticity difference in surface complexes. Colloids Surf. A Physicochem. Eng. 2013, 434, 72–77. [Google Scholar] [CrossRef]
- Den Auwer, C.; Drot, R.; Simoni, E.; Conradson, S.D.; Gailhanou, M.; Mustre de Leon, J. Grazing incidence XAFS spectroscopy of uranyl sorbed onto TiO2 rutile surfaces. New J. Chem. 2003, 27, 648–655. [Google Scholar] [CrossRef]
- Marques Fernandes, M.; Scheinost, A.C.; Baeyens, B. Sorption of trivalent lanthanides and actinides onto montmorillonite: Macroscopic, thermodynamic and structural evidence for ternary hydroxo and carbonato surface complexes on multiple sorption sites. Water Res. 2016, 99, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Konstantinou, M.; Pashalidis, I. Competitive sorption of Cu(II), Eu(III) and U(VI) ions on TiO2 in aqueous solutions—A potentiometric study. Colloids Surf. A Physicochem. Eng. 2008, 324, 217–221. [Google Scholar] [CrossRef]
- Gou, W.; Ji, J.; Li, W. An EXAFS investigation of the mechanism of competitive sorption between Co(II) and Ni(II) at γ-alumina/solution interface. Acta Geochim. 2017, 36, 462–464. [Google Scholar] [CrossRef]
- Sun, Q.; Cui, P.-X.; Fan, T.-T.; Wu, S.; Zhu, M.; Alves, M.E.; Zhou, D.-M.; Wang, Y.-J. Effects of Fe(II) on Cd(II) immobilization by Mn(III)-rich δ-MnO2. Chem. Eng. J. 2018, 353, 167–175. [Google Scholar] [CrossRef]
- Bozena, G.; Zakrzewska, D.; Szymczycha, B. Sorption of Cr, Pb, Cu, Zn, Cd, Ni, and Co to nano-TiO2 in seawater. Water Sci. Technol. 2018, 77, 145–158. [Google Scholar] [CrossRef]
- Polly, R.; Schimmelpfennig, B.; Flörsheimer, M.; Rabung, T.; Kupcik, T.; Klenze, R.; Geckeis, H. Quantum chemical study of inner-sphere complexes of trivalent lanthanide and actinide ions on the corundum (110) surface. Radiochim. Acta 2013, 101, 561–570. [Google Scholar] [CrossRef]
- Polly, R.; Schimmelpfennig, B.; Rabung, T.; Flörsheimer, M.; Klenze, R.; Geckeis, H. Quantum chemical study of inner-sphere complexes of trivalent lanthanide and actinide ions on the corundum (0001) surface. Radiochim. Acta 2010, 98, 627–634. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 1995, 242, 652–660. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 1995, 240, 283–289. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Treutler, O.; Ahlrichs, R. Efficient molecular numerical integration schemes. J. Chem. Phys. 1995, 102, 346–354. [Google Scholar] [CrossRef]
- Von Arnim, M.; Ahlrichs, R. Performance of parallel TURBOMOLE for density functional calculations. J. Comput. Chem. 1998, 19, 1746–1757. [Google Scholar] [CrossRef]
- Sierka, M.; Hogekamp, A.; Ahlrichs, R. Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys. 2003, 118, 9136–9148. [Google Scholar] [CrossRef]
- Moritz, A.; Cao, X.Y.; Dolg, M. Quasirelativistic energy-consistent 5f-in-core pseudopotentials for trivalent actinide elements. Theor. Chem. Acc. 2007, 117, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Kupcik, T.; Rabung, T.; Lützenkirchen, J.; Finck, N.; Geckeis, H.; Fanghänel, T. Macroscopic and spectroscopic investigations on Eu(III) and Cm(III) sorption onto bayerite (β-Al(OH)3) and corundum α-Al2O3). J. Colloid Interface Sci. 2016, 461, 215–224. [Google Scholar] [CrossRef]
- Roques, J.; Veilly, E.; Simoni, E. Periodic density functional theory investigation of the uranyl ion sorption on three mineral surfaces: A comparative study. Int. J. Mol. Sci. 2009, 10, 2633–2661. [Google Scholar] [CrossRef] [Green Version]
- Geckeis, H.; Lützenkirchen, J.; Polly, R.; Rabung, T.; Schmidt, M. Mineral-water interface reactions of actinides. Chem. Rev. 2013, 113, 1016–1062. [Google Scholar] [CrossRef]
- Perron, H.; Vandenborre, J.; Domain, C.; Drot, R.; Roques, J.; Simoni, E.; Ehrhardt, J.J.; Catalette, H. Combined investigation of water sorption on TiO2 rutile (110) single crystal face: XPS vs. periodic DFT. Surf. Sci. 2007, 601, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Kremleva, A.; Krüger, S.; Rösch, N. Uranyl adsorption at (010) edge surfaces of kaolinite: A density functional study. Geochim. Cosmochim. Acta 2011, 75, 706–718. [Google Scholar] [CrossRef]
- Janeček, J.; Netz, R.R.; Flörsheimer, M.; Klenze, R.; Schimmelpfennig, B.; Polly, R. Influence of hydrogen bonding on the structure of the (001) corundum-water interface. Density functional theory calculations and Monte Carlo simulations. Langmuir 2014, 30, 2722–2728. [Google Scholar] [CrossRef]
- Martorell, B.; Kremleva, A.; Krüger, S.; Rösch, N. Density Functional Model Study of Uranyl Adsorption on the Solvated (001) Surface of Kaolinite. J. Phys. Chem. C 2010, 114, 13287–13294. [Google Scholar] [CrossRef]
- Virtanen, S.; Bok, F.; Ikeda-Ohno, A.; Rossberg, A.; Lützenkirchen, J.; Rabung, T.; Lehto, J.; Huittinen, N. The specific sorption of Np(V) on the corundum (α-Al2O3) surface in the presence of trivalent lanthanides Eu(III) and Gd(III): A batch sorption and XAS study. J. Colloid Interface Sci. 2016, 483, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Kretzschmar, J.; Drobot, B. Not just a background: pH buffers do interact with lanthanide ions-a Europium(III) case study. J. Biol. Inorg. Chem. 2022, 27, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Scheinost, A.C.; Claussner, J.; Exner, J.; Feig, M.; Findeisen, S.; Hennig, C.; Kvashnina, K.O.; Naudet, D.; Prieur, D.; Rossberg, A.; et al. ROBL-II at ESRF: A synchrotron toolbox for actinide research. J. Synchrotron Rad. 2021, 28, 333–349. [Google Scholar] [CrossRef] [PubMed]
- George, G.N.; Pickering, I.J. EXAFSPAK: A Suite of Computer Programs for Analysis of X-ray Absorption Spectra; Stanford Synchrotron Radiation Laboratory, Stanford Linear Accelerator Center: Menlo Park, CA, USA, 1995. [Google Scholar]
- Ressler, T. WinXAS: A Program for X-ray Absorption Spectroscopy Data Analysis under MS-Windows. J. Synchrotron Rad. 1998, 5, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Ankudinov, A.L.; Ravel, B.; Rehr, J.J.; Conradson, S.D. Real-space multiple-scattering calculation and interpretation of X-ray-absorption near-edge structure. Phys. Rev. B 1998, 58, 7565–7576. [Google Scholar] [CrossRef] [Green Version]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Cryst. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- David, F.H.; Vokhmin, V. Thermodynamic properties of some tri- and tetravalent actinide aquo ions. New J. Chem. 2003, 27, 1627–1632. [Google Scholar] [CrossRef]
- Dzombak, D.A.; Morel, F.M.M. Surface Complexation Modeling: Hydrous Ferric Oxide; John Wiley & Sons: Hoboken, NJ, USA, 1990. [Google Scholar]
- Baeyens, B.; Bradbury, M.H. A mechanistic description of Ni and Zn sorption on Na-montmorillonite Part I: Titration and sorption measurements. J. Contam. Hydrol. 1997, 27, 199–222. [Google Scholar] [CrossRef]
- Kosmulski, M. The Effect of the Ionic Strength on the Adsorption Isotherms of Nickel on Silica. J. Colloid Interface Sci. 1997, 190, 212–223. [Google Scholar] [CrossRef]
- Bradbury, M.H.; Baeyens, B. Modelling the sorption of Mn(II), Co(II), Ni(II), Zn(II), Cd(II), Eu(III), Am(III), Sn(IV), Th(IV), Np(V) and U(VI) on montmorillonite: Linear free energy relationships and estimates of surface binding constants for some selected heavy metals and actinides. Geochim. Cosmochim. Acta 2005, 69, 875–892. [Google Scholar] [CrossRef]
- Dähn, R.; Baeyens, B.; Marques Fernandes, M. Zn uptake by illite and argillaceous rocks. Geochim. Cosmochim. Acta 2021, 312, 180–193. [Google Scholar] [CrossRef]
- Li, G.G.; Bridges, F.; Booth, C.H. X-ray-absorption fine structure standards: A comparison of experiment and theory. Phys. Rev. B 1995, 52, 6332–6348. [Google Scholar] [CrossRef] [PubMed]
- Aidhy, D.S.; Zhang, Y.W.; Weber, W.J. Radiation damage in cubic ZrO2 and yttria-stabilized zirconia from molecular dynamics simulations. Scr. Mater. 2015, 98, 16–19. [Google Scholar] [CrossRef] [Green Version]
- Purans, J.; Afify, N.D.; Dalba, G.; Grisenti, R.; De Panfilis, S.; Kuzmin, A.; Ozhogin, V.I.; Rocca, F.; Sanson, A.; Tiutiunnikov, S.I.; et al. Isotopic effect in extended x-ray-absorption fine structure of germanium. Phys. Rev. Lett. 2008, 100, 055901. [Google Scholar] [CrossRef] [Green Version]
- Taube, F.; Drobot, B.; Rossberg, A.; Foerstendorf, H.; Acker, M.; Patzschke, M.; Trumm, M.; Taut, S.; Stumpf, T. Thermodynamic and Structural Studies on the Ln(III)/An(III) Malate Complexation. Inorg. Chem. 2019, 58, 368–381. [Google Scholar] [CrossRef]
- Stumpf, T.; Hennig, C.; Bauer, A.; Denecke, M.A.; Fanghänel, T. An EXAFS and TRLFS study of the sorption of trivalent actinides onto smectite and kaolinite. Radiochim. Acta 2004, 92, 133–138. [Google Scholar] [CrossRef]
- Stumpf, S.; Stumpf, T.; Dardenne, K.; Hennig, C.; Foerstendorf, H.; Klenze, R.; Fanghänel, T. Sorption of Am(III) onto 6-line-Ferrihydrite and Its Alteration Products: Investigations by EXAFS. Environ. Sci. Technol. 2006, 40, 3522–3528. [Google Scholar] [CrossRef] [Green Version]
- Morelova, N.; Finck, N.; Lutzenkirchen, J.; Schild, D.; Dardenne, K.; Geckeis, H. Sorption of americium/europium onto magnetite under saline conditions: Batch experiments, surface complexation modelling and X-ray absorption spectroscopy study. J. Colloid Interface Sci. 2020, 561, 708–718. [Google Scholar] [CrossRef]
- Gao, P.; Zhang, D.; Jin, Q.; Chen, Z.; Wang, D.; Guo, Z.; Wu, W. Multi-scale study of Am(III) adsorption on Gaomiaozi bentonite: Combining experiments, modeling and DFT calculations. Chem. Geol. 2021, 581, 120414. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isotherm | c(Eu3+) (mol/L) | c(Y3+) (mol/L) | n(M3+tot)/m(Al2O3) (mol/g) | pHeq |
| 1 | 1.3 × 10−9–1.0 × 10−4 | 0 | 2.6 × 10−9–2.0 × 10−4 | 8.24 ± 0.023 |
| 2 | 1.3 × 10−9–5.0 × 10−5 | 1 × 10−6 | 2.0 × 10−6–1.02 × 10−4 | 8.35 ± 0.012 |
| 3 | 1.3 × 10−9–2.5 × 10−5 | 1 × 10−4 | 2.0 × 10−4–2.5 × 10−4 | 8.26 ± 0.030 |
| EXAFS Sample | c(243Am3+) (mol/L) | c(Y3+) (mol/L) | n(M3+tot)/m(Al2O3) (mol/g) | pHeq |
| 1 | 6 × 10−6 | 0 | 3 × 10−6 | 8.41 |
| 2 | 2 × 10−5 | 0 | 1 × 10−5 | 8.46 |
| 3 | 6 × 10−6 | 2 × 10−5 | 1.3 × 10−5 | 8.47 |
| 4 | 2 × 10−5 | 2 × 10−5 | 2 × 10−5 | 8.48 |
| 5 | 2 × 10−5 | 2 × 10−4 | 1.1 × 10−4 | 8.50 |
| Tetradentate Am Complex (CN 9) | Tridentate Am Complex (CN 8) | ||
|---|---|---|---|
| Am–Owater (Å) | Am–Osurface (Å) | Am–Owater (Å) | Am–Osurface (Å) |
| 2.46 | 2.39 | 2.45 | 2.27 |
| 2.46 | 2.46 | 2.523 | 2.33 |
| 2.52 | 2.57 | 2.57 | 2.44 |
| 2.56 | 2.59 | 2.60 | |
| 2.64 | 2.66 | ||
| Ø 2.53 | Ø 2.50 | Ø 2.56 | Ø 2.35 |
| Øall 2.52 | Øall 2.49 | ||
| Tetradentate Am Complex | Am–Al (Å) | Am–Al (Å) | Am–Al (Å) | Tridentate Am Complex | Am–Al (Å) | Am–Al (Å) |
|---|---|---|---|---|---|---|
| Am–O1 | 3.12 | Am–O1 | 3.32 | 3.73 | ||
| Am–O2 | 3.12 | 4.26 | Am–O2 | 3.32 | 4.23 | |
| Am–O3 | 3.12 | 3.93 | 3.97 | Am–O3 | 3.91 | |
| Am–O4 | 4.20 | |||||
| weighted Ø | 3.67 | weighted Ø | 3.74 | |||
| Sample Composition | Shell | CN | R (Å) | σ2 (Å2) | ΔE0 (eV) | R (%) |
|---|---|---|---|---|---|---|
| Sample 1 6 × 10−6 mol/L Am | Am–O | 9.0 (3) | 2.478 (3) | 0.0109 (6) | 5.07 (17) | 5.06 |
| Am–Al | 2.9 (7) | 3.228 (10) | 0.0122(30) | |||
| Sample 2 2 × 10−5 mol/L Am | Am–O | 8.5 (3) | 2.474 (2) | 0.0107 (5) | 4.67 (15) | 4.18 |
| Am–Al | 2.8 (7) | 3.225 (9) | 0.0140 (30) | |||
| Sample 3 6 × 10−6 mol/L Am + 2 × 10−5 mol/L Y | Am–O | 8.8 (5) | 2.478 (4) | 0.0116 (7) | 4.83 (16) | 7.35 |
| Am–Al | 3.0 (7) | 3.210 (10) | 0.0134 (33) | |||
| Sample 4 2 × 10−5 mol/L Am + 2 × 10−5 mol/L Y | Am–O | 8.4 (3) | 2.475 (2) | 0.0107 (4) | 4.81 (14) | 3.59 |
| Am–Al | 3.2 (8) | 3.235 (9) | 0.0165 (33) | |||
| Sample 5 2 × 10−5 mol/L Am + 2 × 10−4 mol/L Y | Am–O | 8.2 (3) | 2.467 (2) | 0.0105 (5) | 4.01 (15) | 4.17 |
| Am–Al | 2.3 (6) | 3.239 (7) | 0.0115 (27) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huittinen, N.; Virtanen, S.; Rossberg, A.; Eibl, M.; Lönnrot, S.; Polly, R. A Combined Extended X-ray Absorption Fine Structure Spectroscopy and Density Functional Theory Study of Americium vs. Yttrium Adsorption on Corundum (α–Al2O3). Minerals 2022, 12, 1380. https://doi.org/10.3390/min12111380
Huittinen N, Virtanen S, Rossberg A, Eibl M, Lönnrot S, Polly R. A Combined Extended X-ray Absorption Fine Structure Spectroscopy and Density Functional Theory Study of Americium vs. Yttrium Adsorption on Corundum (α–Al2O3). Minerals. 2022; 12(11):1380. https://doi.org/10.3390/min12111380
Chicago/Turabian StyleHuittinen, Nina, Sinikka Virtanen, André Rossberg, Manuel Eibl, Satu Lönnrot, and Robert Polly. 2022. "A Combined Extended X-ray Absorption Fine Structure Spectroscopy and Density Functional Theory Study of Americium vs. Yttrium Adsorption on Corundum (α–Al2O3)" Minerals 12, no. 11: 1380. https://doi.org/10.3390/min12111380