
A Study of Thermal Stability of Hydroxyapatite

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
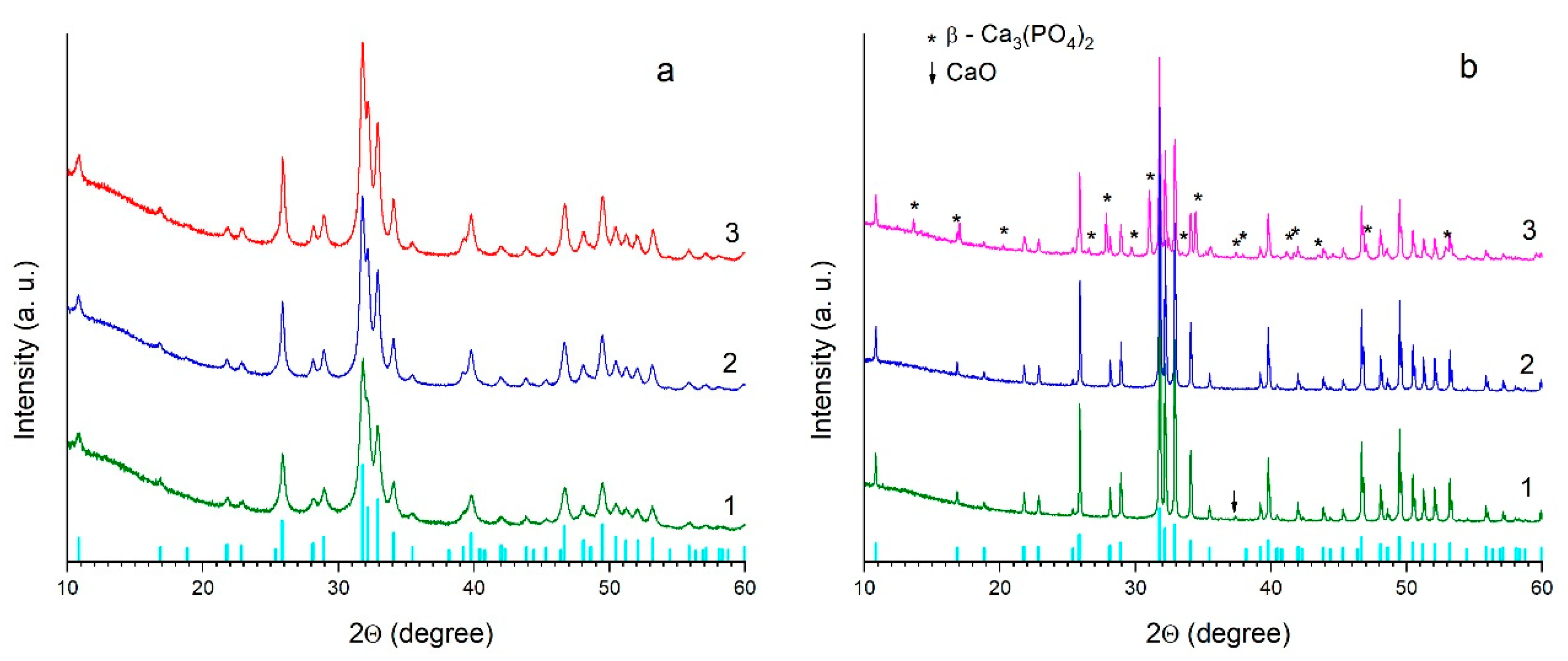
3.1. Effect of the Ca/P Ratio on the Thermal Stability of HA
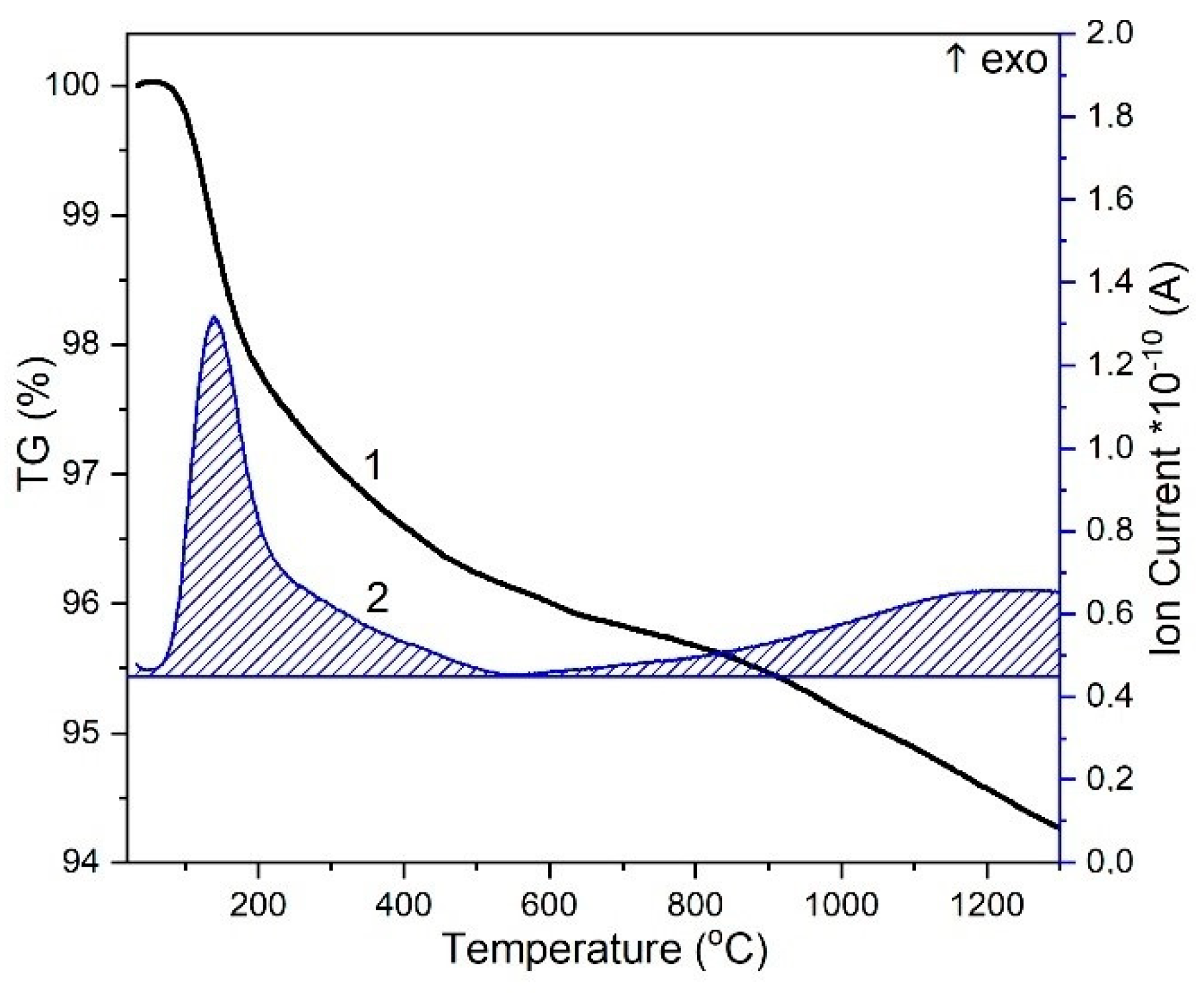
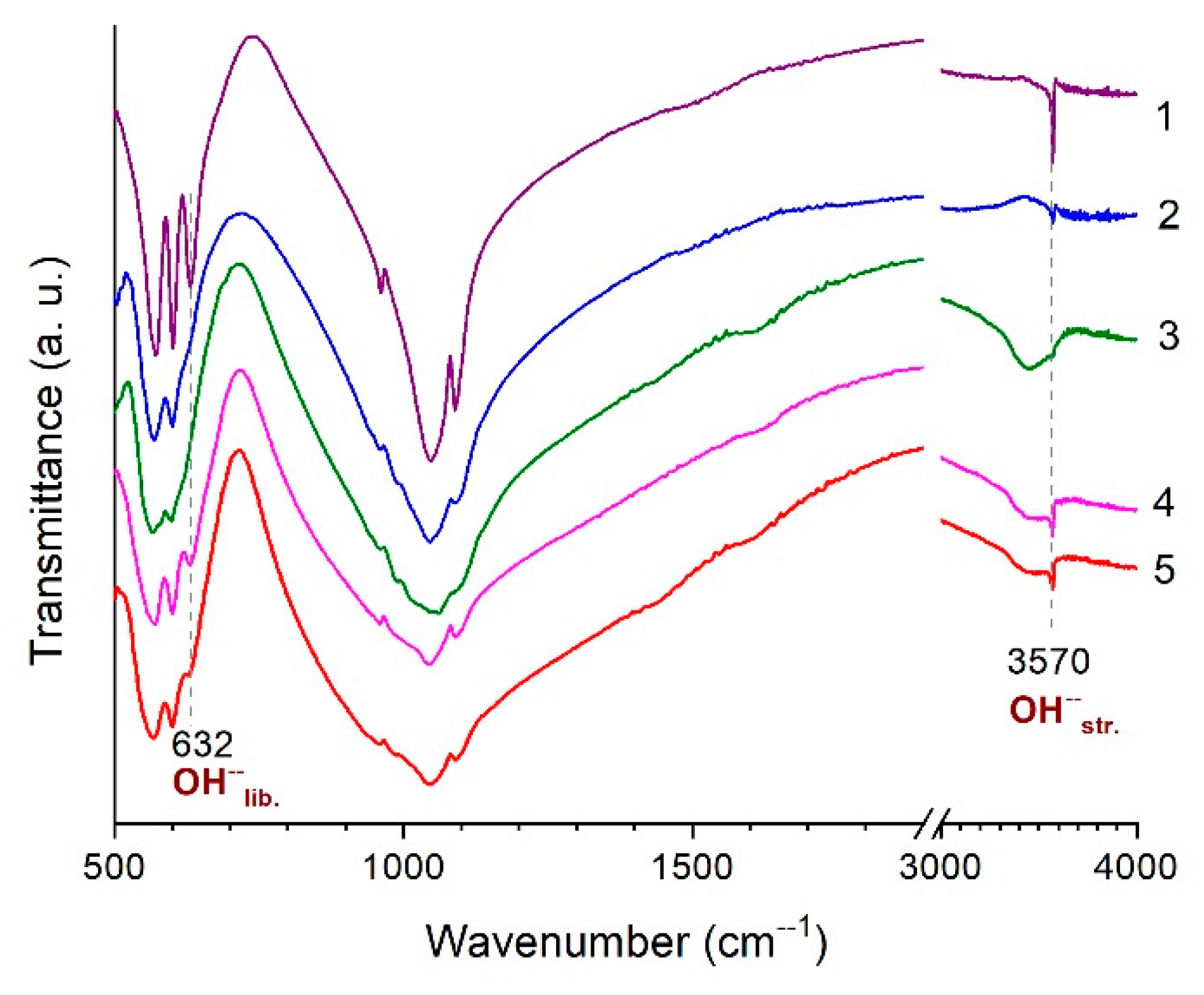
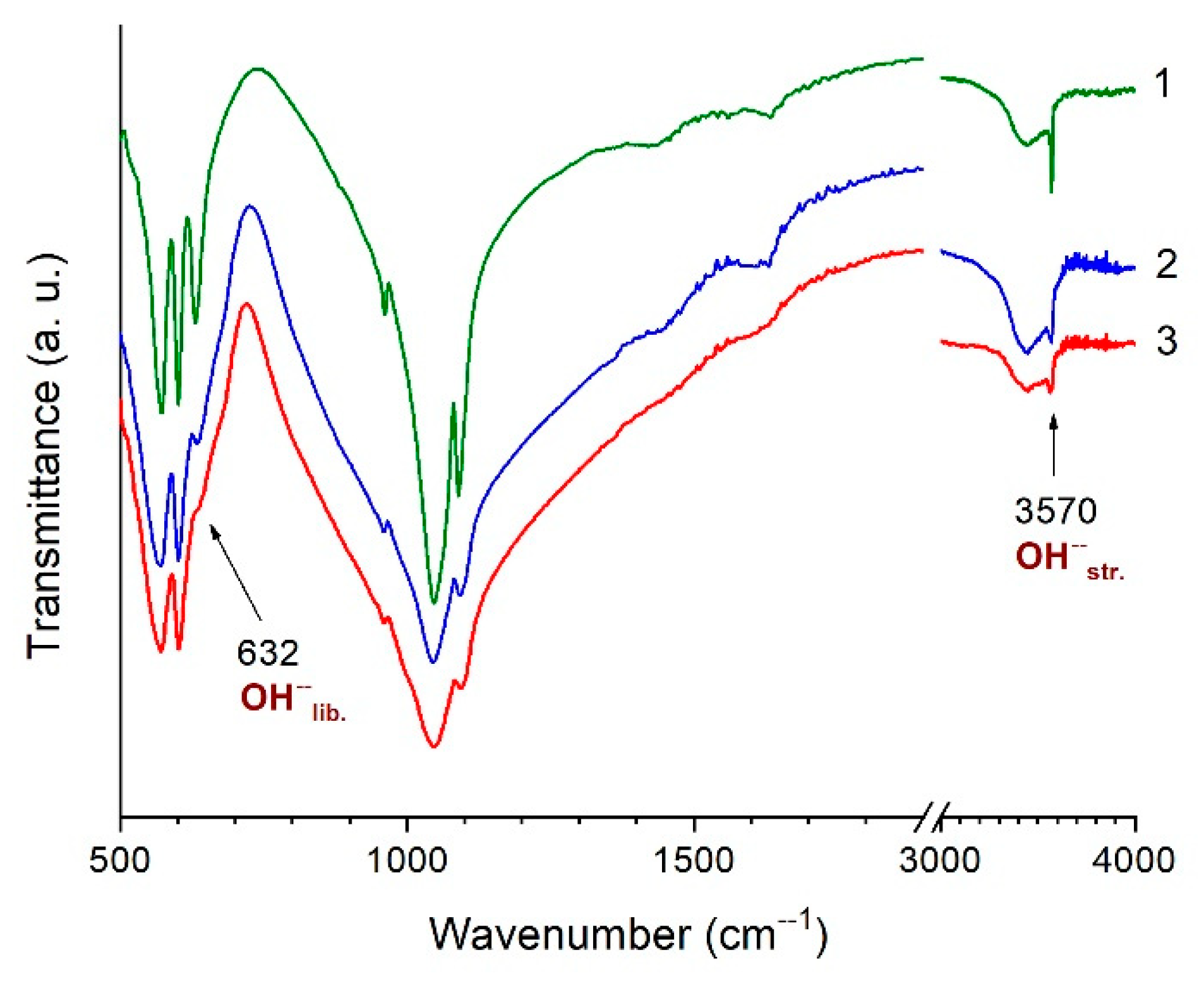
3.2. Thermal Analysis of HA
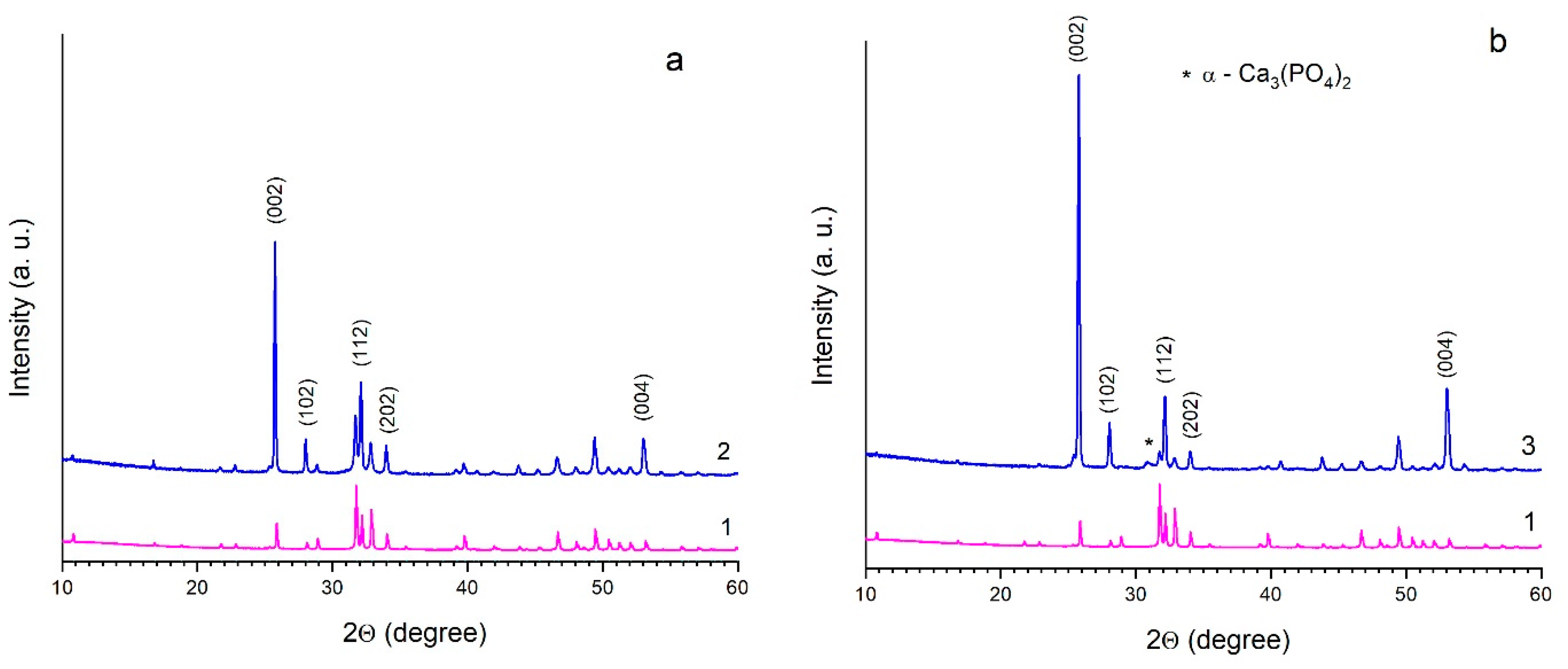
3.3. Sintering of HA in a High-Temperature Furnace
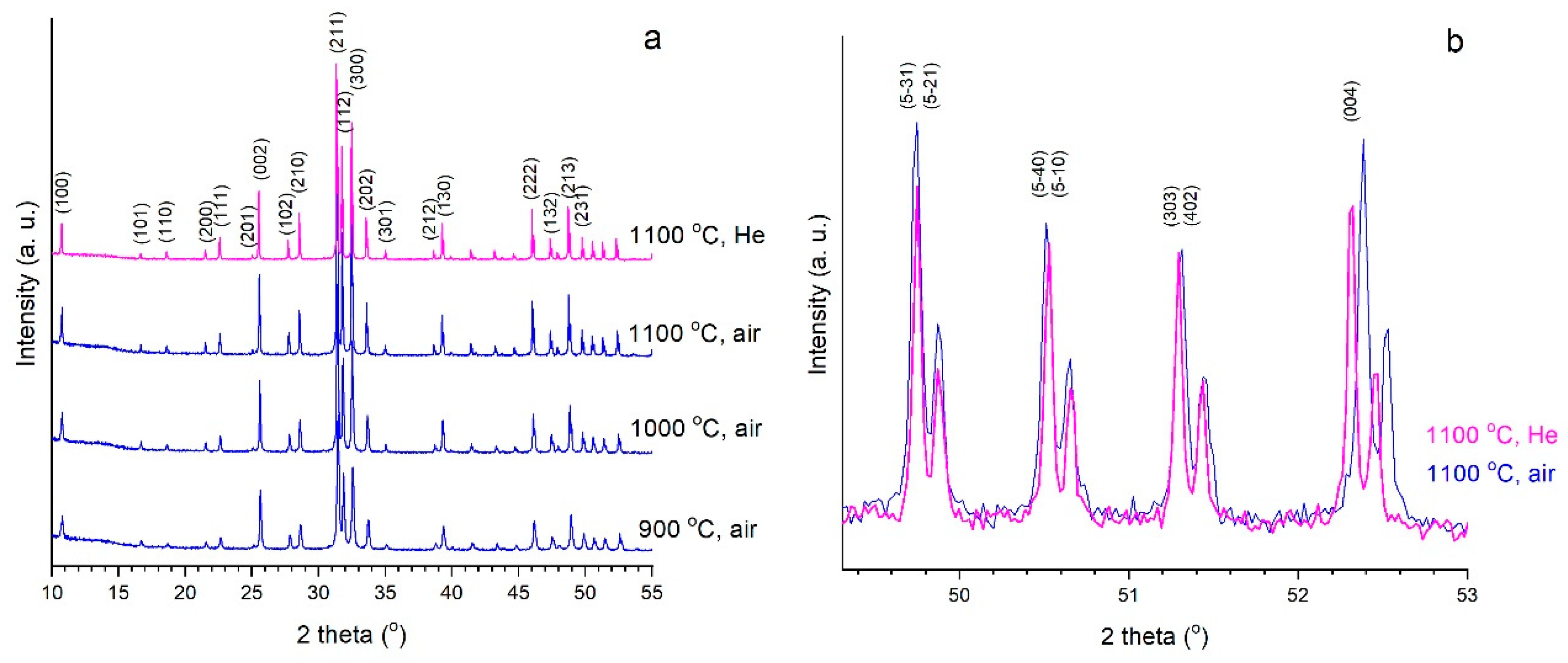
3.4. High-Temperature In Situ Diffraction
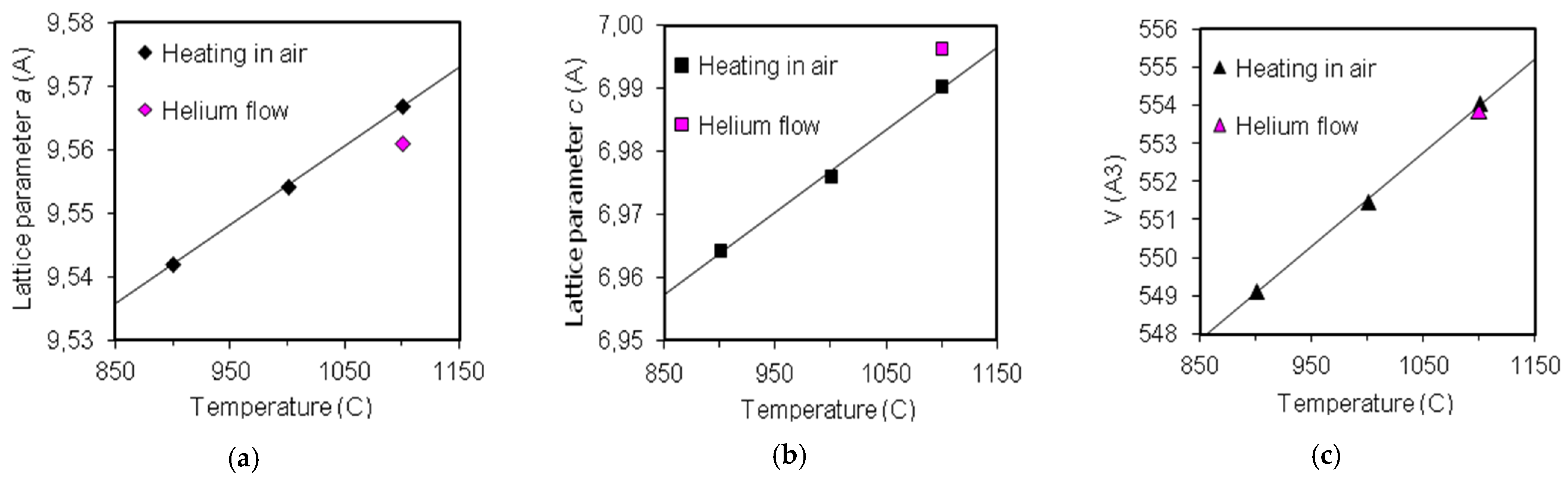
3.5. Extremal Heating Conditions
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Elliott, J.C. Structure and Chemistry of the Apatites and Other Calcium Orthophosphates; Studies in Inorganic Chemistry; Elsevier: Amsterdam, The Netherlands, 1994; pp. 1–404. [Google Scholar]
- Šupová, M. Substituted hydroxyapatites for biomedical applications: A review. Ceram. Int. 2015, 41, 9203–9231. [Google Scholar] [CrossRef]
- White, T.J.; Zhili, D. Structural derivation and crystal chemistry of apatites. Acta Crystallogr. B 2003, 59, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, J.T.B.; Mucalo, M.; Dias, G.J. Substituted hydroxyapatites for bone regeneration: A review of current trends. J. Biomed. Mater. Res. B Appl. Biomater. 2017, 105, 1285–1299. [Google Scholar] [CrossRef] [PubMed]
- Sadat-Shojai, M.; Khorasani, M.-T.; Dinpanah-Khoshdargi, E.; Jamshidi, A. Synthesis methods for nanosized hydroxyapatite with diverse structures. Acta Biomater. 2013, 9, 7591–7621. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Wu, C.; Chang, J. Advances in synthesis of calcium phosphate crystals with controlled size and shape. Acta Biomater. 2014, 10, 4071–4102. [Google Scholar] [CrossRef]
- Chaikina, M.V.; Bulina, N.V.; Vinokurova, O.B.; Prosanov, I.Y.; Dudina, D.V. Interaction of calcium phosphates with calcium oxide or calcium hydroxide during the “soft” mechanochemical synthesis of hydroxyapatite. Ceram. Int. 2019, 45, 16927–16933. [Google Scholar] [CrossRef]
- Ma, G.; Liu, X.Y. Hydroxyapatite: Hexagonal or monoclinic? Cryst. Growth Des. 2009, 9, 2991−2994. [Google Scholar] [CrossRef]
- Pastero, L.; Bruno, M.; Aquilano, D. About the Genetic Mechanisms of Apatites: A Survey on the Methodological Approaches. Minerals 2017, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.M.; Elliott, J.C.; Dowker, S.E.; Rodríguez-Lorenzo, L.M. Rietveld refinements and spectroscopic studies of the structure of Ca-deficient apatite. Biomaterials 2005, 26, 1317–1327. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphates (CaPO4): Occurrence and properties. Prog. Biomater. 2016, 5, 9–70. [Google Scholar] [CrossRef] [Green Version]
- Córdova-Udaeta, M.; Kim, Y.; Yasukawa, K.; Kato, Y.; Fujita, T.; Dodbiba, G. Study on the Synthesis of Hydroxyapatite under Highly Alkaline Conditions. Ind. Eng. Chem. Res. 2021, 60, 4385–4396. [Google Scholar] [CrossRef]
- Mucalo, M. Hydroxyapatite (HAp) for Biomedical Applications; Woodhead Publishing Limited: Waltham, MA, USA, 2015; pp. 1–364. [Google Scholar]
- Kenny, S.M.; Buggy, M. Bone cements and fillers: A review. J. Mater. Sci. Mater. Med. 2003, 14, 923–938. [Google Scholar] [CrossRef] [PubMed]
- Gruselle, M. Apatites: A new family of catalysts in organic synthesis. J. Organomet. Chem. 2015, 793, 93–101. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Current State of Bioceramics. J. Ceram. Sci. Technol. 2018, 9, 353–370. [Google Scholar] [CrossRef]
- Dorozhkin, S. Calcium Orthophosphate (CaPO4) Scaffolds for Bone Tissue Engineering Applications. J. Biotechnol. Biomed. Sci. 2018, 1, 25–93. [Google Scholar] [CrossRef]
- Tõnsuaadu, K.; Gross, K.A.; Plūduma, L.; Veiderma, M. A review on the thermal stability of calcium apatites. J. Therm. Anal. Calorim. 2012, 110, 647–659. [Google Scholar] [CrossRef]
- Liao, C.-J.; Lin, F.-H.; Chen, K.-S.; Sun, J.-S. Thermal decomposition and reconstitution of hydroxyapatite in air atmosphere. Biomaterials 1999, 20, 1807–1813. [Google Scholar] [CrossRef]
- Tanaka, H.; Chikazawa, M.; Kandori, K.; Ishikawa, T. Influence of thermal treatment on the structure of calcium hydroxyapatite. Phys. Chem. Chem. Phys. 2000, 2, 2647–2650. [Google Scholar] [CrossRef]
- Wang, T.; Dorner-Reisel, A. Thermo-analytical investigations of the decomposition of oxyhydroxyapatite. Mater. Lett. 2004, 58, 3025–3028. [Google Scholar] [CrossRef]
- Drouet, C. A comprehensive guide to experimental and predicted thermodynamic properties of phosphate apatite minerals in view of applicative purposes. J. Chem. Thermodyn. 2015, 81, 143–159. [Google Scholar] [CrossRef] [Green Version]
- Riboud, P.V. Comparaison de la stabilite de l’apatite d’oxyde de fer et de l’hydroxyapatite a haute temperature. Bull. Soc. Chim. Fr. 1968, 1701–1703. [Google Scholar]
- Riboud, P.V. Composition et stabilité des phases à structure d`apatite dans le système CaO-P2O5- de fer-H2O à haute température. Ann. Chim. 1973, 8, 381–390. [Google Scholar]
- Bhatnagar, V.M. The melting points of synthetic apatites. Miner. Mag. 1969, 37, 527–528. [Google Scholar] [CrossRef]
- Ma, J.; Ge, X.; Pelate, N.; Lei, S. Numerical investigation of two-dimensional thermally assisted ductile regime milling of nanocrystalline hydroxyapatite bioceramic material. Ceram. Int. 2015, 41, 3409–3419. [Google Scholar] [CrossRef]
- Bulina, N.V.; Baev, S.G.; Makarova, S.V.; Vorobyev, A.M.; Titkov, A.I.; Bessmeltsev, V.P.; Lyakhov, N.Z. Selective Laser Melting of Hydroxyapatite: Perspectives for 3D Printing of Bioresorbable Ceramic Implants. Materials 2021, 14, 5425. [Google Scholar] [CrossRef] [PubMed]
- Bulina, N.V.; Rybin, D.K.; Makarova, S.V.; Dudina, D.V.; Batraev, I.S.; Utkin, A.V.; Prosanov, I.Y.; Khvostov, M.V.; Ulianitsky, V.Y. Detonation Spraying of Hydroxyapatite on a Titanium Alloy Implant. Materials 2021, 14, 4852. [Google Scholar] [CrossRef]
- Trombe, J.; Montel, G. Some features of the incorporation of oxygen in different oxidation states in the apatitic lattice—I On the existence of calcium and strontium oxyapatites. J. Inorg. Nucl. Chem. 1978, 40, 15–21. [Google Scholar] [CrossRef]
- Bessmeltsev, V.P.; Goloshevsky, N.V.; Smirnov, K.K. Specific features of controlling laser systems for micromachining of moving carriers. Optoelectron. Instrum. Data Process. 2010, 46, 79–86. [Google Scholar] [CrossRef]
- Raynaud, S.; Champion, E.; Bernache-Assollant, D.; Thomas, P. Calcium phosphate apatites with variable Ca/P atomic ratio I. Synthesis, characterisation and thermal stability of powders. Biomaterials 2002, 23, 1065–1072. [Google Scholar] [CrossRef]
- Montel, G.; Bonel, G.; Heughebaert, J.; Trombe, J.; Rey, C. New concepts in the composition, crystallization and growth of the mineral component of calcified tissues. J. Cryst. Growth 1981, 53, 74–99. [Google Scholar] [CrossRef]
- Dorozhkin, S. Solid-Phase Conversion of Nonstoichiometric Hydroxoapatite into Two-Phase Calcium Phosphate. Russ. J. Appl. Chem. 2002, 75, 1897–1902. [Google Scholar] [CrossRef]
- Champion, E. Sintering of calcium phosphate bioceramics. Acta Biomater. 2013, 9, 5855–5875. [Google Scholar] [CrossRef]
- Alberius-Henning, P.; Adolfsson, E.; Grins, J.; Fitch, A. Triclinic oxy-hydroxyapatite. J. Mater. Sci. 2001, 36, 663–668. [Google Scholar] [CrossRef]
- Gross, K.; Berndt, C.; Stephens, P.; Dinnebier, R. Oxyapatite in hydroxyapatite coatings. J. Mater. Sci. 1998, 33, 3985–3991. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphate deposits: Preparation, properties and biomedical applications. Mater. Sci. Eng. C 2015, 55, 272–326. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal System | Space Group | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | Reference |
|---|---|---|---|---|---|---|---|---|
| HA | ||||||||
| Hexagonal | P63/m | 9.4302 | 9.4302 | 6.8911 | 90 | 90 | 120 | [9,11] |
| 9.4176 | 9.4176 | 6.8814 | 90 | 90 | 120 | [1] | ||
| Monoclinic | P21/b | 9.4176 | 2a | 6.8804 | 90 | 90 | 120 | [1] |
| 9.4214 | 2a | 6.881 | 90 | 90 | 120 | [9] | ||
| 9.84214 | 2a | 6.8814 | 90 | 90 | 120 | [11] | ||
| OA | ||||||||
| Hexagonal | P63/m | 9.40 | 9.40 | 6.89 | 90 | 90 | 120 | [1] |
| 9.432 | 9.432 | 6.881 | 90.3 | 90 | 119.9 | [3,9,11] | ||
| Ca/P Ratio | Content (wt%) | Structural Parameters of the HA Phase | Relative Density (%) | ||||
|---|---|---|---|---|---|---|---|
| HA | β-Ca3(PO4)2 | CaO | a (Å) | c (Å) | Crystallite Size (nm) | ||
| Before sintering | |||||||
| 1.6 | 100 | – | – | 9.4394(10) | 6.8867(7) | 25.1(2) | 63.9 |
| 1.667 | 100 | – | – | 9.4336(12) | 6.8932(9) | 29.0(4) | 64.9 |
| 1.7 | 100 | – | – | 9.4374(16) | 6.8929(10) | 20.0(2) | 63.9 |
| After sintering at 1000 °C, 2 h | |||||||
| 1.6 | 72 | 28 | – | 9.4249(6) | 6.8813(5) | 99(2) | 77.1 |
| 1.667 | 100 | – | – | 9.4240(2) | 6.8817(1) | 243(5) | 85.6 |
| 1.7 | 99 | – | 1 | 9.4230(2) | 6.8814(2) | 209(4) | 76.9 |
| Temperature (°C) | Content (wt%) | Structural Parameters of the HA Phase | Pycnometric Density (g/cc) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| α-Ca3(PO4)2 | Ca4O(PO4)2 | CaO | HA | a (Å) | c (Å) | Crystallite Size (nm) | Strain | ||
| HA powder, duration of sintering 10 min | |||||||||
| 1100 | – | – | – | 100 | 9.4250(1) | 6.8821(1) | 317(8) | – | – |
| 1200 | – | – | – | 100 | 9.4233(2) | 6.8831(1) | 551(20) | – | – |
| 1300 | – | – | – | 100 | 9.4196(1) | 6.8869(1) | 768(66) | 0.048(4) | – |
| 1400 | 3 | – | – | 97 | 9.4179(2) | 6.8867(2) | 252(12) | 0.057(7) | – |
| 1500 | 26 | 14 | – | 60 | 9.4172(6) | 6.8861(6) | 139(8) | 0.19(1) | – |
| HA powder, duration of sintering 2 h | |||||||||
| 1000 | – | – | – | 100 | 9.4232(2) | 6.8822(2) | 183(6) | – | – |
| 1100 | – | – | – | 100 | 9.4227(3) | 6.8845(1) | 405(20) | – | – |
| 1200 | – | – | – | 100 | 9.4215(1) | 6.8862(1) | 762(50) | 0.024(4) | – |
| 1300 | – | – | – | 100 | 9.4180(1) | 6.8885(1) | 425(26) | 0.053(5) | – |
| 1400 | 8 | – | – | 92 | 9.4182(1) | 6.8878(2) | 372(20) | 0.081(4) | – |
| 1500 | 43 | 22 | – | 35 | 9.4181(1) | 6.8878(1) | 111(9) | 0.22(1) | – |
| HA pellet, duration of sintering 10 min | |||||||||
| 1100 | – | – | – | 100 | 9.4237(1) | 6.8820(1) | 391(20) | – | 3.068(7) |
| 1200 | – | – | – | 100 | 9.4224(3) | 6.8828(2) | 349(24) | 0.122(6) | 3.126(19) |
| 1300 | – | – | – | 100 | 9.4181(2) | 6.8877(2) | 495(36) | 0.118(4) | 3.169(6) |
| 1400 | 2 | – | – | 98 | 9.4118(6) | 6.8903(4) | 441(58) | 0.330(9) | 3.117(19) |
| 1500 | 19 | – | – | 81 | 9.4170(8) | 6.8877(5) | 225(23) | 0.33(1) | 2.905(6) |
| HA pellet, duration of sintering 2 h | |||||||||
| 1500 | 50 | 14 | 1 | 35 | 9.4094(12) | 6.8811(14) | 304(70) | 0.34(4) | 2.909(6) |
| Heat Conditions | Sample State | a (Å) | c (Å) | V (Å3) | Crystallite Size (nm) | Strain |
|---|---|---|---|---|---|---|
| Heating up to 1100 °C in air, cooling in air | Pellet | 9.41663(3) | 6.88542(3) | 528.752(4) | 542(10) | 0.025(1) |
| Heating up to 1100 °C in air, cooling in He flow | Pellet | 9.41497(6) | 6.88443(5) | 528.490(8) | 321(5) | 0.101(1) |
| Selective laser melting | Pellet | 9.4139(8) | 6.8902(6) | 528.82(9) | 197(18) | 0.35(2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bulina, N.V.; Makarova, S.V.; Baev, S.G.; Matvienko, A.A.; Gerasimov, K.B.; Logutenko, O.A.; Bystrov, V.S. A Study of Thermal Stability of Hydroxyapatite. Minerals 2021, 11, 1310. https://doi.org/10.3390/min11121310
Bulina NV, Makarova SV, Baev SG, Matvienko AA, Gerasimov KB, Logutenko OA, Bystrov VS. A Study of Thermal Stability of Hydroxyapatite. Minerals. 2021; 11(12):1310. https://doi.org/10.3390/min11121310
Chicago/Turabian StyleBulina, Natalia V., Svetlana V. Makarova, Sergey G. Baev, Alexander A. Matvienko, Konstantin B. Gerasimov, Olga A. Logutenko, and Vladimir S. Bystrov. 2021. "A Study of Thermal Stability of Hydroxyapatite" Minerals 11, no. 12: 1310. https://doi.org/10.3390/min11121310