
Adsorption of Strontium onto Synthetic Iron(III) Oxide up to High Ionic Strength Systems

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Surface Titrations and Zeta Potentials Measurements
2.3. Batch Adsorption Studies
2.4. Speciation Calculations and Surface Complexation Modelling
3. Results
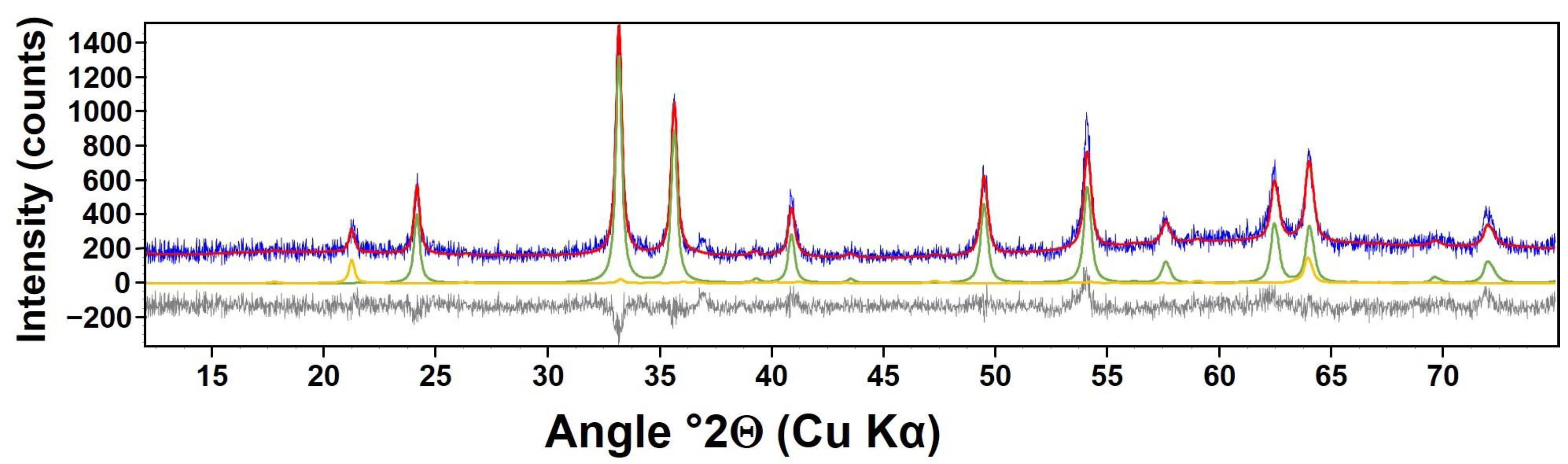
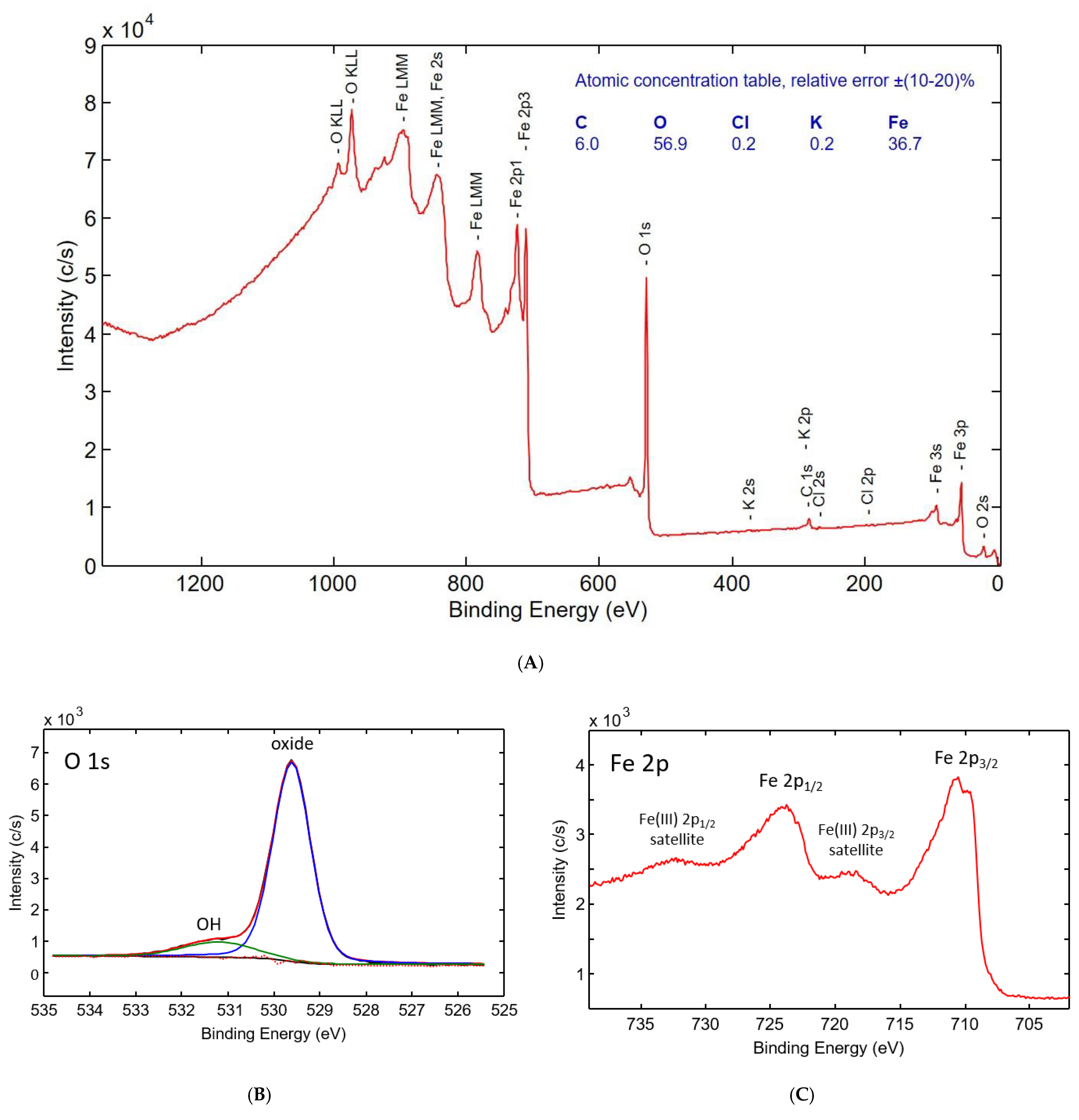
3.1. Solid Characterization
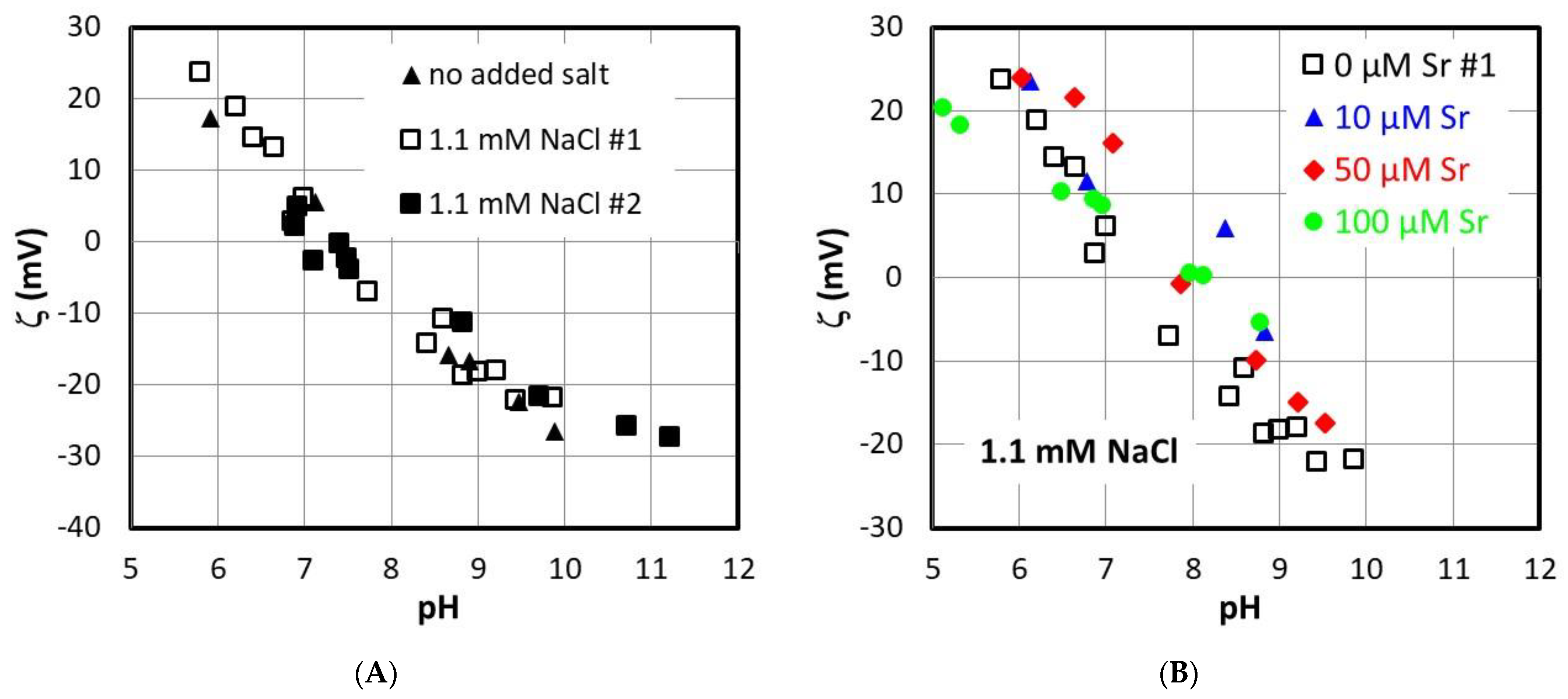
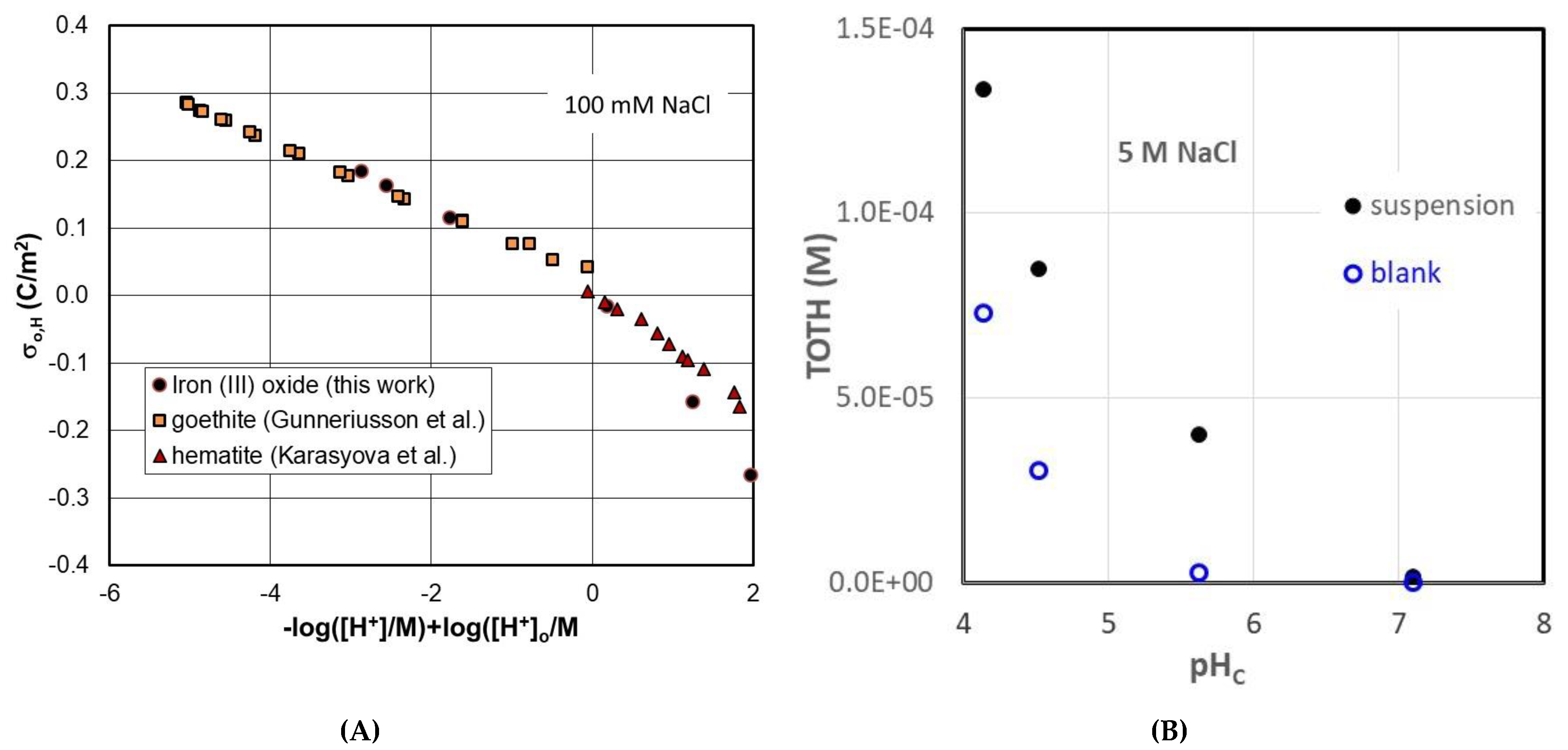
3.2. Electrokinetics and Surface Titrations
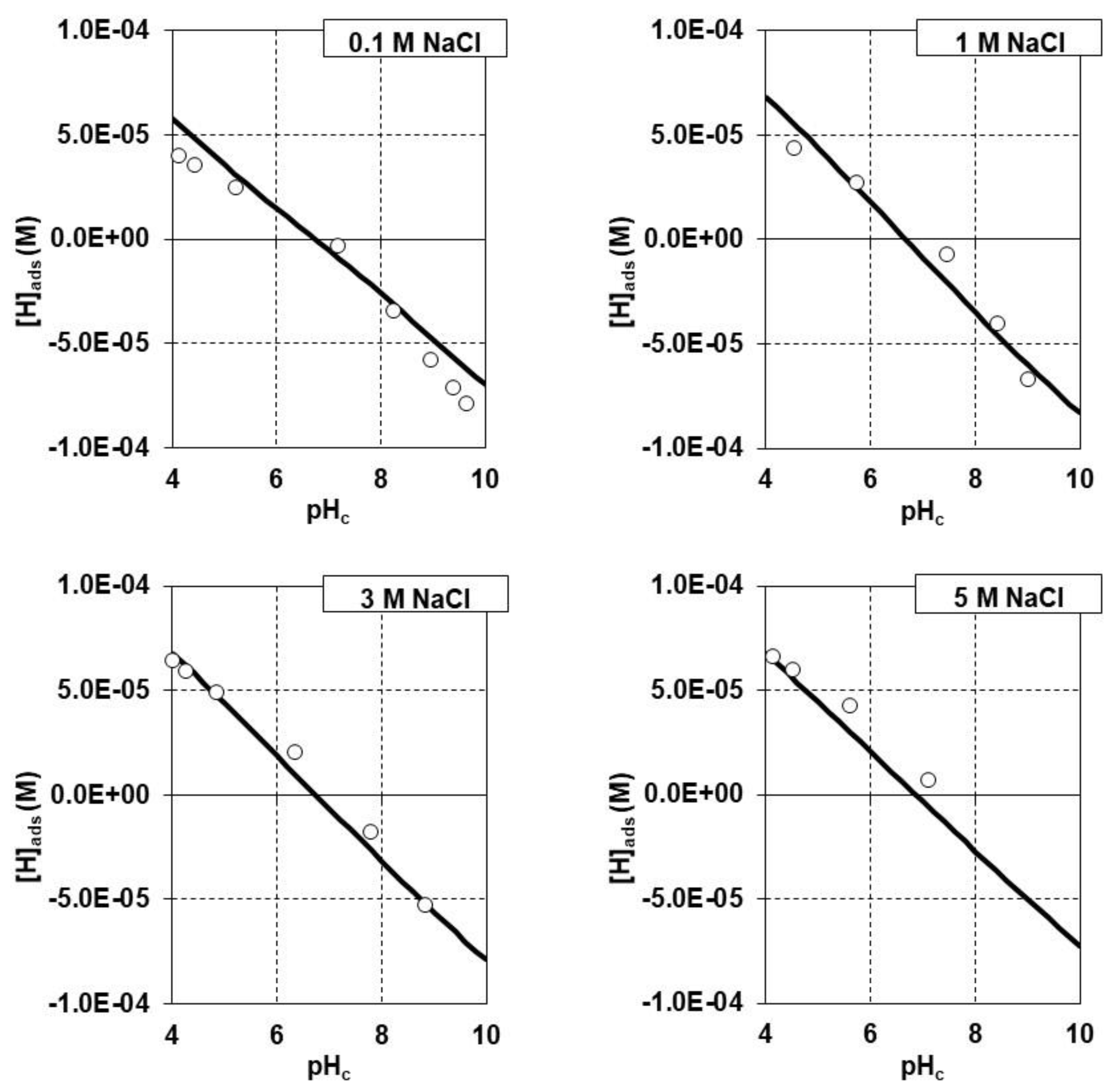
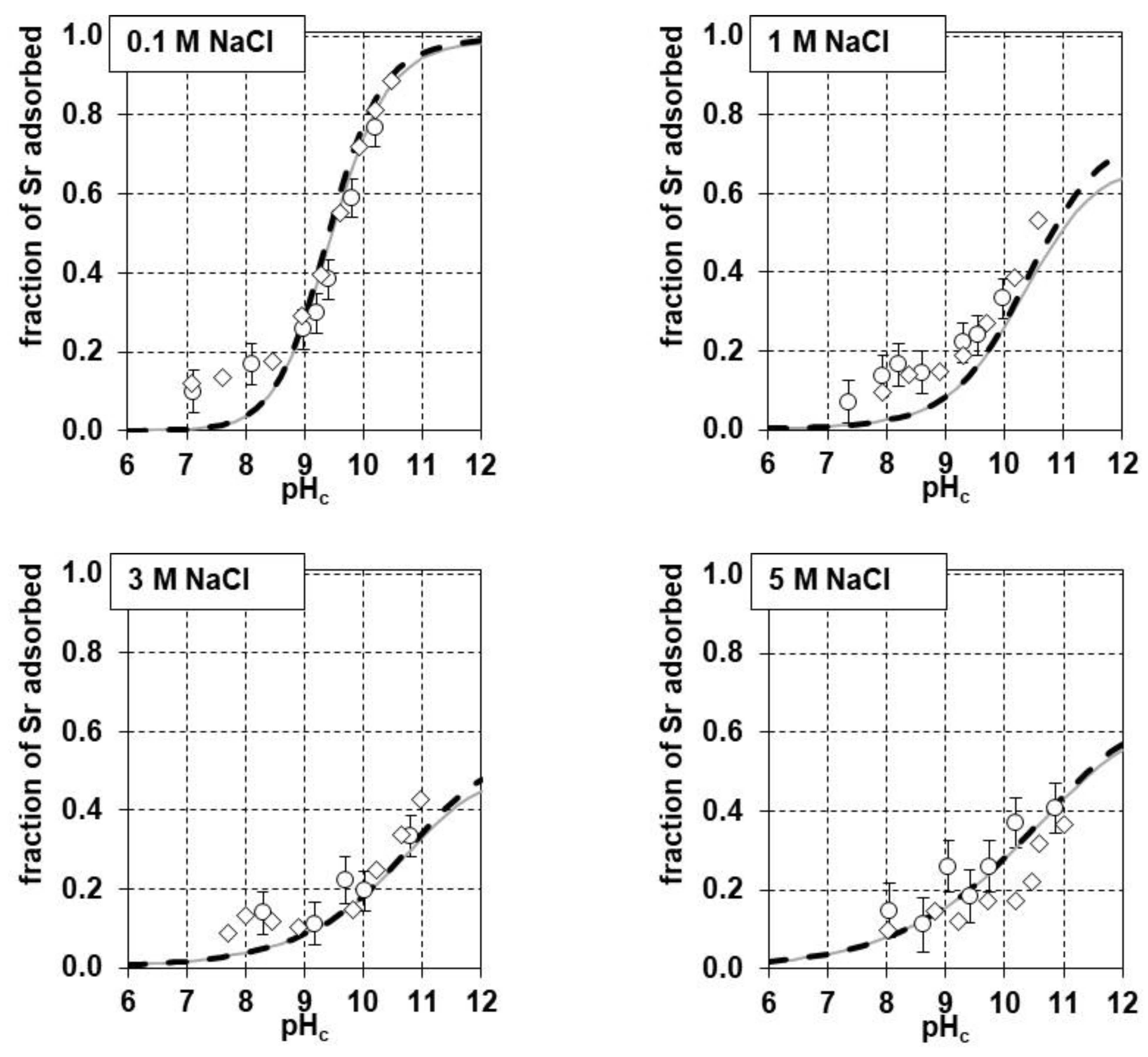
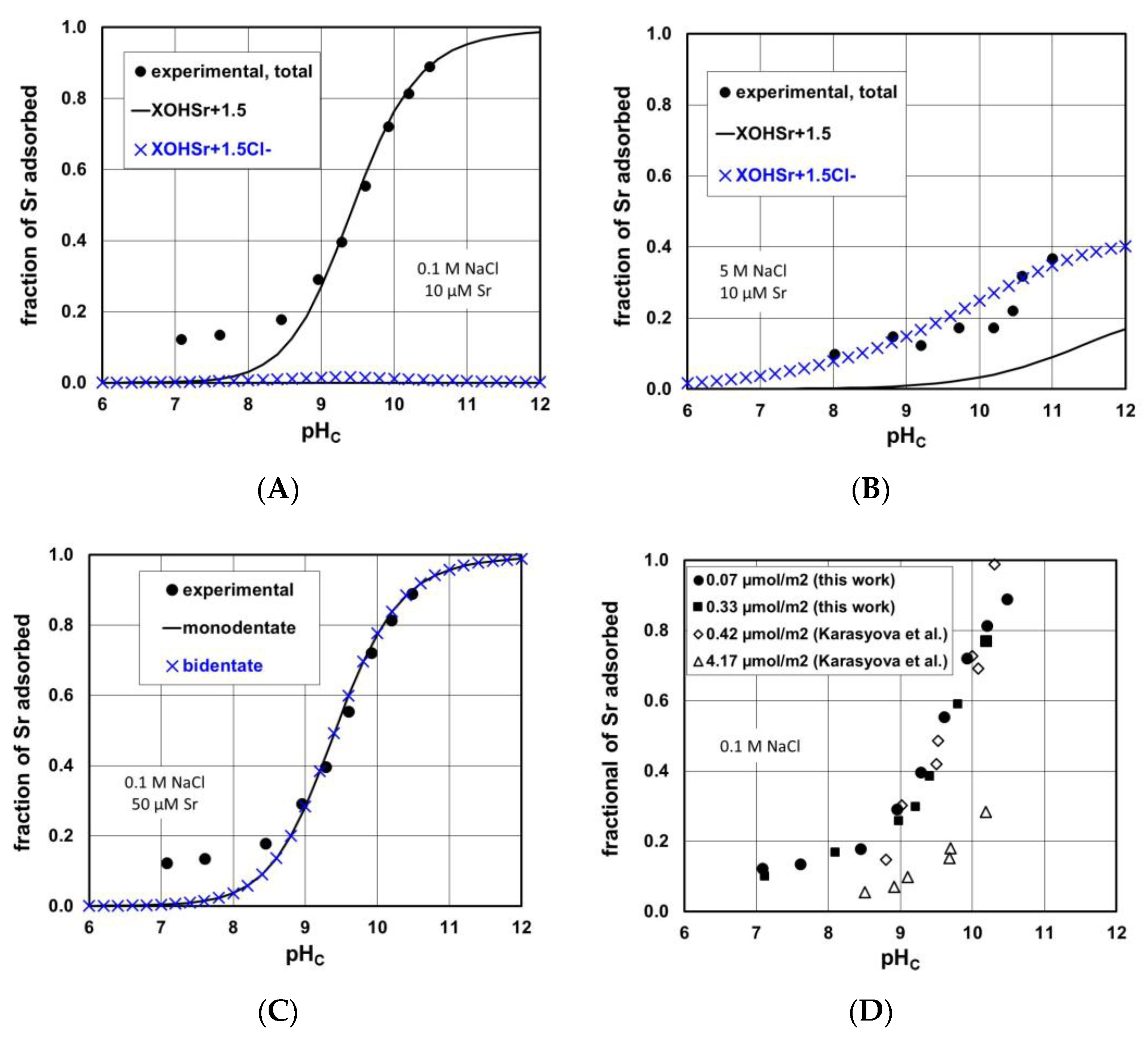
3.3. Sr Adsorption Experiments on the Synthetic Iron(III) Oxide
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IAEA. Geological Disposal Facilities for Radioactive Waste; International Atomic Energy Agency: Vienna, Austria, 2011. [Google Scholar]
- Alley, W.M.; Alley, R. The growing problem of stranded used nuclear fuel. Environ. Sci. Technol. 2014, 48, 2091–2096. [Google Scholar] [CrossRef]
- Birkholzer, J.; Houseworth, J.; Tsang, C.-F. Geologic disposal of high-level radioactive waste: Status, key issues, and trends. Annu. Rev. Environ. Resour. 2012, 37, 79–106. [Google Scholar] [CrossRef]
- Direito, S.O.L.; Clark, S.; Cousins, C.; Fujita, Y.; Gluyas, J.; Harley, S.; Holmes, R.J.; Hutchinson, I.B.; Kudryavtsev, V.A.; Lloyd, J.; et al. Geological repositories: Scientific priorities and potential high-technology transfer from the space and physics sectors. Mineral. Mag. 2018, 79, 1651–1664. [Google Scholar] [CrossRef] [Green Version]
- Mallants, D.; Travis, K.; Chapman, N.; Brady, P.V.; Griffiths, H. The state of the science and technology in deep borehole disposal of nuclear waste. Energies 2020, 13, 833. [Google Scholar] [CrossRef] [Green Version]
- Payer, J.H.; Finsterle, S.; Apps, J.A.; Muller, R.A. Corrosion performance of engineered barrier system in deep horizontal drillholes. Energies 2019, 12, 1491. [Google Scholar] [CrossRef] [Green Version]
- Morelová, N.; Finck, N.; Lützenkirchen, J.; Schild, D.; Dardenne, K.; Geckeis, H. Sorption of americium/europium onto magnetite under saline conditions: Batch experiments, surface complexation modelling and X-ray absorption spectroscopy study. J. Colloid Interface Sci. 2020, 561, 708–718. [Google Scholar] [CrossRef]
- González-Robles, E.; Serrano-Purroy, D.; Sureda, R.; Casas, I.; de Pablo, J. Dissolution experiments of commercial PWR (52 MWd/kgU) and BWR (53 MWd/kgU) spent nuclear fuel cladded segments in bicarbonate water under oxidizing conditions. Experimental determination of matrix and instant release fraction. J. Nucl. Mater. 2015, 465, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Kienzler, B.; Duro, L.; Lemmens, K.; Metz, V.; De Pablo, J.; Valls, A.; Wegen, D.H.; Johnson, L.; Spahiu, K. Summary of the Euratom Collaborative Project FIRST-Nuclides and conclusions for the safety case. Nucl. Technol. 2017, 198, 260–276. [Google Scholar] [CrossRef]
- Labunska, I.; Levchuk, S.; Kashparov, V.; Holiaka, D.; Yoschenko, L.; Santillo, D.; Johnston, P. Current radiological situation in areas of Ukraine contaminated by the Chernobyl accident: Part 2. Strontium-90 transfer to culinary grains and forest woods from soils of Ivankiv district. Environ. Int. 2021, 146, 106282. [Google Scholar] [CrossRef]
- Mangano, J.J.; Sherman, J.D. Elevated in vivo strontium-90 from nuclear weapons test fallout among cancer decedents: A case-control study of deciduous teeth. Int. J. Health Serv. 2011, 41, 137–158. [Google Scholar] [CrossRef]
- Clark, M.W.; Harrison, J.J.; Payne, T.E.; Comarmond, M.J.; Thiruvoth, S.; Wong, H.; Shahin, L.M. Radium and strontium binding by a modified bauxite refinery residue-isotope exchange studies of pH-dependence, reversibility and ageing. Geochem. Explor. Environ. Anal. 2020, 20, 257–267. [Google Scholar] [CrossRef]
- Bots, P.; Comarmond, M.J.; Payne, T.E.; Gückel, K.; Lunn, R.J.; Rizzo, L.; Schellenger, A.E.P.; Renshaw, J.C. Emerging investigator series: A holistic approach to multicomponent EXAFS: Sr and Cs complexation in clayey soils. Environ. Sci. Process. Impacts 2021, 23, 1101–1115. [Google Scholar] [CrossRef]
- Dzombak, D.A.; Morel, F.M.M. Surface Complexation Modeling: Hydrous Ferric Oxide; John Wiley & Sons: Hoboken, NJ, USA, 1990. [Google Scholar]
- Hayes, K.F.; Leckie, J.O. Modeling ionic strength effects on cation adsorption at hydrous oxide/solution interfaces. J. Colloid Interface Sci. 1987, 115, 564–572. [Google Scholar] [CrossRef]
- Montoya, V.; Baeyens, B.; Glaus, M.; Kupcik, T.; Fernandes, M.M.; Van Laer, L.; Bruggeman, C.; Maes, N.; Schäfer, T. Sorption of Sr, Co and Zn on illite: Batch experiments and modelling including Co in-diffusion measurements on compacted samples. Geochim. Cosmochim. Acta 2018, 223, 1–20. [Google Scholar] [CrossRef]
- Missana, T.; García-Gutiérrez, M. Adsorption of bivalent ions (Ca(II), Sr(II) and Co(II)) onto FEBEX bentonite. Phys. Chem. Earth 2007, 32, 559–567. [Google Scholar] [CrossRef]
- King, F.; Hall, D.S.; Keech, P.G. Nature of the near-field environment in a deep geological repository and the implications for the corrosion behaviour of the container. Corros. Eng. Sci. Technol. 2017, 52, 25–30. [Google Scholar] [CrossRef]
- Entwicklungsgemeinschaft, T. F + E-Programm zur Eignungsprüfung der Schachtanlage Konrad für die Einlagerung radioaktiver Abfälle: Zusammenfassender Zwischenbericht <GSF T 114>; Ergebnisse des Untersuchungszeitraumes 1.1.1975 bis 30.6.1980; Ges. f. Strahlen- u.; Umweltforschung: München, Germany, 1980. [Google Scholar]
- Neck, V.; Altmaier, M.; Rabung, T.; Lützenkirchen, J.; Fanghänel, T. Thermodynamics of trivalent actinides and neodymium in NaCl, MgCl2, and CaCl2 solutions: Solubility, hydrolysis, and ternary Ca-M (III)-OH complexes. Pure Appl. Chem. 2009, 81, 1555–1568. [Google Scholar] [CrossRef]
- Rafferty, P.; Shiao, S.Y.; Binz, C.M.; Meyer, R.E. Adsorption of Sr(II) on clay minerals: Effects of salt concentration, loading, and pH. J. Inorg. Nucl. Chem. 1981, 43, 797–805. [Google Scholar] [CrossRef]
- Mahoney, J.J.; Langmuir, D. Adsorption of Sr on kaolinite, illite and montmorillonite at high ionic strengths. Radiochim. Acta 1991, 54, 139–144. [Google Scholar] [CrossRef]
- Bolt, G.H. Determination of the charge density of silica sols. J. Phys. Chem. 1957, 61, 1166–1169. [Google Scholar] [CrossRef]
- Pivovarov, S. Diffuse sorption modeling. J. Colloid Interface Sci. 2009, 332, 54–59. [Google Scholar] [CrossRef]
- Altmaier, M.; Metz, V.; Neck, V.; Müller, R.; Fanghänel, T. Solid-liquid equilibria of Mg(OH)2(cr) and Mg2(OH)3Cl·4H2O(cr) in the system Mg-Na-H-OH-Cl-H2O at 25 °C. Geochim. Cosmochim. Acta 2003, 67, 3595–3601. [Google Scholar] [CrossRef]
- Wiesner, A.D.; Katz, L.E.; Chen, C.-C. The impact of ionic strength and background electrolyte on pH measurements in metal ion adsorption experiments. J. Colloid Interface Sci. 2006, 301, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, A.; Marsac, R.; Rabung, T.; Lützenkirchen, J.; Geckeis, H. Sorption of Cm(III) and Eu(III) onto clay minerals under saline conditions: Batch adsorption, laser-fluorescence spectroscopy and modeling. Geochim. Cosmochim. Acta 2015, 151, 192–202. [Google Scholar] [CrossRef]
- García, D.; Lützenkirchen, J.; Petrov, V.; Siebentritt, M.; Schild, D.; Lefèvre, G.; Rabung, T.; Altmaier, M.; Kalmykov, S.; Duro, L.; et al. Sorption of Eu(III) on quartz at high salt concentrations. Colloids Surf. A Physicochem. Eng. Asp. 2019, 578, 123610. [Google Scholar] [CrossRef] [Green Version]
- Banik, N.L.; Marsac, R.; Lützenkirchen, J.; Marquardt, C.M.; Dardenne, K.; Rothe, J.; Bender, K.; Geckeis, H. Neptunium sorption and redox speciation at the illite surface under highly saline conditions. Geochim. Cosmochim. Acta 2017, 215, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Marsac, R.; Lal Banik, N.; Lützenkirchen, J.; Diascorn, A.; Bender, K.; Marquardt, C.M.; Geckeis, H. Sorption and redox speciation of plutonium at the illite surface under highly saline conditions. J. Colloid Interface Sci. 2017, 485, 59–64. [Google Scholar] [CrossRef]
- Pitzer, K.S. Thermodynamics of electrolytes. 1. theoretical basis and general equations. J. Phys. Chem. 1973, 77, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Brönsted, J.N. Studies on solubility. IV The principle of the specific interaction of ions. J. Am. Chem. Soc. 1922, 44, 877–898. [Google Scholar] [CrossRef] [Green Version]
- Scatchard, G. Concentrated solutions of strong electrolytes. Chem. Rev. 1936, 19, 309–327. [Google Scholar] [CrossRef]
- Guggenheim, E.A. Applications of Statistical Mechanics; Clarendon, P.: Oxford, UK, 1966. [Google Scholar]
- Lützenkirchen, J. Surface complexation models of adsorption: A critical survey in the context of experimental data. In Adsorption: Theory, Modeling, and Analysis; Marcel Dekker, Inc.: New York, NY, USA, 2002; pp. 631–710. [Google Scholar]
- Barrón, V.; Torrent, J. Surface hydroxyl configuration of various crystal faces of hematite and goethite. J. Colloid Interface Sci. 1996, 177, 407–410. [Google Scholar] [CrossRef]
- Maslen, E.; Streltsov, V.; Streltsova, N.; Ishizawa, N. Synchrotron X-ray study of the electron density in α-Fe2O3. Acta Crystallogr. Sect. B Struct. Sci. 1994, 50, 435–441. [Google Scholar] [CrossRef]
- Hazemann, J.-L.; Berar, J.; Manceau, A. Rietveld studies of the aluminium-iron substitution in synthetic goethite. In Materials Science Forum; Trans Tech Publications: Bäch, Switzerland, 1991; pp. 821–826. [Google Scholar]
- Seah, M.P.; Gilmore, I.S.; Beamson, G. XPS: Binding energy calibration of electron spectrometers 5—Re-evaluation of the reference energies. Surf. Interface Anal. 1998, 26, 642–649. [Google Scholar] [CrossRef]
- Moulder, J.; Stickle, W.; Sobol, W.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy; Chastain, J., King, R.C., Jr., Eds.; Ulvac-Phi Inc.: Chigasaki, Japan; Physical Electronics Inc.: Chanhassaen, MN, USA, 1995. [Google Scholar]
- Bethke, C.; Yeakel, S. Geochemist’s Workbench: Release 8.0 Reaction Modeling Guide; RockWare Incorporated: Golden, CO, USA, 2009. [Google Scholar]
- Baes, C.F.; Mesmer, R.E. The Hydrolysis of Cations; John Wiley & Sons: Hoboken, NJ, USA, 1976. [Google Scholar]
- Parkhurst, D.L.; Appelo, C. Description of Input and Examples for Phreeqc Version 3: A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; US Geological Survey: Reston, VI, USA, 2013; pp. 2328–7055. [Google Scholar]
- Hiemstra, T.; Van Riemsdijk, W.H. Effect of different crystal faces on experimental interaction force and aggregation of hematite. Langmuir 1999, 15, 8045–8051. [Google Scholar] [CrossRef]
- Venema, P.; Hiemstra, T.; Weidler, P.G.; van Riemsdijk, W.H. Intrinsic proton affinity of reactive surface groups of metal (Hydr)oxides: Application to Iron (Hydr)oxides. J. Colloid Interface Sci. 1998, 198, 282–295. [Google Scholar] [CrossRef]
- Livi, K.J.T.; Villalobos, M.; Leary, R.; Varela, M.; Barnard, J.; Villacís-García, M.; Zanella, R.; Goodridge, A.; Midgley, P. Crystal Face distributions and surface site densities of two synthetic goethites: Implications for Adsorption capacities as a function of particle size. Langmuir 2017, 33, 8924–8932. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, T.; Van Riemsdijk, W.H. On the relationship between charge distribution, surface hydration, and the structure of the interface of metal hydroxides. J. Colloid Interface Sci. 2006, 301, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Poeter, E.P.; Hill, M.C. Documentation of UCODE. In A Computer Code for Universal Inverse Modeling; DIANE Publishing: Darby, PA, USA, 1998; pp. 98–4080. [Google Scholar]
- Westall, J.C.; Chemistry, O.S.U.D.O. FITEQL: A Computer Program for Determination of Chemical Equilibrium Constants from Experimental Data; Department of Chemistry, Oregon State University: Corvallis, OR, USA, 1982. [Google Scholar]
- Huber, F.; Schild, D.; Vitova, T.; Rothe, J.; Kirsch, R.; Schäfer, T. U(VI) removal kinetics in presence of synthetic magnetite nanoparticles. Geochim. Cosmochim. Acta 2012, 96, 154–173. [Google Scholar]
- Kosmulski, M. Chemical properties of material surfaces; Schick, M.J., Hubbard, A.T., Eds.; Surfactant Science Series; Marcel Dekker, Inc.: New York, NY, USA, 2001; Volume 102, ISBN 0-8247-0560-2. [Google Scholar]
- Tschapek, M.; Wasowski, C.; Torres Sanchez, R.M. The p.z.c. and i.e.p. of γ-Al2O3 and TiO2. J. Electroanal. Chem. Interfacial Electrochem. 1976, 74, 167–176. [Google Scholar] [CrossRef]
- Gunneriusson, L.; Lövgren, L.; Sjöberg, S. Complexation of Pb(II) at the goethite (α-FeOOH)/water interface: The influence of chloride. Geochim. Cosmochim. Acta 1994, 58, 4973–4983. [Google Scholar] [CrossRef]
- Karasyova, O.N.; Ivanova, L.I.; Lakshtanov, L.Z.; Lövgren, L. Strontium sorption on hematite at elevated temperatures. J. Colloid Interface Sci. 1999, 220, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Lützenkirchen, J.; Boily, J.F.; Gunneriusson, L.; Lövgren, L.; Sjöberg, S. Protonation of different goethite surfaces—unified models for NaNO3 and NaCl media. J. Colloid Interface Sci. 2008, 317, 155–165. [Google Scholar] [CrossRef]
- Nie, Z.; Finck, N.; Heberling, F.; Pruessmann, T.; Liu, C.; Lützenkirchen, J. Adsorption of Selenium and strontium on goethite: EXAFS study and surface complexation modeling of the ternary systems. Environ. Sci. Technol. 2017, 51, 3751–3758. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Giammar, D.E. Mass action expressions for bidentate adsorption in surface complexation modeling: Theory and practice. Environ. Sci. Technol. 2013, 47, 3982–3996. [Google Scholar] [CrossRef]
- Lützenkirchen, J.; Marsac, R.; Kulik, D.A.; Payne, T.E.; Xue, Z.; Orsetti, S.; Haderlein, S.B. Treatment of multi-dentate surface complexes and diffuse layer implementation in various speciation codes. Appl. Geochem. 2015, 55, 128–137. [Google Scholar] [CrossRef]
- Carroll, S.A.; Roberts, S.K.; Criscenti, L.J.; O‘Day, P.A. Surface complexation model for strontium sorption to amorphous silica and goethite. Geochem. Trans. 2008, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Lützenkirchen, J. Evaluation of experimental procedures and discussion of two different modelling approaches with respect to long-term kinetics of metal cation Sorption onto (Hydr)oxide Surfaces. Aquat. Geochem. 2001, 7, 217–235. [Google Scholar] [CrossRef]
- Inan, S.; Tel, H.; Altaş, Y. Sorption studies of strontium on hydrous zirconium dioxide. J. Radioanal. Nucl. Chem. 2006, 267, 615–621. [Google Scholar] [CrossRef]
- Samanta, S.K. Hydrated titanium(IV) oxide as a granular inorganic sorbent for removal of radiostrontium.1. Batch equilibration studies. J. Radioanal. Nucl. Chem. Artic. 1996, 209, 235–242. [Google Scholar] [CrossRef]
- Danačíková, E.; John, J.; Motl, A.; Šebesta, F.; Hooper, E.W. Study of sorption properties of various titanium dioxide materials. Czechoslov. J. Phys. 1999, 49, 789–795. [Google Scholar] [CrossRef]
- Trivedi, P.; Axe, L. A comparison of strontium sorption to hydrous aluminum, iron, and manganese oxides. J. Colloid Interface Sci. 1999, 218, 554–563. [Google Scholar] [CrossRef]
- Mayordomo, N.; Alonso, U.; Missana, T. Effects of γ-alumina nanoparticles on strontium sorption in smectite: Additive model approach. Appl. Geochem. 2019, 100, 121–130. [Google Scholar] [CrossRef]
- Mendez, J.C.; Hiemstra, T. High and low affinity sites of ferrihydrite for metal ion adsorption: Data and modeling of the alkaline-earth ions Be, Mg, Ca, Sr, Ba, and Ra. Geochim. Cosmochim. Acta 2020, 286, 289–305. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | log K0 |
|---|---|
| ≡XOH−1/2 + H+ → ≡XOH2+1/2 | 7.10 a |
| ≡XOH−1/2 + Na+ → ≡XOH−1/2…Na+ | 0.42a |
| ≡XOH2+1/2 + Cl− → ≡XOH2+1/2…Cl− | −0.17a |
| Parameter | Parameter Value |
| Site x (sites·nm−2), fixed at this value | 6.36 |
| Capacitance (F·m−2) | 2.59 |
| (A) For Monodentate Surface Complexes | |
|---|---|
| Reaction | log K0 |
| ≡XOH−1/2 + Sr2+ → ≡XOHSr+3/2 | 1.11 a |
| ≡XOHSr+3/2 + Cl− → ≡XOHSr+3/2…Cl− | 1.05 a,b |
| (B) For Bidentate Surface Complexes | |
| Reaction | log K0 |
| (≡XOH−1/2)2 + Sr2+ → (≡XOH)2Sr+1 | 1.11 c,d |
| (≡XOH)2Sr+1 + Cl− → (≡XOH)2Sr+1…Cl− | 1.04 c,d,e |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García, D.; Lützenkirchen, J.; Huguenel, M.; Calmels, L.; Petrov, V.; Finck, N.; Schild, D. Adsorption of Strontium onto Synthetic Iron(III) Oxide up to High Ionic Strength Systems. Minerals 2021, 11, 1093. https://doi.org/10.3390/min11101093
García D, Lützenkirchen J, Huguenel M, Calmels L, Petrov V, Finck N, Schild D. Adsorption of Strontium onto Synthetic Iron(III) Oxide up to High Ionic Strength Systems. Minerals. 2021; 11(10):1093. https://doi.org/10.3390/min11101093
Chicago/Turabian StyleGarcía, David, Johannes Lützenkirchen, Maximilien Huguenel, Léa Calmels, Vladimir Petrov, Nicolas Finck, and Dieter Schild. 2021. "Adsorption of Strontium onto Synthetic Iron(III) Oxide up to High Ionic Strength Systems" Minerals 11, no. 10: 1093. https://doi.org/10.3390/min11101093