1. Introduction
The linear combination of multiple biomarkers is often used in clinical practice [
1] for disease diagnosis due to its ease of interpretation and performance [
2], which is usually superior to considering each biomarker separately [
3,
4,
5,
6,
7,
8]. These new biomarkers are key for disease screening or understanding the evolution of a disease after diagnosis. As an example, Prostate-specific antigen (PSA) is the most used biomarker to diagnose prostate cancer, although it lacks the necessary sensitivity and specificity. The prostate health index (PHI) and 4Kscore are new biomarkers with greater predictive ability derived from linear models that include PSA [
9].
To assess diagnostic accuracy, statistics derived from the receiver operating characteristic (ROC) curve, such as the area under the ROC curve (AUC) [
10] or the Youden index [
11], are often used. In this context, the development of binary classification model approaches that maximise the AUC has been extensively studied in the literature. Many of these studies have been the basis for the formulation of subsequently published improved approaches under ROC-curve-derived optimality criteria.
Su and Liu [
12] formulated the optimal linear model that maximises the AUC under the assumption of multivariate normality. This normality assumption is often not easy to observe in real clinical practice, being too demanding in part due to the symmetry that biomarkers must meet. For many diseases, the progression or advanced stages of them are associated with high values of the diagnostic tests, so these types of variables tend to follow asymmetric distributions. Results from a prostate cancer screening cohort show a clear asymmetry of PSA in the Canadian population [
13]. This limitation was solved by Pepe et al. [
14,
15], who proposed a distribution-free approach for the estimation of the linear model that maximises AUC based on the Mann–Whitney U-statistic [
16]. This approach is based on discrete optimisation under extensive search on the parameter vector of biomarkers coefficients. Although the statistical foundation underpinning the approach proposed by Pepe et al. has been the basis for subsequent approaches, it has the drawback of being computationally infeasible when the number of biomarkers is greater than or equal to three. To address this computational limitation, Pepe et al. [
14,
15] suggested the use of stepwise algorithms based on selecting and estimating, at each step, the best linear combination of two biomarkers, including at each step a new biomarker. This proposal for partial optimisations at each step was later implemented by Esteban et al. [
17] and Kang et al. [
18]. Esteban et al. included tie-handling strategies, and Kang et al. proposed a simpler and less demanding approach by setting the order of biomarker inclusion at the beginning of the algorithm. Liu et al. [
19] proposed an approach, called the min–max approach, which is computationally tractable regardless of the number of biomarkers. This is because it is based on the linear combination of the minimum and maximum values of biomarkers under the optimisation of the Mann–Whitney U-statistic of the AUC, involving the search for a single optimal coefficient. Despite its computational advantage, it has been shown to generally achieve lower accuracy than other approaches that use information from all biomarkers, such as stepwise approaches, but shows superiority in some scenarios [
3,
4,
18].
In diagnostic or binary classification problems where combinations of continuous biomarkers are estimated, dichotomisation of the resulting continuous value, i.e., establishing a cut-off point, is often key, as it provides a classification rule that allows this classification of patients into groups [
20]. In this sense, the Youden index is a good criterion for choosing the best cut-off point to dichotomise a biomarker [
21] and is an appropriate summary of the performance of the diagnostic model [
22]. For example, the Youden index takes a cut-off value of 45.9 for PHI in nonfused biopsies [
23]. The Youden index maximises the sum of sensitivity and specificity, giving equal weight to both metrics, so that it can be considered as the symmetrical point that maximises both metrics simultaneously.
Therefore, although there are different metrics that provide the optimal cut-off point, in the absence of consensus, with no clear reason to optimise either sensitivity or specificity, the Youden index provides that optimal balance, being the most used parameter to choose a threshold.
Although the area under the ROC curve is the most studied diagnostic assessment statistic in the literature, other statistics such as the Youden index are also used in different clinical studies and provide accurate categorisation. The algorithms under AUC optimality cited above were used as a basis for the formulation of subsequent approaches under Youden index maximisation. Based on the stepwise approach of Kang et al. [
18], Yin and Tian [
24] conducted a study under Youden index optimisation. Aznar-Gimeno et al. [
25] developed the stepwise algorithm suggested by Pepe et al. [
14,
15] under Youden index maximisation and compared its performance with other approaches in the literature, modified under Youden index maximisation, such as Yin and Tian’s stepwise approach [
24], the min–max approach [
19], logistic regression [
26], a parametric method with multivariate normality and a non-parametric kernel smoothing method. Although Aznar-Gimeno et al. demonstrated that their proposed approach achieved acceptable performance, superior in some scenarios, it has the computational limitation of being difficult to approach when the number of biomarkers increases. The min–max approach, which solves this computational problem through the linear combination of the minimum and maximum values, did not prove to be sufficient in terms of discrimination, except in a few specific scenarios.
Maintaining the advantage of not being subject to any distributional assumptions, being computationally tractable regardless of the number of original biomarkers while incorporating more information through a new summary statistic (the median or the interquartile range), Aznar-Gimeno et al. [
27] proposed the so-called min–max–median and min–max-IQR approaches. These approaches are based on estimating the linear combination of these three variables using the proposed stepwise algorithm [
25]. Aznar-Gimeno et al. compared the proposed algorithms with the min–max algorithm and logistic regression. The aim was to compare computationally tractable methods, regardless of the number of biomarkers. In this sense, cancer shows a substantial clinical heterogeneity, and the max–min derived approach tries to capture the potential variation underlying the biological heterogeneity.
Machine learning algorithms have been increasingly used in various fields of application [
28] and, in particular, in clinical practice and medical research [
29,
30,
31,
32,
33,
34], due to their performance potential and efficiency. There are different machine learning and deep learning techniques that have been applied in the area of health from different sources of information covering different formats ranging from numerical data to text or images [
35]. Numerous studies have applied and evaluated these techniques in recent years with different objectives in the healthcare domain [
36], such as predicting events, diagnosing or prognosing diseases or cancers [
37,
38,
39,
40]. Analysing their association with patient biomarkers such as demographic data, clinical data, pharmacology, genetics, medical imaging or wearable sensors, [
41] (among others), is a challenge that needs to be addressed in a way that prevents or detects the disease early.
Deep learning has been used to assist in the identification of genes and associated proteomics and metabolomics profiles to detect cancers at early stages [
42,
43,
44]. Concerning the early detection of breast cancer, Mahesh et al. [
45] evaluated the Naive Bayes classifier, the Decision Tree classifier, Random Forest and their ensembles. Botlagunta et al. [
46] assessed nine machine learning methods for breast cancer metastasis classification, including logistic regression, k-nearest neighbours, decision trees, random forest, gradient boosting, and eXtreme Gradient Boosting (XGBoost) [
47]. Rustam et al. [
48] compared the performance of Support Vector Machine (SVM) and Naive Bayes for prostate cancer patient classification. Huo et al. [
49] also evaluated the effectiveness of machine learning models for prostate cancer prediction, including SVM, decision tree, random forest, XGBoost, and adaptive boosting (Adaboost). Sabbagh et al. [
50] applied logistic regression and XGBoost techniques to the prediction of lymph node metastasis in prostate cancer patients using clinicopathologic features. Khan et al. [
51] propose a self-normalised multiview convolutional neural network model with adaptive boosting (AdaBoost-SNMV-CNN) for lung cancer nodule detection in computed tomography scans. Regarding diabetes, Saheb-Honar et al. [
52] examined the classification ability of logistic regression, decision tree, and random forest in identifying the relationship between type 2 diabetes and its risk factors. Budholiya et al. [
53] present a diagnostic system that employs an optimised XGBoost classifier with the aim of predicting the occurrence of heart disease. Ensemble models, combining machine learning and deep learning approaches, provide personalized patient treatment strategies based on medical histories and diagnostics [
54]. The versatility of deep learning models is clear, with applications for omics data types, as well as histopathology-based genomic inference, providing perspectives on the integration of different data types to develop decision support tools [
55], but few of them have yet demonstrated real-world medical utility [
56].
The primary drawback of one of these algorithms compared to techniques based on linear models is the lack of explainability and interpretability of the models. One of the key reasons why these tools may not be effectively implemented and integrated into routine clinical practice is due to the lack of transparency and explainability of the models. Explainable artificial intelligence (XAI) is attracting much interest in medicine [
57] and, fortunately, in recent years, work has been carried out on the concept of XAI, which provides techniques that also offer explainability and transparency of these models.
XGBoost [
47] is one of the most widely used machine learning algorithms of recent times. This is due to its ease of implementation and good results, proving to be a leader in many competitions and state-of-the-art studies [
58]. The XGBoost algorithm assumes no normality and combines several weak prediction models, which are usually decision trees, improving its predictivity and accuracy. This type of model shows versatility as it depends on some parameters relating to the building trees that can be optimized.
In terms of machine learning algorithms, our work focuses on analysing the predictive capacity of logistic regression and XGBoost. Numerous studies have compared the performance of logistic regression and XGBoost in the health domain in recent years [
59,
60,
61,
62,
63,
64,
65,
66]. Unlike logistic regression or other statistical approaches based on linear models, XGBoost allows capturing non-linear relationships, one of the main reasons for its popularity. However, although XGBoost is an effective tool in healthcare and has been in demand in recent years, demonstrating good performance, it does not always outperform conventional statistical methods such as logistic regression [
62,
66,
67]. The choice of the optimal model will depend on the problem and the type of data. Therefore, it is always necessary to conduct a comprehensive comparative study to analyse the performance of algorithms in different scenarios in order to obtain useful information and establish certain guidelines.
Due to the enormous number of data available nowadays by the advances in technology, it has been shown that it is essential to develop non-parametric biomarker combination models that are computationally tractable, regardless of the number of initial biomarkers. In this sense, our proposed approaches (min–max–median/IQR approach) reduce the dimensional problem by capturing the heterogeneity of the information through summary statistics. Although studies comparing the performance of different machine learning techniques have increased in the literature in recent years, so far, there are no studies comparing the performance of our proposed approaches with machine learning models such as XGBoost, which has been in high demand in recent years and which can capture more complex relationships than the statistical linear methods compared in other studies [
25,
27]. The aim of our work was to compare the performance of our proposed min–max–median/IQR approaches with the min–max approach and the machine learning algorithms known as logistic regression and XGBoost, maximising the Youden index. For this purpose, they were compared on a wide range of simulated symmetric or asymmetric data scenarios, as well as on real clinical diagnostic datasets.
We provide a novel approach based on three main basic characteristics of the set of predictor variables, the maximum, minimum and median or IQR to capture the larger discrimination ability to summarize in these three parameters. On the other hand, from a different perspective, we train and validate additive tree models trying to capture the sum of the predictive ability of all predictor variables. The results of this work provide the reader with useful information that can serve as a guide for the choice of the most suitable algorithm for binary classification problems depending on the characteristics and behaviour of the data.
2. Materials and Methods
This section introduces some notations and the non-parametric approach of Pepe et al. [
14,
15], which forms the basis for our min–max–median/IQR approaches. In the following, we explain our proposed approaches (min–max–median/IQR) and the algorithms with which we compare performance: min–max approach, logistic regression and XGBoost. These algorithms were adapted by optimising the Youden index. Finally, the simulated scenarios and real datasets are detailed, as well as the validation procedure. The entire study was conducted using the free software R (The R Foundation for statistical computing, Vienna, Austria) [
68]. The code of the whole study can be found in
Supplementary Material.
2.1. Background
Consider the following binary classification problem where p is the number of biomarkers, is the number of case individuals (with disease) and is the number of control individuals (healthy individuals). If denotes the value of the variable or biomarker () for the individual of group k = 1, 2 (disease and non-disease), then is the vector of biomarkers for the individual of group k = 1, 2 and and . Therefore, the linear combination of each group is expressed as , where denotes the parameter vector.
Defined in the above notation, by definition, the Youden index (
J) of the linear combination is expressed as:
where
c denotes the cut-off point and
the cumulative distribution function of random variable
. Denoting by
the optimal cut-off point, the expression of the empirical estimate of the Youden index is:
where
I denotes the indicator function.
Pepe et al.’s Approach
Pepe and Thompson [
14] proposed a distribution-free approach (without any distribution assumptions) to estimate the linear model that maximizes the AUC based on the Mann–Whitney U-statistic [
16]. The basis on which their proposed approach lies is mainly in the property of invariance of the ROC curve to any monotonic transformation.
Specifically, Pepe and Thompson propose the following linear model:
where
p denotes the number of biomarkers,
the biomarker
and
the parameter to be estimated. Observe that they did not include an intercept in the linear model (
3), and the coefficient associated with the first variable
is 1. This is because the ROC curves for
(
3) and
are the same, so it is enough to consider (
3). Thus, considering the optimal parameter vector, the maximum empirical AUC based on the Mann–Whitney U statistic would be given by the following expression:
Note that searching the entire possible parameter vector space
and possible coefficient-variable combinations is computationally intractable. To overcome this limitation, Pepe et al. suggested estimating the parameter vector through a discrete optimisation over 201 equally spaced values between −1 and 1. This is because selecting
in
is equivalent to covering the range
since the AUC of
for
and
is the same as
for
. Even so, this optimisation is computationally costly for dimensions
. To address this, Pepe et al. [
14,
15] suggested the use of stepwise algorithms, in which a new variable is included in each step, selecting the best combination of two variables. In this way, the problem is transformed into a computationally tractable problem by estimating a single parameter
times using a linear combination of two variables.
Both the model formulation and the empirical search are the basis for the formulation of the min–max approach and our proposed algorithms (min–max–median/IQR) that extend the min–max approach, which are explained below.
2.2. Min–Max Approach
Liu et al. [
19] proposed the so-called min–max approach (
MM), which is a distribution-free approach, as proposed by Pepe et al. [
14,
15], but with the advantage of being computationally tractable, regardless of the number of original biomarkers. The idea of this approach is to calculate the minimum and maximum values of the
p biomarkers and to consider the optimal linear combination of these two markers, involving the search for a single optimal coefficient. Specifically, the original aim is to estimate the
parameter such as the combination
which maximizes AUC based on the Mann–Whitney U statistic, where
and
are the minimum and maximum values of the original
p biomarkers for each individual, respectively.
Considering the Youden index as our target metric to maximise, the min–max approach can be adapted by selecting the optimal parameter
and cut-off point
that maximises the following expression
where
and
for
and each
, and
, following Pepe et al’s. suggestion of the empirical search of
.
The procedure can be summarised as follows:
- 1.
For each i individual, the biomarkers with minimum and maximum values ( and ) are considered as the new 2 markers (for simplicity, and ).
- 2.
For each of the 201 possible values of , the value of the linear combination () is calculated for each i individual and the optimal cut-off point is chosen, i.e., the one that maximises the Youden index.
- 3.
The linear combination that achieves the highest Youden index is the optimal combination.
2.3. Min–Max–Median/IQR Approach
Aznar-Gimeno et al. proposed new non-parametric approaches, so-called min–max–median (
MMM) and min–max-IQR (
MMIQR) [
27], which extend the idea of the min–max approach by applying our proposed stepwise algorithm [
25], following the suggestion of Pepe et al. [
14,
15]. The aim was to include more information in the model while remaining computationally affordable, although more intensive.
Specifically, the idea behind the approaches is to reduce the dimension of the problem by reducing the number of original p biomarkers to three, considering the summary statistic information of the original variables, i.e., the minimum, maximum, median, or interquartile range (IQR). Our approach extends the min–max approach as it incorporates a new summary statistic, turning the problem into a three-variable linear combination optimisation problem. As suggested by Pepe et al., a stepwise algorithm that we developed is used in this case, where the best linear combination of two variables is selected, including a new variable in each step.
Below, we provide a detailed description of the procedure for the min–max–median approach (note that the min–max-IQR approach follows the same steps).
- 1.
Firstly, for each
i individual, the minimum, maximum, and median values of
p biomarkers are calculated:
where
and
These values are considered as the three new variables (
,
and
, for simplicity). Specifically, from now on, the problem is to estimate the optimal linear combination of these three variables using the proposed stepwise algorithm.
- 2.
The first step of the stepwise approach is to choose the combination(s) of the two variables that maximises the Youden index such that
using empirical search proposed by Pepe et al. In other words, for each variable pair, for each value of the 201 (
values), the linear combination is calculated and the optimal cut-off point that maximises the Youden index is selected. That linear combination for which the optimal cut-off point has obtained the maximum Youden index is chosen in this step. Suppose, for simplicity, the optimal linear combination
.
- 3.
The last step is to include the remaining variable (
) and select the optimal linear combination(s). Specifically, the previously chosen linear combination (
) is considered as a new variable and the idea of the previous point (2) is re-applied. Therefore, either combination (
8) or (
9) that maximizes the Youden index is chosen as the final optimal combination of the linear model.
For ease, a single optimal linear combination is considered in steps 2 and 3. However, the maximum Youden index can be reached for different linear combinations. Our algorithm considers all ties, which can be broken in the last stage (step 3) or not.
Our proposed approaches are openly available to the scientific community through the R library
SLModels [
69]. The library also incorporates the min–max algorithm adapted for the optimisation of the Youden index (previous section).
2.4. Logistic Regression
The logistic regression (
LR) (or logit regression) [
26] is a statistical model that provides the probability of an observation/individual
i belonging to an output category, given its set of independent variables
, through the logistics function:
where
is the vector of parameters to estimate by means of the maximum likelihood method.
2.5. Extreme Gradient Boosting (XGBoost)
XGBoost (eXtreme Gradient Boosting,
XGB) is a scalable tree boosting system that was developed by Chen and Guestrin [
47]. It is a specific optimised implementation of gradient boosting and is therefore based on the principle of sequential order ensemble learning, where errors are minimized (loss function) using a gradient descent algorithm. Specifically, XGBoost is a decision tree ensemble based on the idea of training several weak learners (base learners) sequentially in order to create a strong learner with higher accuracy. During training, the parameters of each weak model are adjusted by minimising the objective function, and each new model is trained to correct the errors of the previous ones. Correctly and incorrectly predicted results receive different scores that are finally weighted to obtain a final result.
The XGBoost algorithm, unlike those presented above, has both parameters and hyperparameters. Hyperparameters are values of model settings that must be set during the training process to control the behaviour and performance of the model.
Considering the XGBoost algorithm as an ensemble base learners of decision trees, the loss function at iteration
t to minimise has the following expression:
where
l is the loss term and
is the regularisation term, which penalizes the complexity of the model, avoiding over-fitting.
indicates the real output,
the prediction of the
individual at the
iterations,
f denotes the base learners,
T the number of leaves of the tree and
the weights of the leaves.
represents the minimum loss reductions needed to split a leaf node of the tree. The larger
is, the more conservative the algorithm will be.
The complexity of the model can also be limited through the maximum-depth hyperparameter, which specifies the maximum number of levels of the tree, where each level represents a division of the data based on a variable. Another possible regularisation hyperparameter is shrinkage, which reduces the step size to make the boosting process more conservative. In other words, it decreases the influence of each individual tree and allows future trees to improve the model. Random subsampling is another regularisation technique that can be used. In the case of a column subsample, the hyperparameter specifies the subsample fraction of columns to be used to construct each tree. The same idea is for rows, where, if the value is less than 1, a random subset of rows (observations/individuals) is selected for each tree.
The XGBoost model was applied using the free software R library xgboost. Specifically, in this study, the following hyperparameters were adjusted over a set of possibilities:
nrounds: Number of decision trees in the final model.
gamma (): Minimum loss reduction required to split a node.
eta (shrinkage, learning rate): Step size shrinkage.
max_depth: Maximum depth of the tree.
colsample_bytree: Subsample ratio of columns.
subsample: Subsample ratio of the training instances.
Table 1 shows the hyperparameter possibilities space explored in the study. The explored values of maximum tree depth for datasets with fewer variables were lower than those with higher dimensions. For the selection of the best combination of hyperparameters, the grid search technique was used, and 5-fold cross-validation was performed on the training set. Finally, the model was trained on the entire training dataset with the selected optimal hyperparameters. The early stopping technique was used as an additional technique to avoid over-fitting by stopping the training if there was no improvement in 10 iterations in a row.
2.6. Simulations
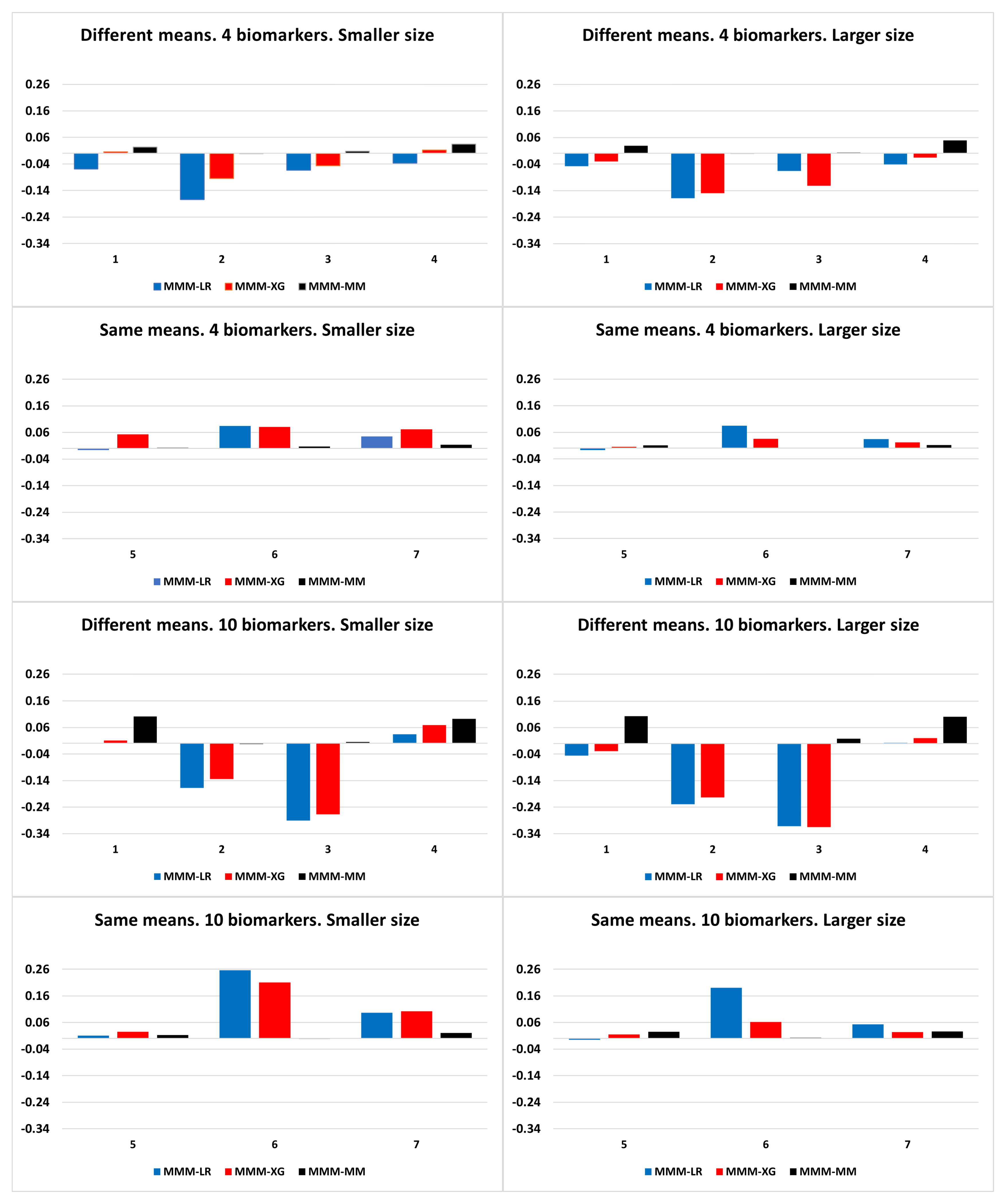
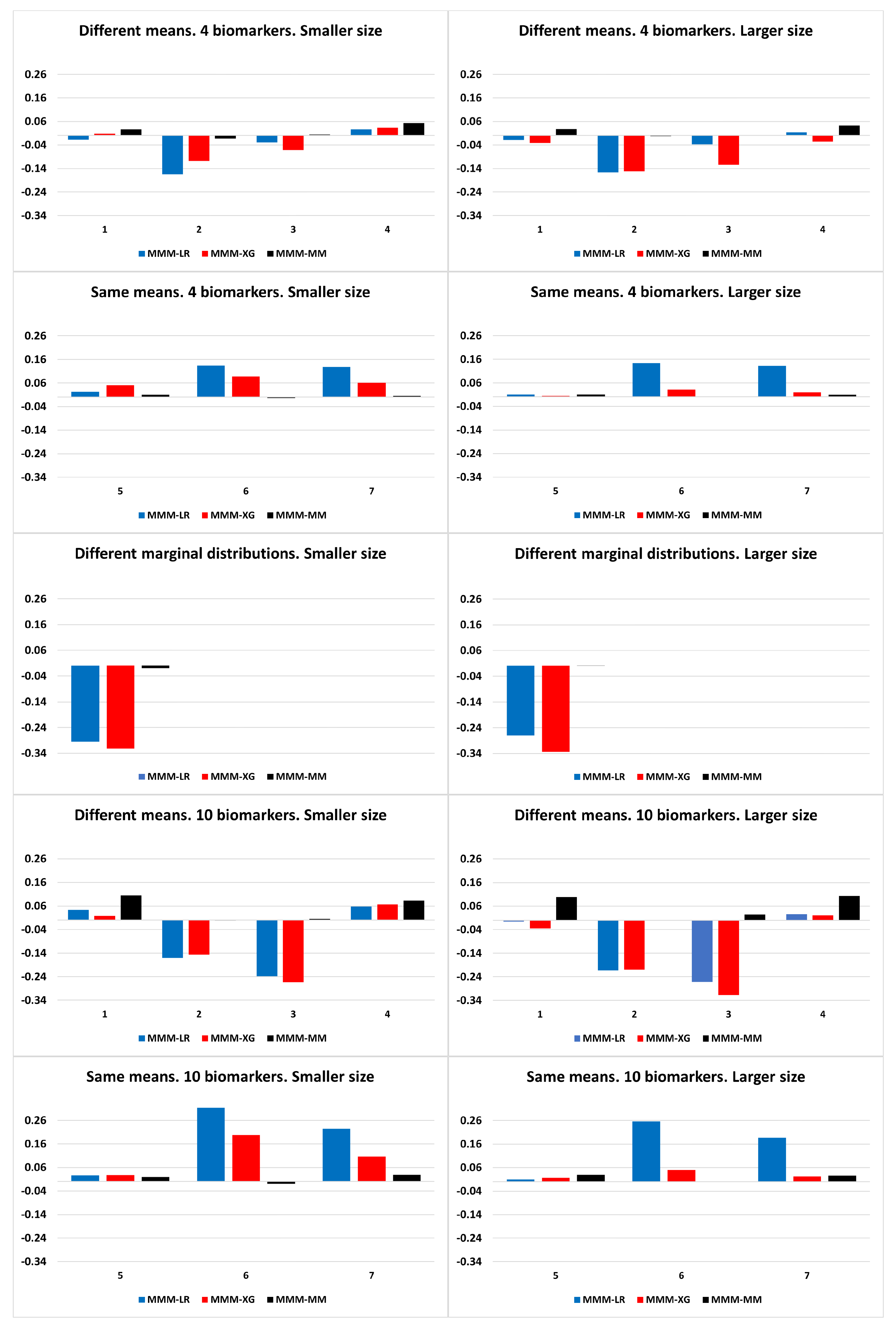
A wide range of simulated data were explored in order to analyse and compare the performance of the algorithms previously discussed. Specifically, scenarios simulating different biomarker distributions, discrimination capabilities, and correlation between them were analysed, considering and biomarkers, and smaller ( = 50) and larger ( = 500) sample sizes. As for the biomarker distributions, both symmetric distributions (normal distributions) and asymmetric distributions (different marginal distributions and multivariate log-normal skewed distribution) were simulated.
The scenarios with different marginal distributions were simulated with 4 biomarkers following chi-square, normal, gamma and exponential distributions via normal copula with a dependence parameter between biomarkers of 0.7 for the case population (patients; diseased population) and 0.3 for the control population (healthy; non-diseased population). More specifically, the biomarkers for the control population were considered to be marginally distributed as , , , and , , , for case population. Scenarios under log-normal distribution were generated from the configurations of the simulated scenarios under a normal distribution and then exponentiated.
Concerning the scenarios of normal distributions, the null vector was considered as the mean vector of the non-diseased population. With respect to the mean vector of the diseased population (), scenarios with the same means , i.e., the same predictive ability, and different means , were explored. For simplicity, the variance of each biomarker was set to 1, so that covariances are equivalent to correlations. The same correlation value was considered for all pairs of biomarkers. Let and be the variance–covariances matrices for diseased and non-diseased populations, respectively. The following scenarios with different biomarker means were analysed:
- –
Independents ().
- –
High correlation ().
- –
Different correlation between groups ().
- –
Negative correlation ().
- –
Low correlation ().
- –
Different correlation between groups ().
- –
Different correlation between groups with biomarkers independents in the non-diseased population ().
2.7. Application in Real Datasets
The methods being examined were also applied in two real clinical datasets: for the diagnosis of Duchenne muscular dystrophy and for maternal mortality risk.
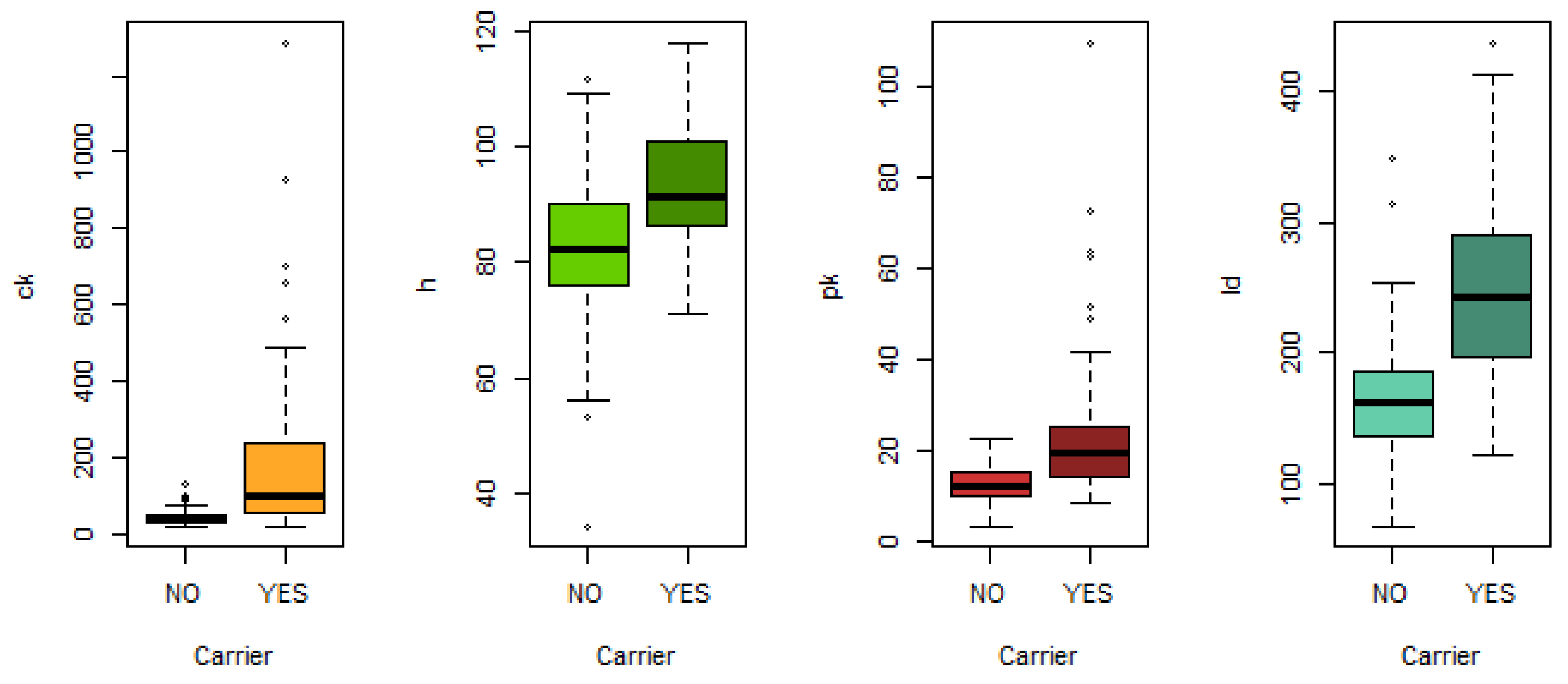
Duchenne muscular dystrophy (DMD) is a genetic disorder passed down from a mother to her children, causing progressive muscle weakness and wasting. Percy et al. [
70] analysed the effectiveness of detecting this disorder using four biomarkers extracted from blood samples: serum creatine kinase (CK), haemopexin (H), pyruvate kinase (PK) and lactate dehydrogenase (LD). The dataset was obtained at
https://hbiostat.org/data/, accessed on 30 January 2023. After removing observations with missing data, the dataset used contains information on the four biomarkers of 67 women who are carriers of the progressive recessive disorder DMD and 127 women who are not carriers.
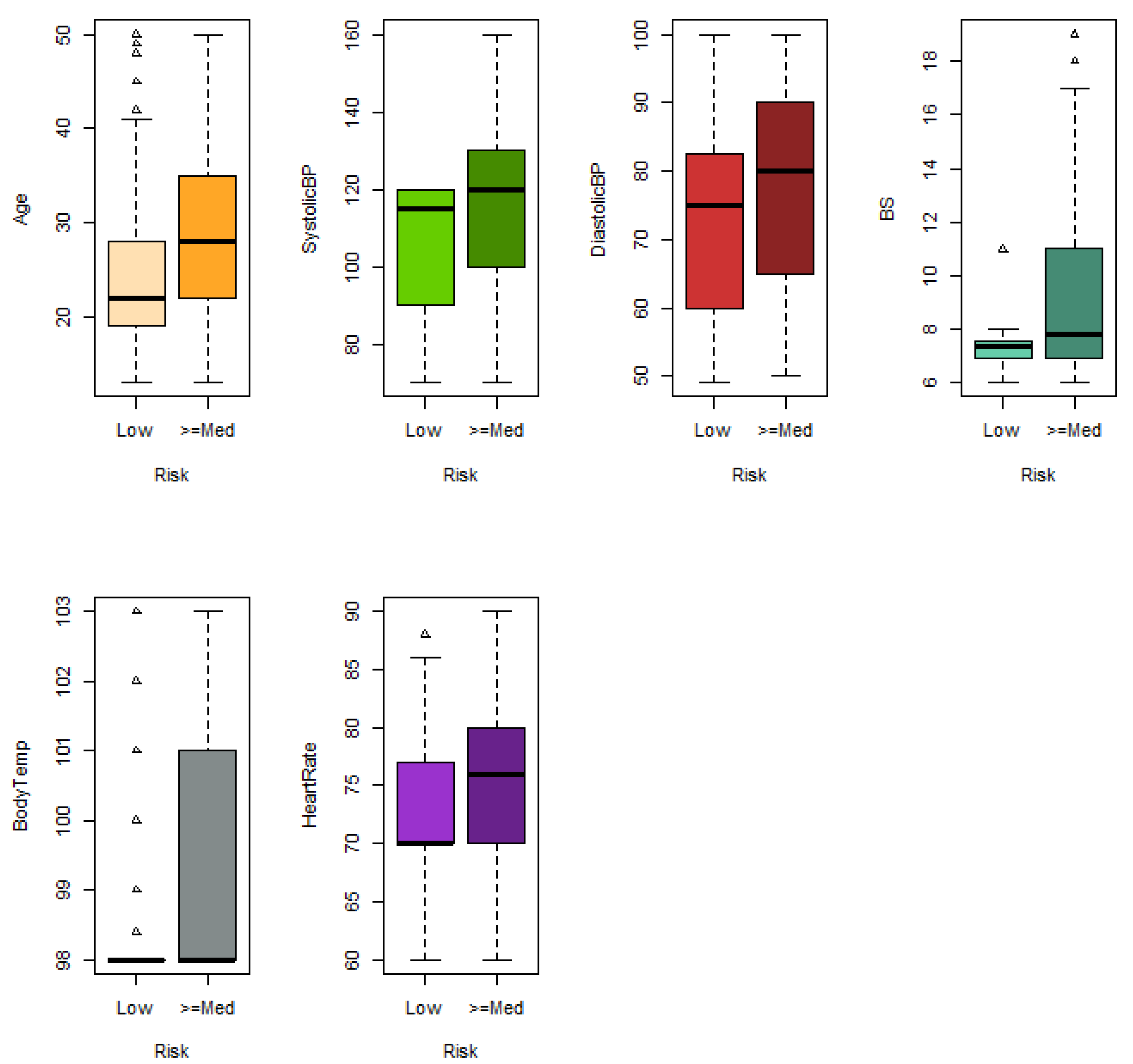
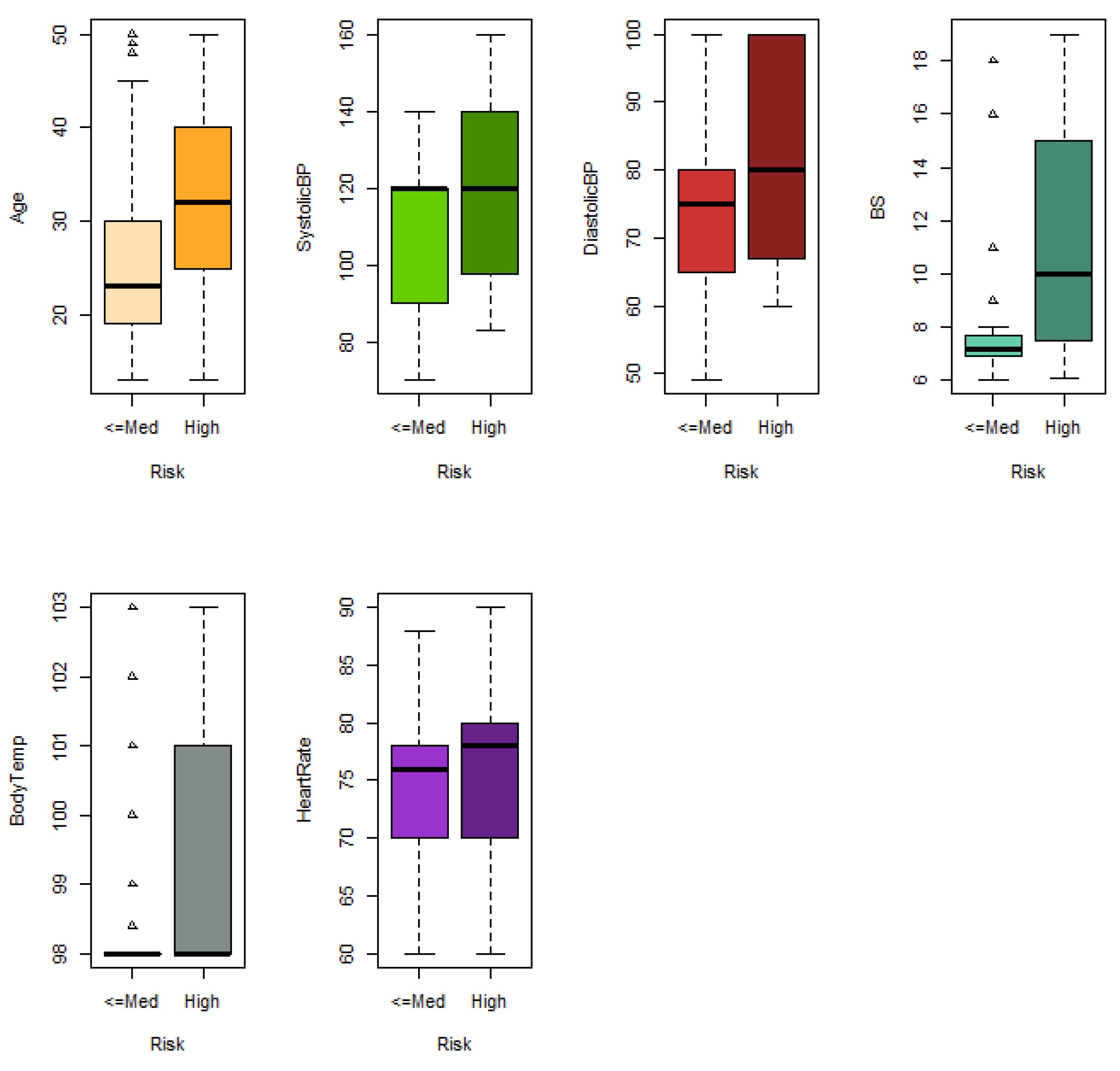
Maternal mortality refers to the death of a woman due to a pregnancy-related cause. It is one of the main concerns of the Sustainable Development Goals (SDG) of the United Nations. The dataset used for analysing maternal mortality was obtained at [
71] (Maternal Health Risk), which contains information on the following six risk factors for maternal mortality: age in years during pregnant, upper value of blood pressure in mmHg (SystolicBP), lower value of blood pressure in mmHg (DiastolicBP), blood glucose levels in mmol/L (BS), body temperature in ºF (BodyTemp) and a normal resting heart rate in beats per minute (HeartRate). An IoT-based risk monitoring system was used to gather this information from various hospitals, community clinics, and maternal healthcare centres in the rural areas of Bangladesh. The level of risk intensity was also provided by differentiating three categories: low (406 women), medium (336 women) and high (272 women). To adapt data to our study, the following binary problems were considered: (i) predicting high or medium risk versus low risk and (ii) predicting high risk versus medium or low risk. The original dataset contains repeated data, outliers and anomalous data that may be due to errors in data retrieval. In our study, data from women aged 13–50 years were considered, duplicate rows were removed, and two observations were removed with heart rate values of 7 beats per minute, which is an erroneous value. Finally, the dataset used in the study contained 196 low-risk, 86 medium-risk and 95 high-risk observations.
2.8. Validation
A total of 59 simulated data scenarios were explored, considering different sample sizes, number of biomarkers, distributions, discriminatory ability, and correlations. For each simulated scenario, each method was trained considering random samples from the underlying distribution (100) and validated using new data simulated with the same configuration (100). For real data, a 10-fold cross-validation procedure was performed.
During training, the model parameters were estimated, and the optimal cut-off point that maximises the Youden index was obtained. The estimated model and the cut-off point selected were applied to the validation set. The Youden indices obtained in the validation set are shown in the tables in the following sections.
4. Discussion and Conclusions
In binary classification problems in healthcare, the choice of thresholds to dichotomise the model output into groups of patients is crucial and can aid decision-making in clinical practice. In the absence of consensus on the benefits of optimising the classification of one group or another, the Youden index is a standard criterion that provides good performance for the model.
Models combining biomarkers for binary classification have received sufficient attention in the literature. The parametric approach has the limitation of meeting the assumption of normality; by contrast, other authors propose non-parametric approaches without assumptions of biomarker distributions but with the limitation of being computationally intractable when the number of biomarkers increases.
Liu et al. [
19] proposed the min–max approach, which is a non-parametric and computationally tractable approach regardless of the number of biomarkers, based on the linear combination of minimum and maximum values of biomarkers. The idea behind this proposal is that the maximum and minimum values chosen among the biomarkers can allow the best discrimination between sick and healthy patients. However, this approach may not be sufficient in terms of discrimination when the number of biomarkers grows, because it is not enough to capture all the discrimination abilities of the set of predictor variables. To improve the min–max algorithm, we proposed the min–max–median/IQR approach under a Youden index maximisation that incorporates a new summary statistic with reasonably good performance. This approach uses a stepwise algorithm that we proposed in [
25], which is based on the work of Pepe et al. [
14,
15].
The use of machine learning algorithms, such as XGBoost, has become increasingly popular in recent years due to their ease of implementation and good results. However, the choice of the optimal approach depends on the problem and the data to be processed. It is therefore essential to make a thorough comparison before providing some guidelines for the selection of the optimal algorithm.
The aim of this paper is to present a comprehensive comparison of our min–max–median/IQR approaches with the min–max approach and machine learning algorithms such as logistic regression and XGBoost, in order to optimise the Youden index. For this purpose, the algorithms were compared on 59 different simulated data scenarios with symmetric and non-symmetric distributions, as well as on two real-world datasets.
The results of the simulated scenarios showed that the machine learning approaches outperformed our approaches, in particular in scenarios with biomarkers with different predictive abilities and in biomarker scenarios with different marginal distributions. However, our approaches outperformed them in scenarios with biomarkers with normal and log-normal distributions with the same predictive ability and different correlations between groups.
Regarding the real datasets, XGBoost outperformed the other algorithms in predicting maternal health risk, while logistic regression achieved the best performance in predicting Duchenne dystrophy, with our proposed approaches closely following. The data show that the problem of predicting Duchenne dystrophy is simpler than that of predicting maternal death risk. In the former, linear combination approaches outperform XGBoost. However, XGBoost outperforms the others on the more complex problem. This may be due to its ability to capture non-linear relationships.
In summary, regardless of the symmetry assumption, non-parametric approaches are always a good alternative for modelling data, but their performance is not guaranteed. Therefore, the modelling process requires a combination of techniques and the optimization of hyperparameters, as we have demonstrated with extensive simulations and application on real data. This work provides the scientific community with a comparison of the performance of our approaches (min–max–median/IQR) and machine learning algorithms, that can be applied and explored in different binary classification problems, such as cancer diagnosis. We proposed a non-parametric approach, addressing the limitations of previous linear biomarker combinations that assume multivariate normality. It also addresses the limitation of the computational burden of certain approaches in the literature, being always approachable regardless of the number of initial biomarkers. This is achieved thanks to the formulation of our algorithm that linearly combines the minimum, maximum, and median or interquartile range biomarkers, thereby converting the n-biomarker combination problem into a three-biomarker combination problem. Although there are several techniques that reduce the dimensionality of the problem for subsequent classification algorithm application, our proposed approach provides a different perspective in this regard. The way our approach is formulated allows the three biomarkers considered (minimum, maximum and median or interquartile range) to correspond to different original biomarkers for each patient. This offers the possibility of capturing biomarker heterogeneity in the data. Subsequently, our approach applies a stepwise algorithm, which we published in [
25] , and which demonstrated acceptable performance in the comparison study. Therefore, our approach proposes a novel formulation in the state of the art that addresses certain limitations in the literature. Furthermore, a comparison of our approach with other approaches, such as the XGBoost algorithm, provides performance results with other approaches that also help to capture biomarker heterogeneity, albeit from a different perspective. XGBoost is a decision tree ensemble algorithm that builds multiple trees and combines their predictions to produce the final output. For the construction of each tree, a random subset of biomarkers/variables is selected and used to partition the data. In this way, each tree is constructed with information from different biomarkers, thus helping to avoid overfitting.
Our approaches have been shown to be superior to other algorithms, including machine learning algorithms, in scenarios with biomarkers having the same predictive capacity and different correlations between groups. These results are not surprising, as there is a variety of health problems in which the combination of the minimum and maximum of biomarkers provides the best classification. In prostate cancer, the worst diagnosis corresponds to a higher value in PSA and a lower value in prostate volume. PSA density is defined by the division of PSA and prostate volume, and it shows a better predictive ability than PSA. Thus, we can choose as a biomarker for prostate cancer the PSA density or the combination provided by PSA and prostate volume. Moreover, as with PSA, there is a variety of competing biomarkers such as PCA3, SelectMdx, and 4Kscore, in which high values correspond to a greater probability of cancer. The min–max derived approaches gives the opportunity to choose from them the one which takes the highest value. Similar to prostate volume, the free PSA takes lower values with a worse diagnosis of prostate cancer; therefore, choosing the minimum and maximum marker for a group of candidates with similar performance can contribute to the best discrimination ability. In addition, the third parameter, median or interquartile range, informs about the performance of the set of biomarkers. The cost-effectiveness of a set of biomarkers to diagnose a unique disease can be controversial, but molecular or metabolomic markers are associated with a variety of cancers, and their analysis has been increasing in recent years. The stratification of cancer or its prognosis will be derived from biomarkers built from information derived from different perspectives.
Although our work includes an exhaustive comparison study in various real and simulated data scenarios, yielding interesting results, the conclusions must be considered within the framework of our study. All conclusions derived from our study are limited to the scenarios and algorithms explored. One of the limitations of the study is the variety of machine learning algorithms considered. While the XGBoost algorithm and logistic regression have been widely used in recent years and have proven efficient, a comparative study that includes additional machine-learning techniques would provide more consistent conclusions. In future work, we propose exploring other machine learning algorithms, deep learning and ensemble models, to compare their performance with our approaches, particularly in scenarios where they are optimal.
Another limitation of the study is the variety of real datasets used, where in no case did our approach achieve the best performance. As future work, we propose evaluating the performance of our approach on real datasets that meet the conditions of the optimal simulation scenarios. One example could be the dataset used in [
19], where the authors demonstrated that the min–max combination of three growth hormones (IGFBP3, IGF1, and GHBP) was superior to the other linear combinations for identifying autism. The aim of this evaluation would be to determine whether our approaches outperform min–max and the other algorithms studied.
In addition to scenarios combining multiple biomarkers with the same predictive capability, our approach could also be applied in scenarios where repeated measurements of a single biomarker are recorded, converting the temporal information into three summary measurements. Readers are encouraged to evaluate our approaches in such problems, for example, the detection of events or neurodegenerative diseases from gait information retrieved from wearable sensor measurements. Another line of future work could involve adapting the models and the study to other objective metrics, such as the weighted Youden index.
In conclusion, our study presents a comprehensive comparison of various approaches, presenting our proposed approach (min–max–Median/IQR) as an alternative to machine learning models such as logistic regression and XGBoost, in certain scenarios where it has demonstrated superior performance. We believe that the results of this research will provide valuable insights for the development and application of classification algorithms in the field of medicine, such as cancer diagnosis.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}