
Interface Asymmetry Induced and Surface Pressure Controlled Valence Tautomerism in Monolayers of bis-Phthalocyaninates of Lanthanides

 , and
, and 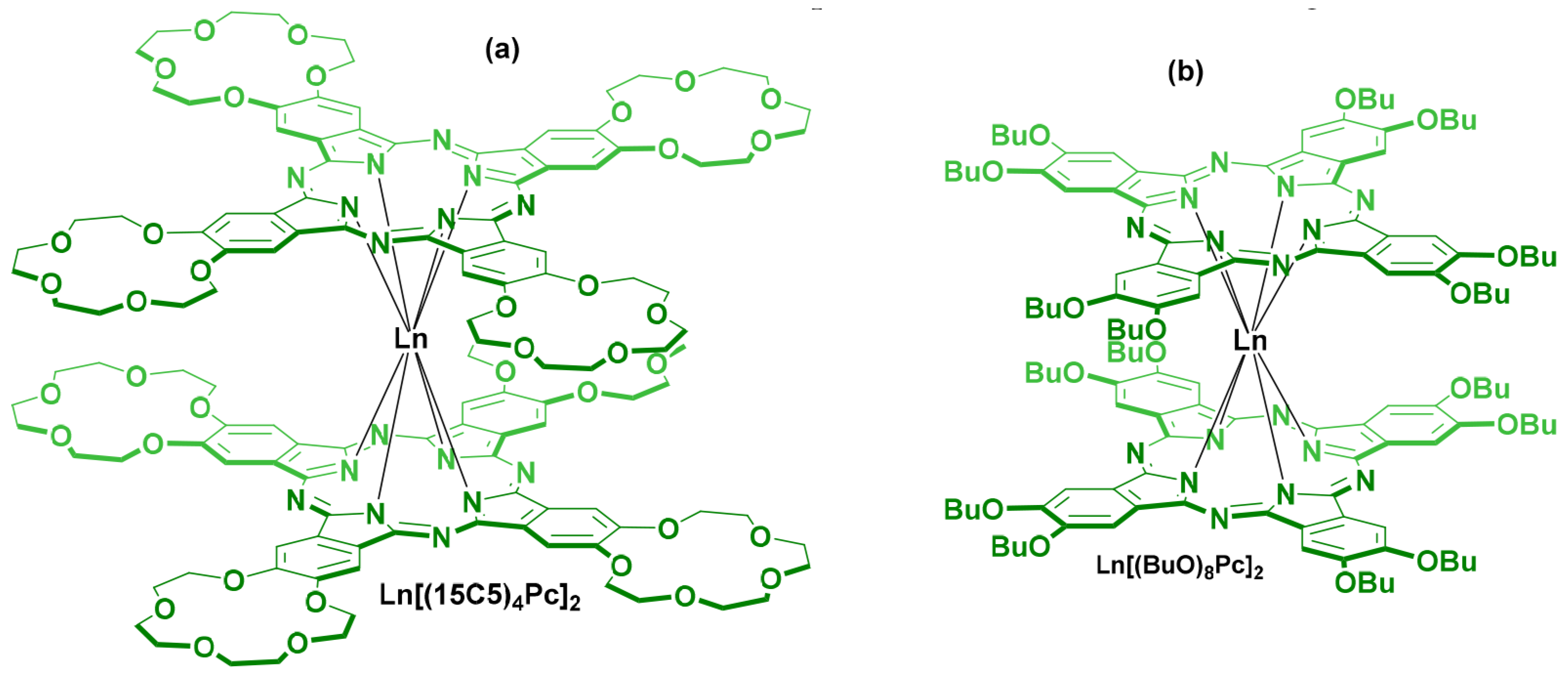{kind=link}
{kind=link}
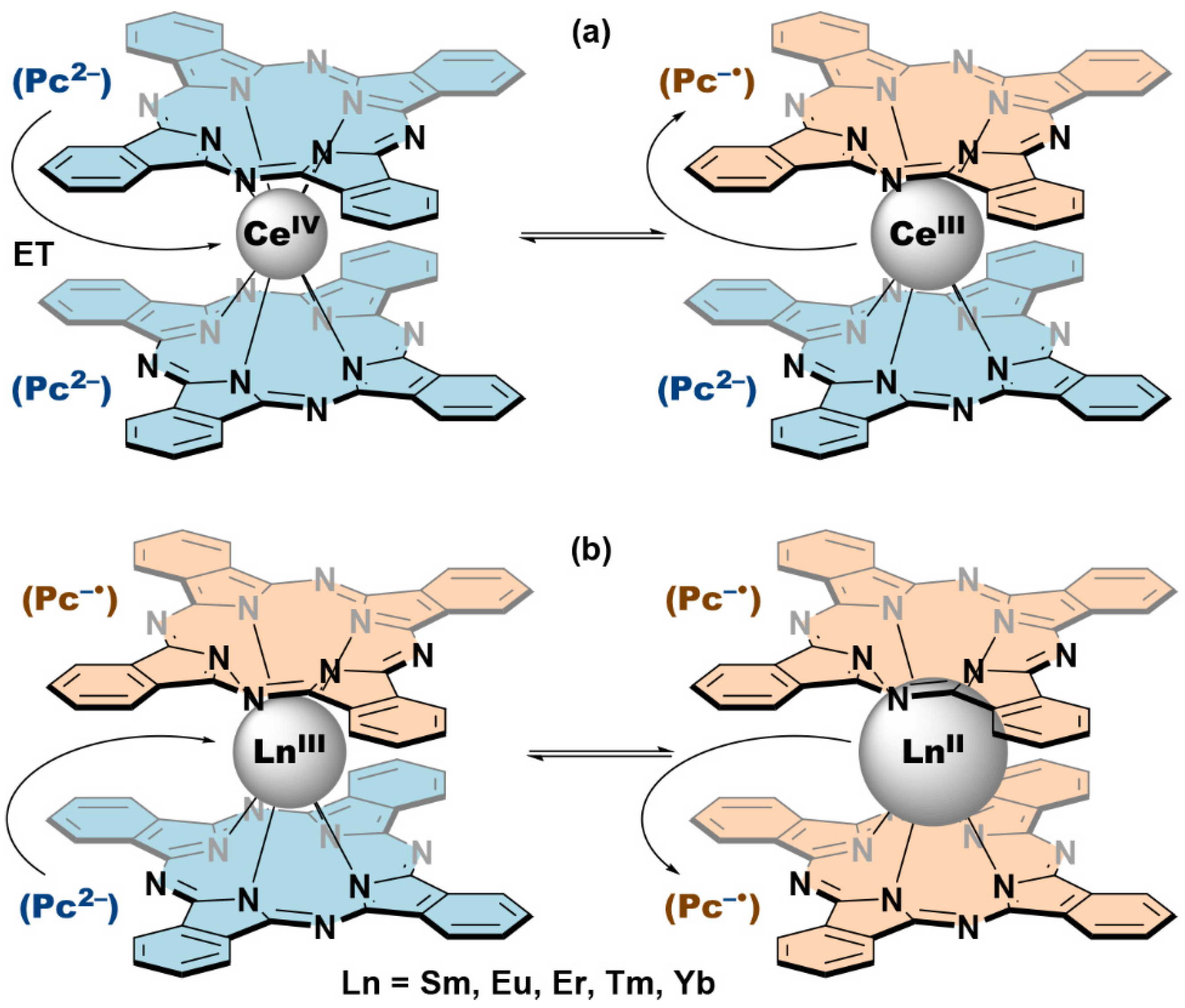
{kind=link}
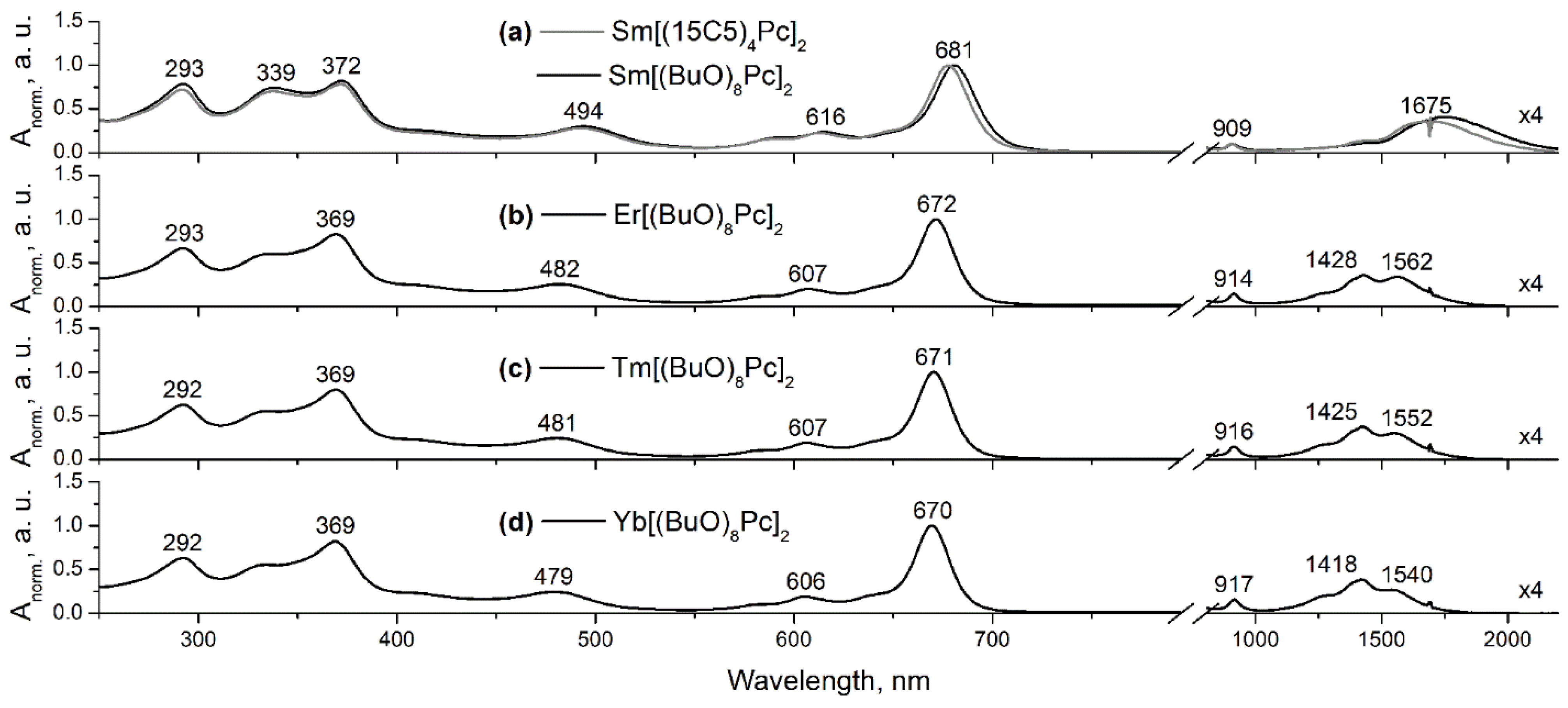
{kind=link}
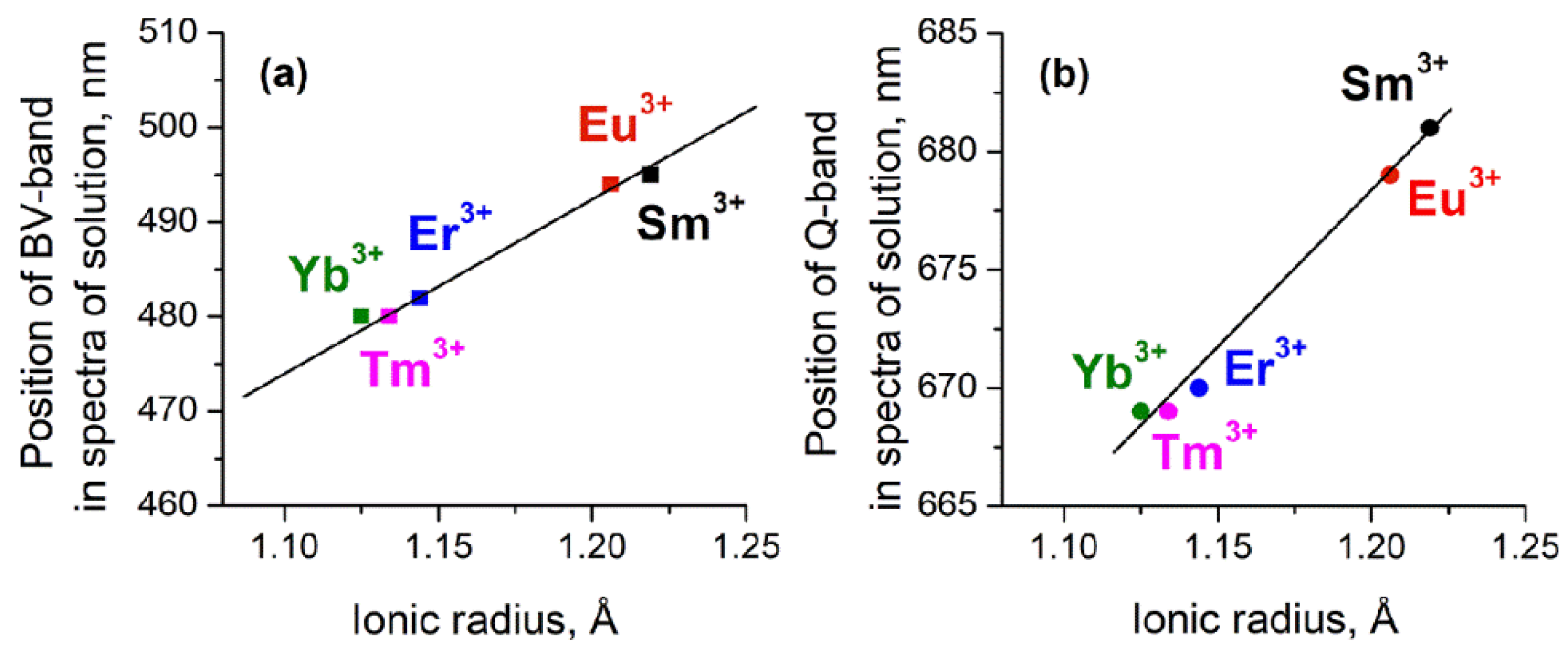
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Physical–Chemical Measurements
2.2. Synthesis and Characterization of Phthalocyanines
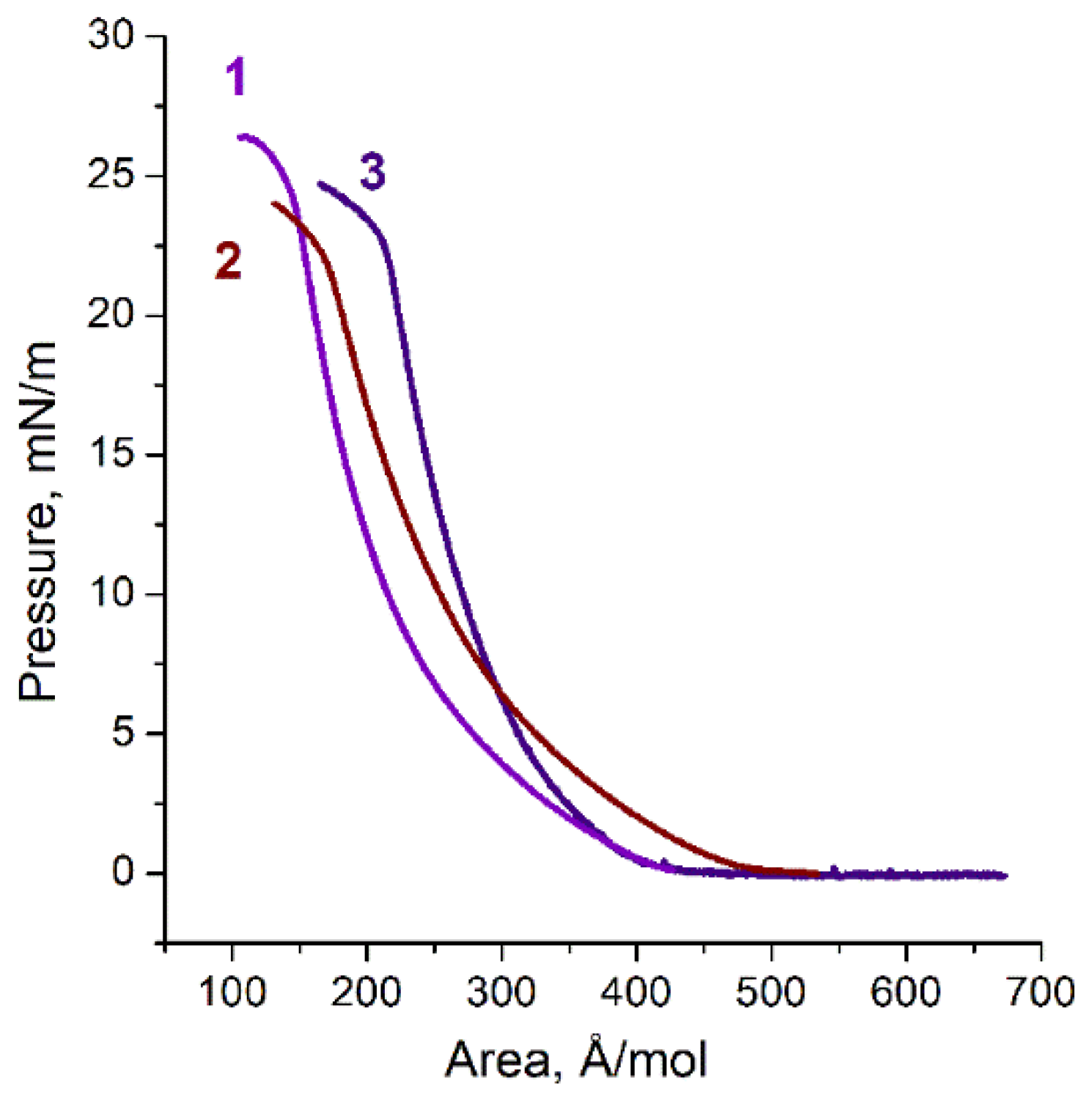
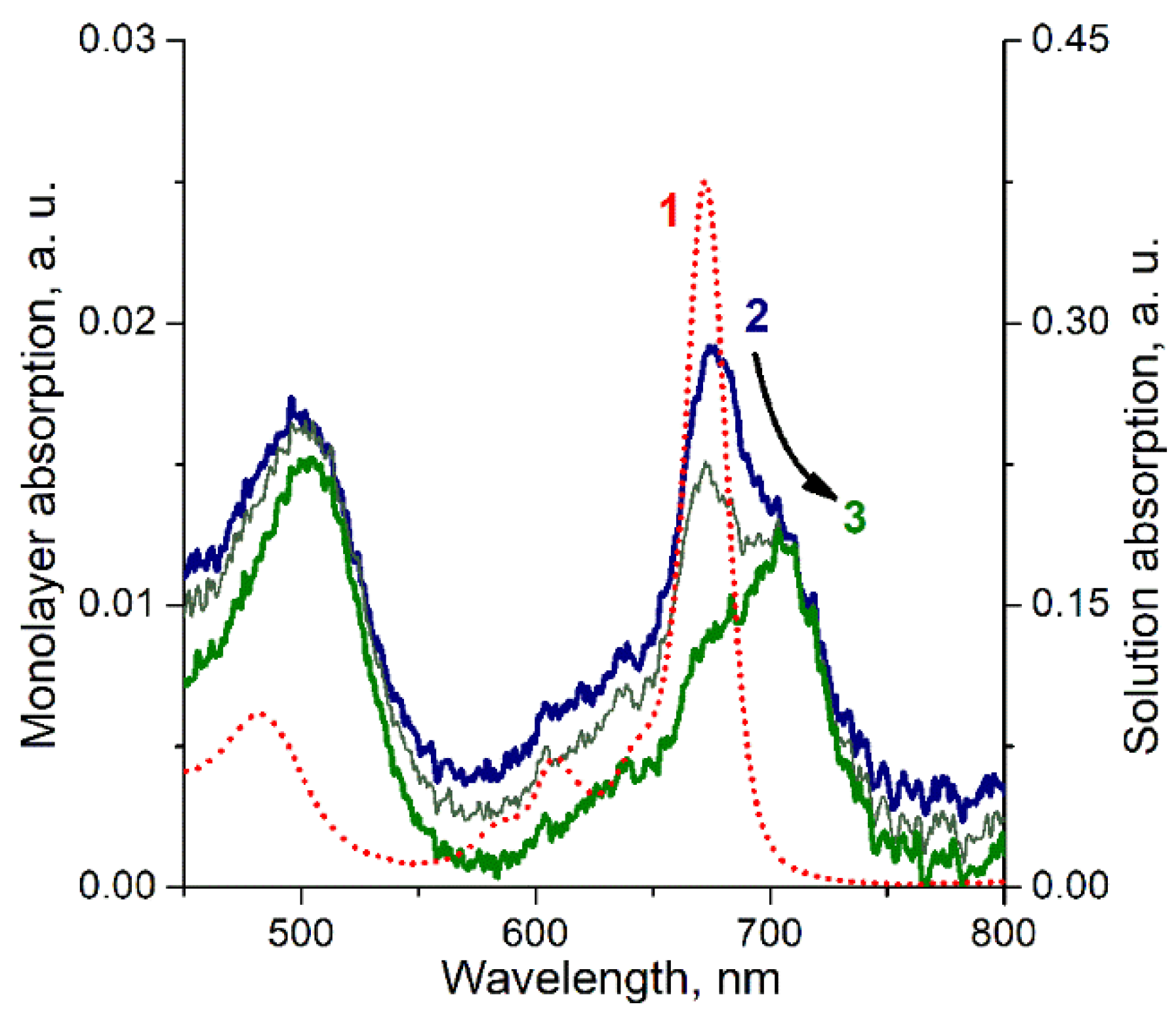
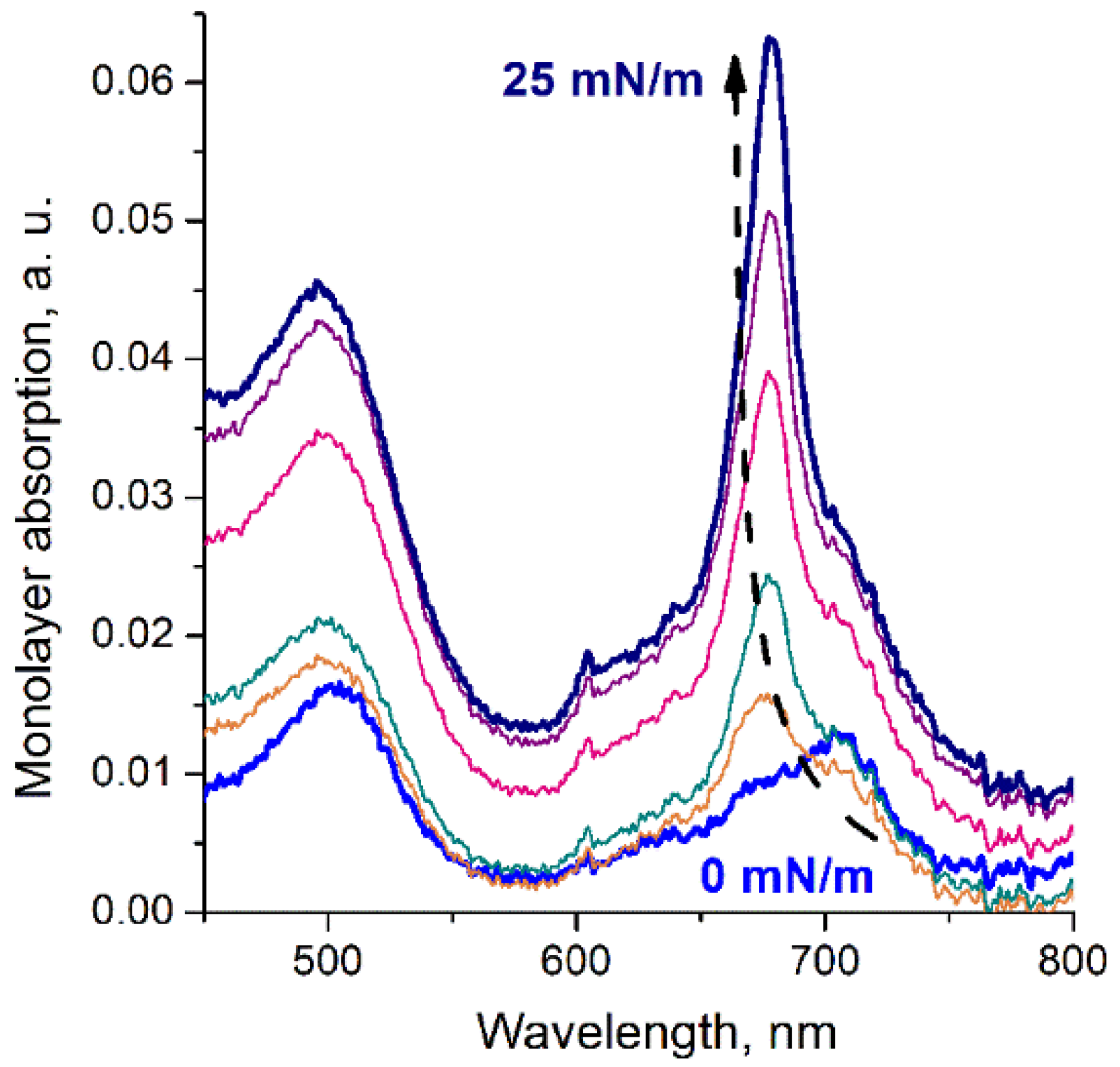
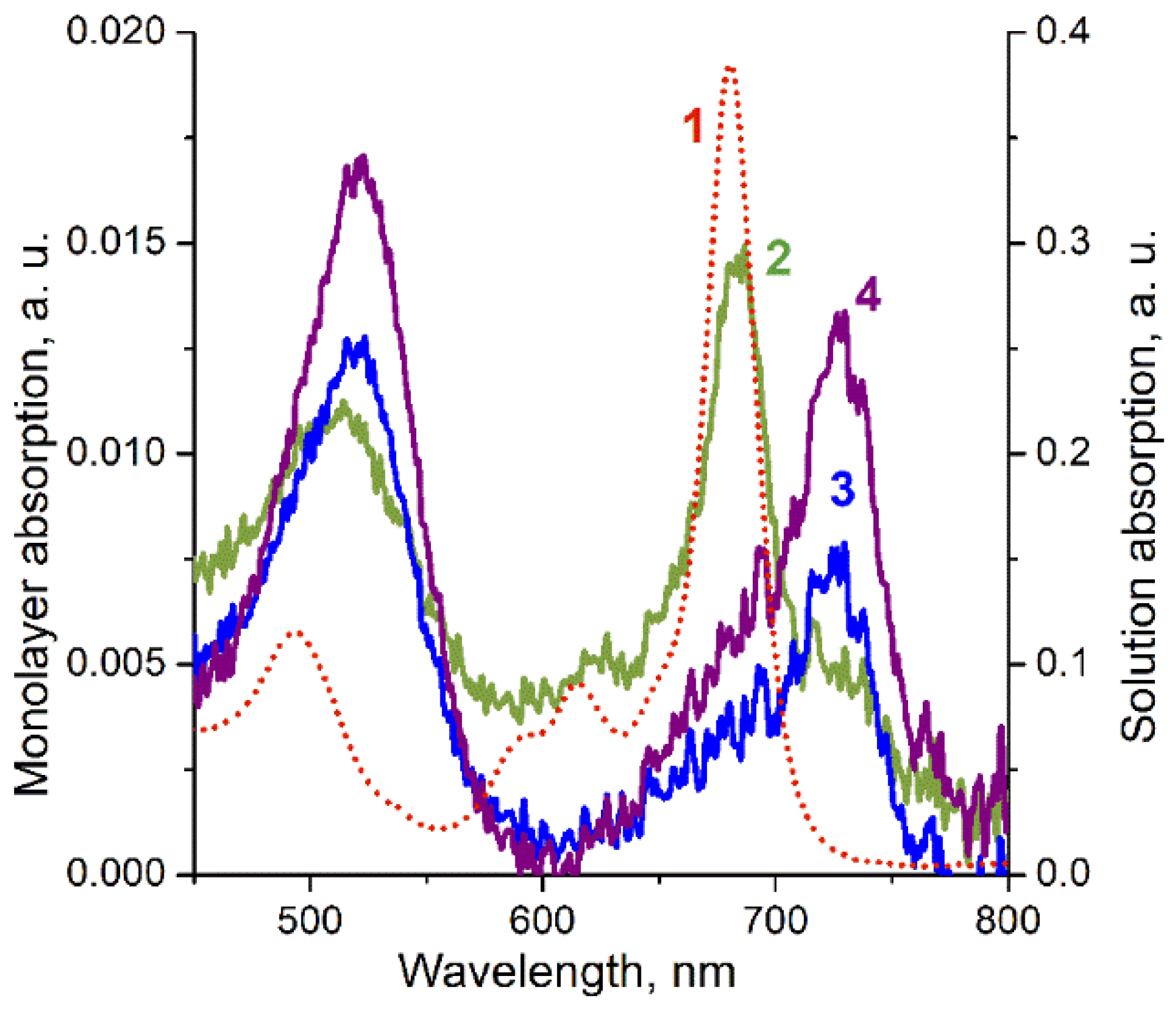
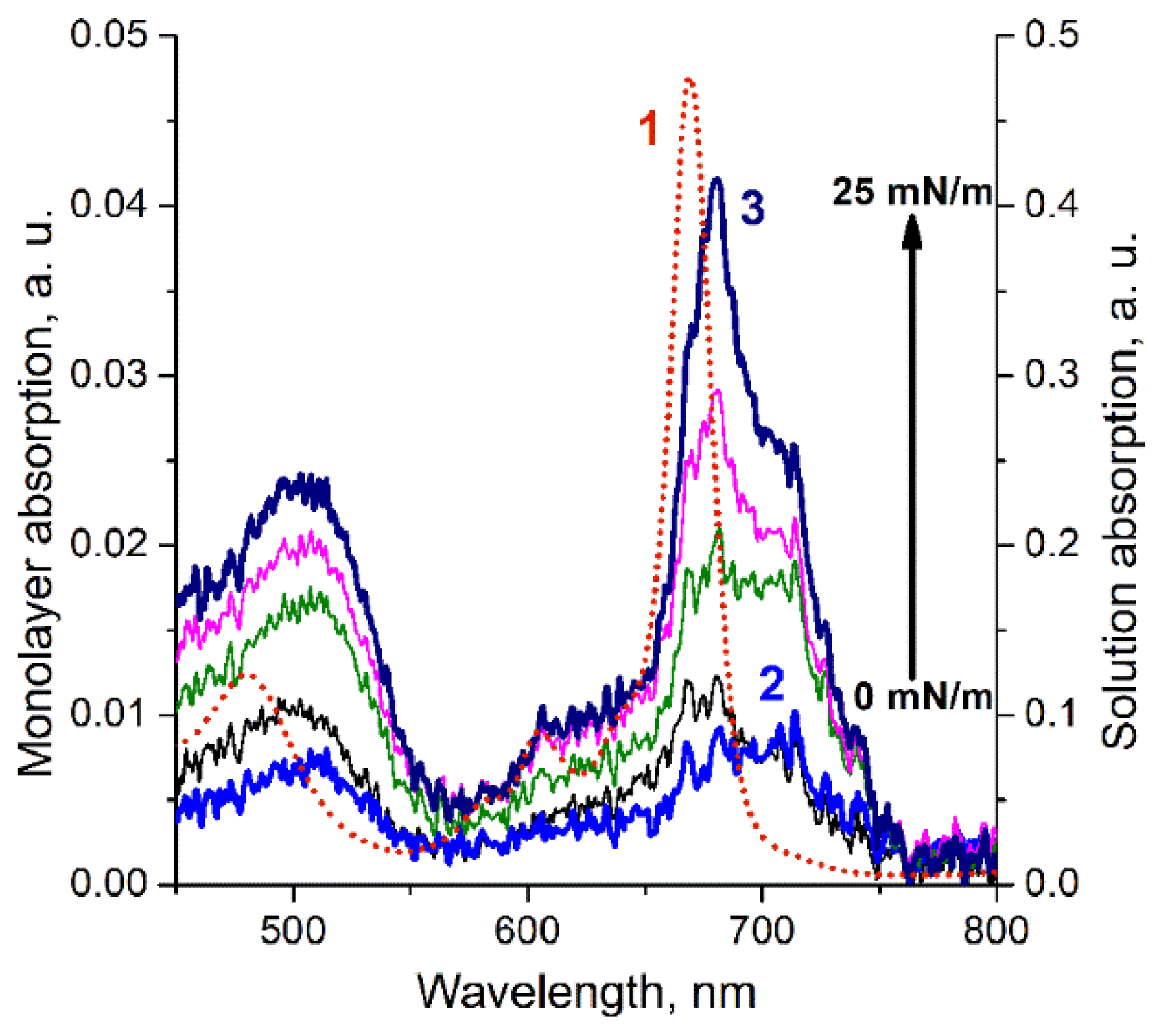
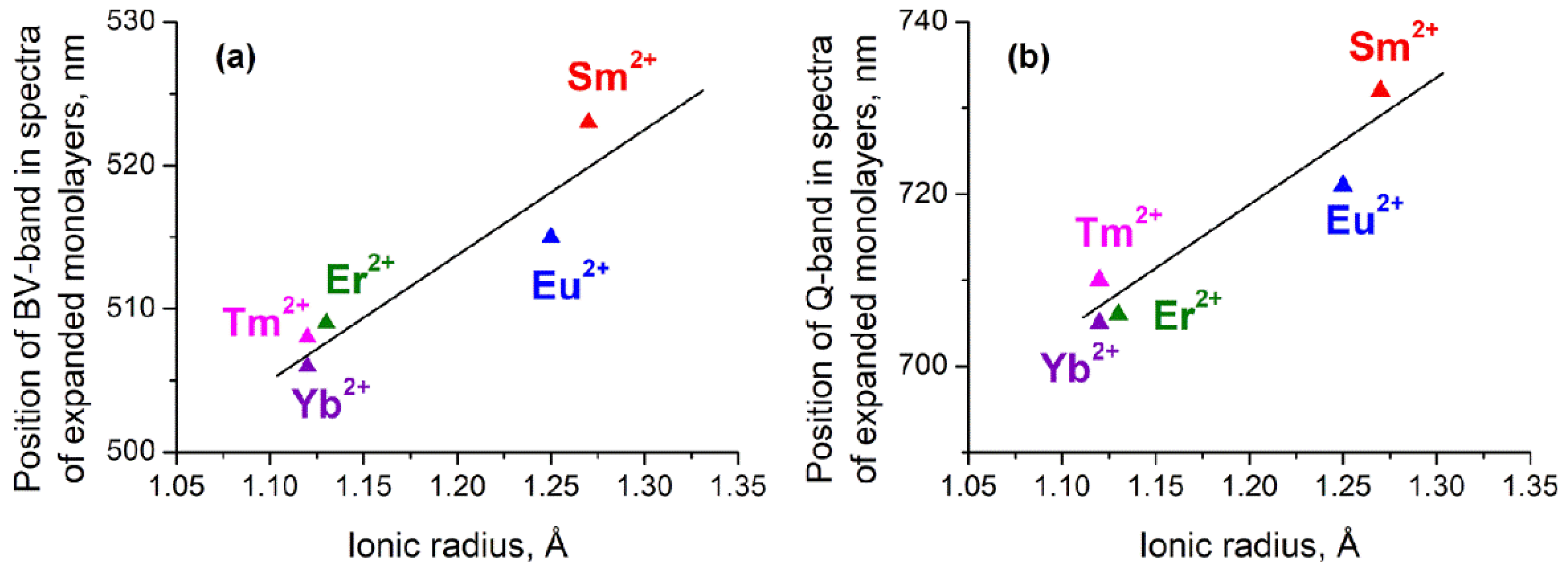
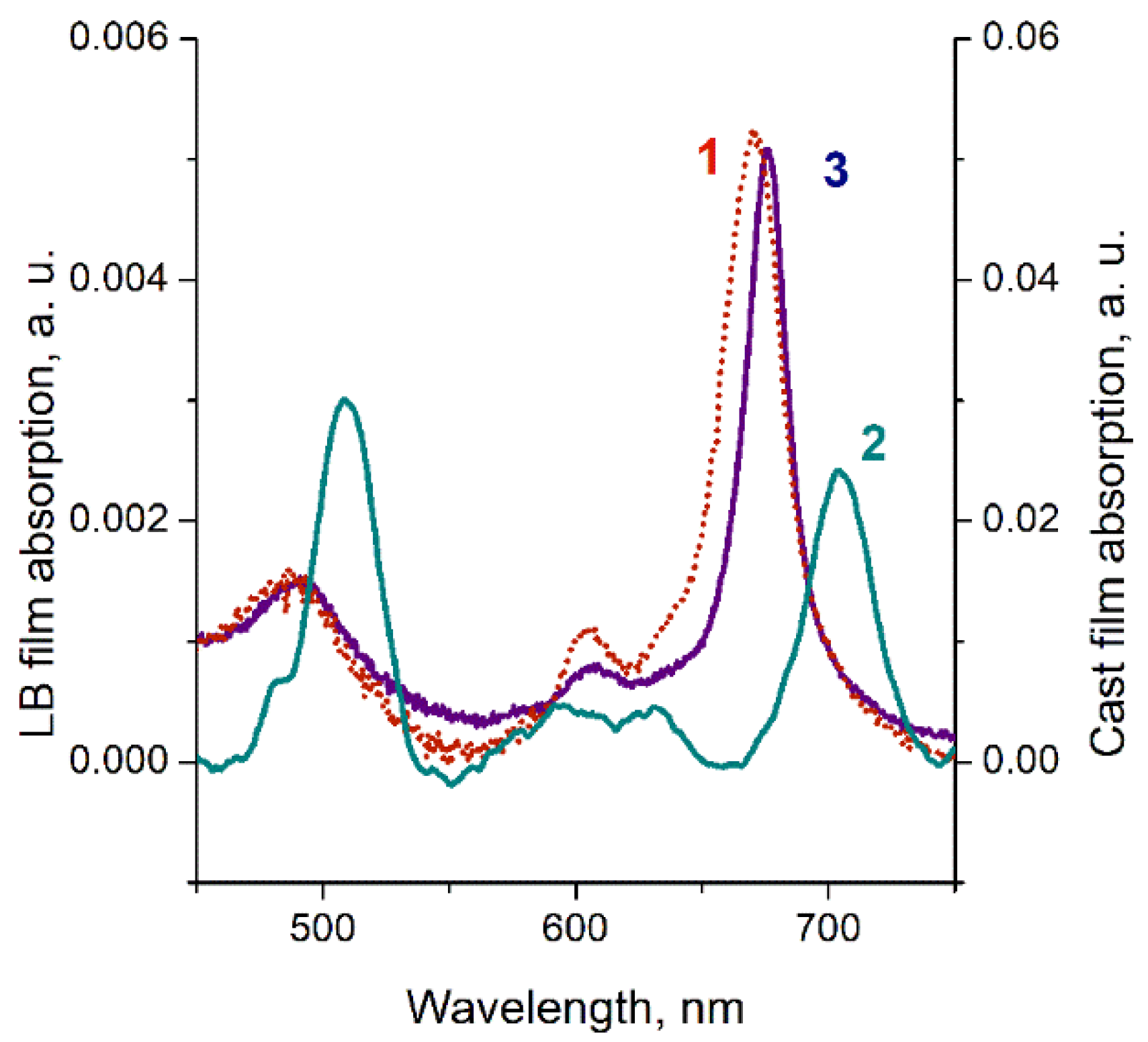
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nicoli, F.; Paltrinieri, E.; Tranfić Bakić, M.; Baroncini, M.; Silvi, S.; Credi, A. Binary logic operations with artificial molecular machines. Coord. Chem. Rev. 2021, 428, 213589. [Google Scholar] [CrossRef]
- Tasbas, M.N.; Sahin, E.; Erbas-Cakmak, S. Bio-inspired molecular machines and their biological applications. Coord. Chem. Rev. 2021, 443, 214039. [Google Scholar] [CrossRef]
- Andreoni, L.; Baroncini, M.; Groppi, J.; Silvi, S.; Taticchi, C.; Credi, A. Photochemical Energy Conversion with Artificial Molecular Machines. Energy Fuels 2021, 35, 18900–18914. [Google Scholar] [CrossRef]
- Feng, Y.; Ovalle, M.; Seale, J.S.W.; Lee, C.K.; Kim, D.J.; Astumian, R.D.; Stoddart, J.F. Molecular Pumps and Motors. J. Am. Chem. Soc. 2021, 143, 5569–5591. [Google Scholar] [CrossRef] [PubMed]
- Martynov, A.G.; Safonova, E.A.; Tsivadze, A.Y.; Gorbunova, Y.G. Functional molecular switches involving tetrapyrrolic macrocycles. Coord. Chem. Rev. 2019, 387, 325–347. [Google Scholar] [CrossRef]
- Kutsybala, D.S.; Shokurov, A.V.; Selektor, S.L. Molecular Machines in 3D and 2D Systems: Movement, Mechanical Work, and Switching. A Review. Prot. Met. Phys. Chem. Surf. 2021, 57, 917–942. [Google Scholar] [CrossRef]
- Guda, A.A.; Chegerev, M.; Starikov, A.G.; Vlasenko, V.G.; Zolotukhin, A.A.; Bubnov, M.P.; Cherkasov, V.K.; Shapovalov, V.V.; Rusalev, Y.V.; Tereshchenko, A.A.; et al. Valence tautomeric transition of bis(o-dioxolene) cobalt complex in solid state and solution. J. Phys. Condens. Matter 2021, 33, 215405. [Google Scholar] [CrossRef]
- Zolotukhin, A.A.; Bubnov, M.P.; Arapova, A.V.; Fukin, G.K.; Rumyantcev, R.V.; Bogomyakov, A.S.; Knyazev, A.V.; Cherkasov, V.K. Valence–Tautomeric Interconversion in a Bis(dioxolene)cobalt Complex with Iminopyridine Functionalized by a TEMPO Moiety. Phase Transition Coupled with Monocrystal Destruction. Inorg. Chem. 2017, 56, 14751–14754. [Google Scholar] [CrossRef]
- Chegerev, M.G.; Piskunov, A.V.; Starikova, A.A.; Kubrin, S.P.; Fukin, G.K.; Cherkasov, V.K.; Abakumov, G.A. Redox Isomerism in Main-Group Chemistry: Tin Complex with o-Iminoquinone Ligands. Eur. J. Inorg. Chem. 2018, 2018, 1087–1092. [Google Scholar] [CrossRef]
- Creutz, C.; Taube, H. Direct approach to measuring the Franck-Condon barrier to electron transfer between metal ions. J. Am. Chem. Soc. 1969, 91, 3988–3989. [Google Scholar] [CrossRef]
- Buchanan, R.M.; Pierpont, C.G. Tautomeric catecholate-semiquinone interconversion via metal-ligand electron transfer. Structural, spectral, and magnetic properties of (3,5-di-tert-butylcatecholato)(3,5-di-tert-butylsemiquinone)(bipyridyl)cobalt(III), a complex containing mixed-valence. J. Am. Chem. Soc. 1980, 102, 4951–4957. [Google Scholar] [CrossRef]
- Pierpont, C.G. Studies on charge distribution and valence tautomerism in transition metal complexes of catecholate and semiquinonate ligands. Coord. Chem. Rev. 2001, 216–217, 99–125. [Google Scholar] [CrossRef]
- Abakumov, G.A.; Cherkasov, V.K.; Nevodchikov, V.I.; Kuropatov, V.A.; Yee, G.T.; Pierpont, C.G. Magnetic properties and redox isomerism for 4,4’-bis(semiquinone) complexes of copper. Inorg. Chem. 2001, 40, 2434–2436. [Google Scholar] [CrossRef] [PubMed]
- Nitahara, S.; Akiyama, T.; Inoue, S.; Yamada, S. A photoelectronic switching device using a mixed self-assembled monolayer. J. Phys. Chem. B 2005, 109, 3944–3948. [Google Scholar] [CrossRef]
- Poneti, G.; Mannini, M.; Sorace, L.; Sainctavit, P.; Arrio, M.-A.; Otero, E.; Cezar, J.C.; Dei, A. Soft-X-ray-induced redox isomerism in a cobalt dioxolene complex. Angew. Chem. 2010, 49, 1954–1957. [Google Scholar] [CrossRef]
- Gütlich, P.; Gaspar, A.B.; Ksenofontov, V.; Garcia, Y. Pressure effect studies in molecular magnetism. J. Phys. Condens. Matter 2004, 16, S1087–S1108. [Google Scholar] [CrossRef]
- Roux, C.; Adams, D.M.; Itié, J.P.; Polian, A.; Hendrickson, D.N.; Verdaguer, M. Pressure-Induced Valence Tautomerism in Cobalt o-Quinone Complexes: An X-ray Absorption Study of the Low-Spin [Co III (3,5-DTBSQ)(3,5-DTBCat)(phen)] to High-Spin [Co II (3,5-DTBSQ) 2 (phen)] Interconversion. Inorg. Chem. 1996, 35, 2846–2852. [Google Scholar] [CrossRef]
- Caneschi, A.; Dei, A.; de Biani, F.F.; Gütlich, P.; Ksenofontov, V.; Levchenko, G.; Hoefer, A.; Renz, F. Pressure- and Temperature-Induced Valence Tautomeric Interconversion in ao-Dioxolene Adduct of a Cobalt-Tetraazamacrocycle Complex. Chem.-A Eur. J. 2001, 7, 3926–3930. [Google Scholar] [CrossRef]
- Deal, A.M.; Rapf, R.J.; Vaida, V. Water–Air Interfaces as Environments to Address the Water Paradox in Prebiotic Chemistry: A Physical Chemistry Perspective. J. Phys. Chem. A 2021, 125, 4929–4942. [Google Scholar] [CrossRef]
- Shokurov, A.V.; Kutsybala, D.S.; Kroitor, A.P.; Dmitrienko, A.A.; Martynov, A.G.; Enakieva, Y.Y.; Tsivadze, A.Y.; Selektor, S.L.; Gorbunova, Y.G. Spin Crossover in Nickel(II) Tetraphenylporphyrinate via Forced Axial Coordination at the Air/Water Interface. Molecules 2021, 26, 4155. [Google Scholar] [CrossRef]
- Zhao, W.; Tong, B.; Pan, Y.; Shen, J.; Zhi, J.; Shi, J.; Dong, Y. Fabrication, electrochemical, and optoelectronic properties of layer-by-layer films based on (phthalocyaninato)ruthenium(II) and triruthenium dodecacarbonyl bridged by 4,4’-bipyridine as ligand. Langmuir 2009, 25, 11796–11801. [Google Scholar] [CrossRef]
- Selektor, S.L.S.L.; Shokurov, A.V.; Arslanov, V.V.; Gorbunova, Y.G.; Birin, K.P.; Raitman, O.A.; Morote, F.; Cohen-Bouhacina, T.; Grauby-Heywang, C.; Tsivadze, A.Y. Orientation-Induced Redox Isomerism in Planar Supramolecular Systems. J. Phys. Chem. C 2014, 118, 4250–4258. [Google Scholar] [CrossRef] [Green Version]
- Shokurov, A.V.; Kutsybala, D.S.; Martynov, A.G.; Bakirov, A.V.; Shcherbina, M.A.; Chvalun, S.N.; Gorbunova, Y.G.; Tsivadze, A.Y.; Zaytseva, A.V. Long-Sought Redox Isomerization of the Europium(III/II) Complex Achieved by Molecular Reorientation at the Interface. Langmuir 2020, 36, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Tezgerevska, T.; Alley, K.G.; Boskovic, C. Valence tautomerism in metal complexes: Stimulated and reversible intramolecular electron transfer between metal centers and organic ligands. Coord. Chem. Rev. 2014, 268, 23–40. [Google Scholar] [CrossRef]
- Drath, O.; Boskovic, C. Switchable cobalt coordination polymers: Spin crossover and valence tautomerism. Coord. Chem. Rev. 2018, 375, 256–266. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Maslova, O.V.; Baranov, E.V.; Shavyrin, A.S. Redox Isomerism in the Lanthanide Complex [(dpp-Bian)Yb(DME)(μ-Br)] 2 (dpp-Bian = 1,2-Bis[(2,6-diisopropylphenyl)imino]acenaphthene). Inorg. Chem. 2009, 48, 2355–2357. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Maslova, O.V.; Morozov, A.G.; Dechert, S.; Demeshko, S.; Meyer, F. Genuine redox isomerism in a rare-earth-metal complex. Angew. Chem. Int. Ed. 2012, 51, 10584–10587. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Ohno, O.; Kaizu, Y. Electronic states of bis(phthalocyaninato)lutetium radical and its related compounds: The application of localized orbital basis set to open-shell phthalocyanine dimers. J. Phys. Chem. 1993, 97, 1004–1010. [Google Scholar] [CrossRef]
- Shokurov, A.V.; Kutsybala, D.S.; Martynov, A.G.; Yagodin, A.V.; Gorbunova, Y.G.; Novikov, D.; Bakirov, A.V.; Shcherbina, M.A.; Chvalun, S.N.; Arslanov, V.V.; et al. Fluorescence Mode XANES Spectroscopy as a Powerful Tool for Redox-Isomerism Studies in Ultrathin Films. Macroheterocycles 2019, 12, 264–267. [Google Scholar] [CrossRef]
- Stuchebryukov, S.D.; Selektor, S.L.; Silantieva, D.A.; Shokurov, A.V. Peculiarities of the reflection-absorption and transmission spectra of ultrathin films under normal incidence of light. Prot. Met. Phys. Chem. Surf. 2013, 49, 189–197. [Google Scholar] [CrossRef]
- Martynov, A.G.; Berezhnoy, G.S.; Safonova, E.A.; Polovkova, M.A.; Gorbunova, Y.G.; Tsivadze, A.Y. Aromatic Nucleophilic Substitution as a Side Process in the Synthesis of Alkoxy- and Crown-Substituted (Na)phthalocyanines. Macroheterocycles 2019, 12, 75–81. [Google Scholar] [CrossRef]
- Oluwole, D.O.; Yagodin, A.V.; Mkhize, N.C.; Sekhosana, K.E.; Martynov, A.G.; Gorbunova, Y.G.; Tsivadze, A.Y.; Nyokong, T. First Example of Nonlinear Optical Materials Based on Nanoconjugates of Sandwich Phthalocyanines with Quantum Dots. Chem. Eur. J. 2017, 23, 2820–2830. [Google Scholar] [CrossRef]
- Takahashi, K.; Tomita, Y.; Hada, Y.; Tsubota, K.; Handa, M.; Kasuga, K.; Sogabe, K.; Tokii, T. Preparation and Electrochemical Properties of the Green Ytterbium(III) and Lutetium(III) Sandwich Complexes of Octabutoxy-Substituted Phthalocyanine. Chem. Lett. 1992, 759–762. [Google Scholar] [CrossRef]
- Nefedova, I.V.; Gorbunova, Y.G.; Sakharov, S.G.; Tsivadze, A.Y. Synthesis and spectroscopic study of terbium (III) and neodymium (III) complexes with tetra-15-crown-5-phthalocyanine. Russ. J. Inorg. Chem. 2005, 50, 165–173. [Google Scholar]
- Markovitsi, D.; Tran-Thi, T.-H.; Even, R.; Simon, J. Near infrared absorption spectra of lanthanide bis-phthalocyanines. Chem. Phys. Lett. 1987, 137, 107–112. [Google Scholar] [CrossRef]
- Rousseau, R.; Aroca, R.; Rodríguez-Méndez, M.L. Extended Hückel molecular orbital model for lanthanide bisphthalocyanine complexes. J. Mol. Struct. 1995, 356, 49–62. [Google Scholar] [CrossRef]
- May, A.; Majumdar, P.; Martynov, A.G.; Lapkina, L.A.; Troyanov, S.I.; Gorbunova, Y.G.; Tsivadze, A.Y.; Mack, J.; Nyokong, T. Optical limiting properties, structure and simplified TD-DFT calculations of scandium tetra-15-crown-5 phthalocyaninates. J. Porphyr. Phthalocyanines 2020, 24, 589–601. [Google Scholar] [CrossRef]
- Gorbunova, Y.G.; Rodríguez-Méndez, M.L.; Kalashnikova, I.P.; Tomilova, L.G.; de Saja, J.A.; Rodríguez-Morgade, M.S. Langmuir−Blodgett Films of Bis(octakispropyloxy) Samarium Bisphthalocyanine. Spectroscopic and Gas-Sensing Properties. Langmuir 2001, 17, 5004–5010. [Google Scholar] [CrossRef]
- Shokurov, A.V.; Kutsybala, D.S.S.; Martynov, A.G.; Raitman, O.A.A.; Arslanov, V.V.V.; Gorbunova, Y.G.G.; Tsivadze, A.Y.; Selektor, S.L.L. Modulation of transversal conductivity of europium(III) bisphthalocyaninate ultrathin films by peripheral substitution. Thin Solid Film. 2019, 692, 137591. [Google Scholar] [CrossRef]
- Ayhan, M.M.; Singh, A.; Jeanneau, E.; Ahsen, V.; Zyss, J.; Ledoux-Rak, I.; Gürek, A.G.; Hirel, C.; Bretonnière, Y.; Andraud, C. ABAB Homoleptic Bis(phthalocyaninato)lanthanide(III) Complexes: Original Octupolar Design Leading to Giant Quadratic Hyperpolarizability. Inorg. Chem. 2014, 53, 4359–4370. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutsybala, D.S.; Shokurov, A.V.; Martynov, A.G.; Yagodin, A.V.; Arslanov, V.V.; Gorbunova, Y.G.; Selektor, S.L. Interface Asymmetry Induced and Surface Pressure Controlled Valence Tautomerism in Monolayers of bis-Phthalocyaninates of Lanthanides. Symmetry 2022, 14, 340. https://doi.org/10.3390/sym14020340
Kutsybala DS, Shokurov AV, Martynov AG, Yagodin AV, Arslanov VV, Gorbunova YG, Selektor SL. Interface Asymmetry Induced and Surface Pressure Controlled Valence Tautomerism in Monolayers of bis-Phthalocyaninates of Lanthanides. Symmetry. 2022; 14(2):340. https://doi.org/10.3390/sym14020340
Chicago/Turabian StyleKutsybala, Daria S., Alexander V. Shokurov, Alexander G. Martynov, Alexey V. Yagodin, Vladimir V. Arslanov, Yulia G. Gorbunova, and Sofiya L. Selektor. 2022. "Interface Asymmetry Induced and Surface Pressure Controlled Valence Tautomerism in Monolayers of bis-Phthalocyaninates of Lanthanides" Symmetry 14, no. 2: 340. https://doi.org/10.3390/sym14020340