
Phosphorus Compounds of Natural Origin: Prebiotic, Stereochemistry, Application


V.P. Kukhar’ Institute of Bioorganic Chemistry and Petrochemistry, NAS of Ukraine, Murmanska st., 1, 02094 Kyiv, Ukraine
Symmetry 2021, 13(5), 889; https://doi.org/10.3390/sym13050889
Submission received: 2 April 2021
/
Revised: 28 April 2021
/
Accepted: 1 May 2021
/
Published: 17 May 2021
(This article belongs to the Collection Feature Papers in Chemistry)
Abstract
:Organophosphorus compounds play a vital role as nucleic acids, nucleotide coenzymes, metabolic intermediates and are involved in many biochemical processes. They are part of DNA, RNA, ATP and a number of important biological elements of living organisms. Synthetic compounds of this class have found practical application as agrochemicals, pharmaceuticals, bioregulators, and othrs. In recent years, a large number of phosphorus compounds containing P-O, P-N, P-C bonds have been isolated from natural sources. Many of them have shown interesting biological properties and have become the objects of intensive scientific research. Most of these compounds contain asymmetric centers, the absolute configurations of which have a significant effect on the biological properties of the products of their transformations. This area of research on natural phosphorus compounds is still little-studied, that prompted us to analyze and discuss it in our review. Moreover natural organophosphorus compounds represent interesting models for the development of new biologically active compounds, and a number of promising drugs and agrochemicals have already been obtained on their basis. The review also discusses the history of the development of ideas about the role of organophosphorus compounds and stereochemistry in the origin of life on Earth, starting from the prebiotic period, that allows us in a new way to consider this most important problem of fundamental science.
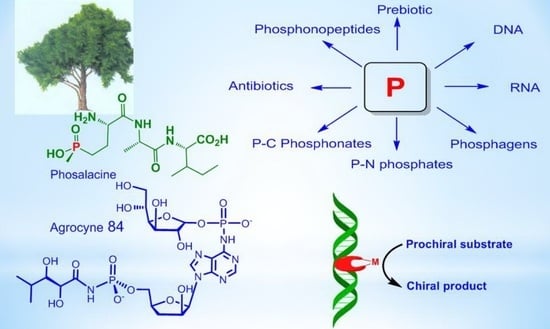
1. Introduction
Phosphorus compounds are essential for modern biological systems, and their diverse biological properties testify to their importance in the world of living organisms. They provide stable ligation necessary for fixing information in RNA and DNA, contribute to cellular structure in phospholipids, serve as the main source of biochemical energy (e.g., ATP, phosphoenolpyruvate, creatinephosphate), and are present in a large number of metabolites. Phosphates retain a central place in biological systems, which allows us to conclude that they played an important role in the emergence of life on Earth [1,2,3,4]. In recent years, a large number of natural phosphorus compounds have been isolated from living organisms and significant advances have been made in understanding the effects of phosphates on prebiotic chemistry. A number of natural phosphorus compounds have practical applications (Figure 1) [2].
Natural phosphorus compounds are invaluable sources for modeling and creating new biologically active substances on their basis. A number of organophosphorus compounds that are C-P analogs of natural compounds are used in the clinic as drugs for the treatment of various diseases. Among the natural phosphorus compounds, effective pharmaceuticals, antibiotics, herbicides, insecticides, and various bioregulators were found.
Research on such important for the vital functions of substances containing phosphorus as phosphagens, especially ATP or creatine phosphate, is of great importance for physiology. The study of polyphosphates in the structure of DNA and RNA allows a better understanding of the functioning of these polymeric molecules, vital for life. The purpose of this review is to show the role of phosphorus in the processes occurring in nature and primarily in living organisms, starting from the early period of prebiotics and the emergence of life, as well as the role of stereochemistry in these processes. In addition, we want to show the high practical value of natural phosphorus compounds in pharmacology, agrochemistry, and various technological processes.
2. The Role of Phosphorus at the Origine and Evolution of Life
Phosphorus is one of the main elements of living organisms. Despite its low content in organisms, phosphorus plays an important role in biochemical reactions. In the human body, phosphorus is the main component of bones and teeth, and acts as a link between DNA and the RNA scaffold, in which the phosphorus content is 9%. Phospholipids are the main component of cell membranes, which are involved in the regulation of membrane permeability participate in the transport of fats, fatty acids and cholesterol. ATP acts as a form of energy conversion in living cells, while protein phosphorylation/dephosphorylation is a way of transmitting the life signal. There are reasons to assert that under the conditions of ancient Earth, phosphorus could contribute to the formation of amino acids, nucleosides/nucleotides, and basic groups and peptides. It is believed that phosphorus was the catalyst for the evolution of the ancient Earth. A series of experiments were reported, the authors of which concluded that phosphorus could have an effect on the prebiotic synthesis of small biomolecules (amino acids, nucleosides, and basic groups) and biomacromolecules (peptides and nucleotides) [5]. When considering the chemistry of natural organophosphorus compounds and their biological significance, it is important to discuss their formation during the prebiotic period. Phosphorus is undoubtedly the central element of living organisms, and along with carbon, nitrogen, oxygen, it undoubtedly played a decisive role in the origin of life [5]. Westheimer in his article “Why nature chose phosphorus” showed that phosphates are ionized at physiological pH due to the low first pKa value (pKa = 2.2, 7.2, 12.3 [1,6]). The advantage of tribasic phosphorus acid over organic tribasic acids, for example citric acid, can be inferred from a comparison of the ratios of successive ionization constants of these acids. The sequential ionization constants of phosphoric acid differ by more than 105 times, while the constants for citric acid differ by less than 50 times, even taking into account statistical factors.
The negative charges of phosphoric acid diesters in DNA help retain molecules within the cell membrane and give nucleic acid the stability necessary for genetic reproducibility. The ionic nature makes phosphates hydrophilic and promotes their retention in the cell membrane. It is important to note that in the case of RNA and DNA ligation, the ionic structure of phosphates allows the ligation of two nucleosides whilst retaining a negative charge at the phosphodiester.The data of Table 1 is taken from [1,6] and https://ru.wikipedia.org, accessed on 2 January 2021.
The charge carried by the bonds of phosphodiesters between nucleotides provides an important solubilizing element and, importantly, protects the phosphodiesters from hydrolysis. Unlike carboxylic acid anhydrides, phosphoric anhydrides are protected by their negative charges from the rapid impact of water and other nucleophiles, so they can persist in an aqueous environment, even if they are thermodynamically unstable, and thus, can control chemical processes (Figure 2) [7].
Phosphorus plays an important role in genetics and in the energy systems of living cells, as well as in the cell membranes. An alternative hypothesis is that a protometabolism could have emerged prior to the incorporation of phosphates. It has been suggested that thioether-based metabolism could have pre-dated the incorporation of phosphates and an RNA-based genetic system [8].
Under the control of DNA ligase, covalent cross-linking of DNA strands in a duplex occurs during replication, repair and recombination. They form phosphodiester bridges between the 5′-phosphoryl and 3′-hydroxyl groups of adjacent deoxynucleotides at DNA breaks or between two DNA molecules. For the formation of these bridges, ligases use the energy of hydrolysis of the pyrophosphoryl bond of ATP, in particular, under the influence of DNA ligase of bacteriophage T4. Considering the influence of phosphates on the most important life processes, it is important to take into account the role of phosphate in the formation of primary organisms [8], lipids and genetics [9,10,11], considering phosphates as the main element in the energy system of nascent organisms. Therefore, the role of inorganic phosphates in the prebiotic period of the early Earth attracted considerable attention and was widely discussed. Various theories have been proposed explaining the role of phosphorus in the formation of primary organisms that led to the emergence of life on Earth. For example, the availability of phosphorus has been widely discussed, due to the fact that orthophosphate (PO43−), a common form of phosphorus on Earth [12]. The mineral apatite, which is the significant source of phosphate on Earth, has long been thought to be problematical in this respect due to its low solubility and reactivity. The insolubility and thus, the insufficient availability of phosphate minerals has been called the “phosphate problem” [13,14], for the phosphate to react and has been used in prebiotic chemistry. The use of phosphates requires an aqueous medium with a low content of dissolved calcium, which can precipitate orthophosphates, making it difficult their participation in the formation of prebiotic organisms [15,16,17,18]. Fernandez-Garcia et al. [1] described several prebiotic pathways for the synthesis of nucleotides, amino acids and basic metabolites, which are controlled by phosphate, which acts as an acid-base catalyst or chemical buffer. The mechanisms that use phosphates for organizing selective prebiotic chemistry, combined with the central and universal role of phosphates in biochemistry, provide an increasingly clear idea that phosphates were a key element in the origin of life on Earth.
The mineral schreibersite (Fe,Ni)3P is a reactive source of phosphorus capable of forming phosphorylated molecules [16,18]. These molecules could be an important component of prebiotics. The authors suggest two ways of using phosphorus in the formation of living organisms in the prebiotic period: The first is the delivery of phosphides by meteorites, and the second is the reduction of phosphates to phosphides under conditions of high temperature and low redox potential. It is estimated that about 1–10% of all phosphorus in the earth’s crust was in schreibersite during Hadean eon, making this mineral likely to react with organic-rich water. Ultimately, such conditions could presumably contribute to the formation of life in the form in which we know it today [18]. The mechanisms of aqueous electrophilic and aqueous nucleophilic phosphorylation, as well as phosphorylation in the anhydrous state, which is carried out by dissociative phosphoryl transfer, were discussed. The mechanisms by which phosphate controls the reactivity of prebiotics through catalysis or buffering effects have been discussed, promoting selective conversions in neutral water. Several prebiotic pathways have been described for the synthesis of nucleotides, amino acids and basic metabolites, which are facilitated or controlled by phosphates, which act as a basic acid-base catalyst, pH buffer or chemical buffer. These mechanisms lead to the conclusion that understanding phosphate chemistry is a key element in elucidating the origin of life on Earth [12].
It has been suggested that most potential prebiotic phosphorylation reactions are based on phosphate anhydrides as sources of phosphorus. In addition, it is quite possible that the geochemistry of phosphorus on the early Earth was controlled by phosphorus compounds with a reduced oxidation state, such as hypophosphite (H2PO2−), which are more soluble and reactive than orthophosphates. It was suggested that phosphorus with a reduced oxidation state was introduced from extraterrestrial space [15,16].
The most important criterion used to define a living organism is the ability to metabolism, constant renewal and reproduction, the ability to convert molecules that have absorbed solar energy into building blocks of their own structures. These functions, like many others, require an energy system in which energy can be stored, transported and released in a controlled and timely manner. The most common concept of the origin of nucleic acids and life is the hypothesis of the RNA world supported by many scientists. There are other concepts as well. For example, the progene hypothesis proposed by Altstein, which is a development of the chemical evolution hypothesis [7]. According to this hypothesis, the bimolecular genetic system does not arise from mononucleotides and monoamino acids, but from precursors, namely trinucleotides aminoacylated at the 3′-end with a non-random amino acid (NpNpNp~pX~Aa, where N-deoxyribo- or ribonucleoside, p—phosphate, X—a bifunctional agent, for example ribose, Aa—an amino acid, ~macroerge bond. Progenes are used as substrates for the interconnected synthesis of a polynucleotide and a polypeptide. The growth of the “polynucleotide-polypeptide” system is controlled by the enzymatic properties of the growing polypeptide. The mechanism of progen formation (NpNp + Np~pX~Aa) explains the emergence of the prebiotic physicochemical genetic code of the group, as well as the selection of organic compounds for the future genetic system from the racemic heterogeneous environment. The system is reproduced on a progenetic basis by means of replication-transcription-translation (the first molecular genetic process). The weak point of this theory is the impossibility of explaining how the stereoselective selection of enantiomers from the racemic mixture is carried out, since in this case the process should occur with violation of chiral parity, which is unlikely.
The definition of life as a way of existence, evolution and transmission of information recorded on an organic matrix attracts attention. For example, it was proposed to consider life as “a highly stable state of matter, using information encoded by the states of individual molecules to develop conserving reactions”. That is the Life is a stable informational state capable of independent existence, development and self-reproduction. An elementary representative of such an information state is a living cell, which is essentially a biocomputer filled with a huge volume of information concentrated in DNA or RNA. DNA can carry out its activity only inside the cell with its full complex of vital processes. A cell without DNA cannot support the metabolic process and the work of the energy block, which is performed by phosphagens. Therefore, in order to be viable, all elements of the organism must be formed simultaneously and within a short time, which excludes the possibility of chemical evolution over billions of years. The elements of the body that support metabolism are very unstable, which is why the metabolic process can easily be disrupted. RNA, DNA, proteins are polymeric homochiral molecules consisting of thousands of chiral units that must be constantly renewed. It should be borne in mind that racemization of only one unit of chain will disrupt the configuration of the polymer molecule, as a result of which the double helix of DNA molecule will be impossible. For example, the half-life for ribose decomposition is 73 min at 100 °C and pH 7, and 44 years at 0 °C and pH 7. Other sugars are similarly unstable at 100 °C and pH 7. Examples are ribose 5-phosphate (t1/2 = 9 min), deoxyribose (t1/2 = 225 min) and ribose-2,4-diphosphate (t1/2 = 31 min) [19].
Geological research suggests that the first forms of life on Earth were microorganisms that appeared about 4 billion years ago. The process of the primary organism formation was short-lived. This conclusion is confirmed by biochemical studies. For example, Lazcano and Miller [3] came to the conclusion that Archean cells had a random rate of duplicons fixation and a rate of spontaneous gene duplications comparable with the present values of 10−5–10−3 gene duplications. Hence, it requires a relatively short time for the genome of a heterotrophic DNA/protein into 7000 genes of filamentous cyanobacteria. It is well-known that directed evolution experiments take only a few weeks to rapidly proliferate in bacterial populations under stressful conditions. The results on organophosphate and phosphonate hydrolysis of phosphotriesterase from Pseudomonas diminuta and other soil eubacteria suggest that this new enzyme, as a result of duplication, separated from its family and reached the diffusion limit in just 40 years. Therefore, the rate of duplication and fixation of new genes can be very high on the geological time scale [19]. There are additional mechanisms that can increase the rate of metabolic evolution, including modular assembly of new proteins, gene fusion, and horizontal gene transfer. Experimentally, it was shown that, after a few weeks, existing enzymes acquire new substrate features as a result of recombination within the gene. Perhaps the widespread belief that the origins and early evolution of life were slow processes that took billions of years stems from the classical Darwinian approach, according to which major changes occur slowly and in stages over long periods of time. Directed evolution experiments, conducted by Hall and Zuzel [20], showed that after a few weeks, existing enzymes acquire new specific substrates as a result of recombination within the gene. In accordance with the theory of Oparin-Haldane chemical evolution, for the formation of coacervates originating in the primary broth, chiral molecules of high chemical purity (homochiral molecules) are required. Modern life is homochiral, and this is reflected in macromolecules, DNA, RNA and proteins. So far remains an unsolved mystery how homochirality originally arose, and several physical and chemical processes of “symmetry breaking (Parity violation in chiral molecules)” have been proposed. However, even the event of a violation of chirality does not solve the problem of enantiomeric cross-inhibition. First of all, biopolymers, such as DNA, RNA and protein must be homochiral, in order to adopt well-defined secondary and tertiary structures oriented to the sequence that is a prerequisite for their biological function. Moreover, in the course of chemical polymerization of D-ribonucleotide-RNA using a homochiral matrix, the building blocks of D-ribonucleotide phosphate effectively inhibit the formation of incorrect building blocks of L-ribonucleotide phosphate by chain termination. Homochirality of organic substances is inherent only in living organisms. Homochiral molecules of abiotic origin are absent in the nature of Earth. In turn, it can be noted that chiral molecules of high stereochemical purity are interconnected with a living organism, because in nature, only living organisms are capable of producing them. Consequently, the initial chiral substances that are necessary for the construction of DNA, RNA, and proteins should have been formed directly during the formation of a living organism. Therefore, summarizing the above data, it can be assumed that in nature there is a programmed process that, under certain conditions, leads to the formation of a primary Life. It is not entirely clear how this program works, but the origin and evolution of life is the subject of intense scientific research, the results of which, unfortunately, still cannot satisfy the discerning scientist [21]. Attempts to explain the emergence of chirality on Earth using chiral amino acid molecules brought by meteorites from space do not look convincing enough. Molecules of chemical compounds, including amino acids, that are delivered to Earth by meteorites, such as the Murchison meteorite, or formed under action of cosmic radiation, have very low enantiomeric excesses, while absolute chirality (homochirality) is required. In addition to meteorites, there is also the theory of panspermia, that is, the emergence of life in other worlds, in distant galaxies and the travel of living spores to Earth over tens and hundreds of light years. However, this theory also looks unconvincing. Although it he not been refuted, but equally it has not been proven. Geological studies have established that life on Earth appeared about 4 billion years ago, in time period between the Hadean and Archean eons, almost immediately as soon as the Earth cooled down to an acceptable temperature. At that time, there was no oxygen atmosphere on Earth yet, but the first anaerobic bacteria appeared, which formed many of the currently existing deposits of minerals: Sulfur, graphite, iron and nickel. The first living microorganisms could appear in limited quantities and therefore there was no deficiency in building material, including phosphorus. However, they later multiplied, changing the ecology of the Earth. Anaerobic bacteria obtain energy in the absence of oxygen through substrate-level phosphorylation, i.e., reaction that results in the production of ATP or GTP by the transfer of a phosphate group from a substrate directly to ADP or GDP. During the oxidation of organic compounds in living cells, inorganic phosphate is transferred to organic matter to form energy-rich molecules (Figure 3) [22].
It is not entirely clear whether anaerobic bacteria were the original organisms in the subsequent development of life on Earth. It is also unclear whether anaerobic bacteria evolved into subsequent species of living organisms, or these new organisms appeared independently of them, like other later forms of life, that is, the origin of life occurred repeatedly. To a certain extent, this is consistent with the theory of punctuated equilibrium in evolution [21]. Nevertheless, in any case, in all forms of life, the phosphorus played a primary role as an irreplaceable binding element of nucleotides, and also as a source of energy for maintaining vital functions of organisms.
Summing up this section, we can conclude that the emergence of life is an extremely complex, so far unresolved and at the same time important scientific problem [22]. This problem requires the close attention of many scientists in various fields of science. Its importance is determined by the fact that if we do not know the origins of life, then it is impossible to foresee what awaits life on Earth in the future. Additional information on the role and influence of naturally occurring phosphorus compounds on life processes will be presented in the following sections of review.
3. Stereochemistry of DNA and RNA
The most important and most amazing phosphorus-containing molecules is deoxyribonucleic acid (DNA)—a macromolecule (one of the three life-forming polymer molecules, along with RNA and proteins) that provides storage, transmission from generation to generation and the implementation of the genetic program for the development and functioning of living organisms [23,24]. A DNA molecule stores biological information in the form of a genetic code consisting of a sequence of nucleotides, conformations, and a convolution of a polymer molecule. DNA contains information about the structure of various types of RNA and proteins. DNA is made up of repeating blocks called nucleotides. Each nucleotide is composed of a nitrogenous base, deoxyribose, and a phosphate group. The connections between nucleotides in the DNA chain are formed by phosphodiester bonds (Figure 4) [24].
An example of a nucleotide is adenosine monophosphate, in which the base attached to phosphate and ribose is adenine. The most important energy molecule ATP (adenosine triphosphate) consists of an adenine base attached to sugar ribose and three phosphate groups. Depending on the structure of the pentose, ribonucleotides and deoxyribonucleotides are distinguished, which are monomers of biological polymers (polynucleotides)—RNA, or DNA, respectively. In nucleotides, the phosphate residue usually forms an ester bond with the 2′-, 3′- or 5′-hydroxyl groups of ribonucleosides, and in the case of 2′-deoxynucleosides, 3′- or 5′-hydroxyl groups are esterified
In the overwhelming majority of cases (except for some viruses containing single-stranded DNA), a DNA macromolecule consists of two chains oriented by nitrogenous bases to each other. This double-stranded molecule is twisted along a helical line, which can be right (A- and B-forms of DNA) or left (Z-form of DNA) (Figure 5) [25]. Phosphates have a significant effect on DNA conformation and twist. Phosphates in Z-DNA are not equivalent to each other and are located at different distances from the axis of the helix; for guanine nucleotides this distance is 0.62 nm, and for cytosine nucleotides it is 0.76 nm. At the same time, neighboring sugars “look” in opposite directions, and because of this, the line that sequentially connects phosphorus atoms in the chain becomes zigzag (hence the name—Z-DNA). B- and Z-forms are able to transform into each other. This occurs when the ionic strength of the solution or the concentration of cations that neutralize the negative charge of the phosphodiester framework changes. Deoxyribonucleic acid (DNA) is a biopolymer (polyanion), the monomer of which is a nucleotide.
The incorporation of D- and/or L-amino acids into nucleopeptide molecules makes it possible to investigate the role of backbone stereochemistry in determining the formation of DNA and RNA hybrids. The characteristic helical structure and chiral environment of DNA was studied by measuring the spectra of induced CD and X-ray diffraction of both natural and synthetic DNA, which showed that it is a long polymer molecule consisting of nucleotides, bonds which form deoxyribose and a phosphate group (phosphodiester bonds). Circular dichroism (CD) studies have shown that the nucleopeptide forms stable hybrid complexes with RNA molecules, fully formed in the L-skeleton configuration. Molecular dynamics modeling suggests a potential structure of the complex resulting from the interaction between the L-nucleopeptide and the RNA chain. These studies have shown that both the backbone (ionic and H-bonds) and nucleotide bases (pairing and π-stacking) of the chiral nucleopeptide are involved in the hybrid complex formation, highlighting the key role of the backbone stereochemistry in the formation of the nucleopeptide/RNA complexes. (Figure 6) [25,26,27].
Helical junctions are important architectural elements in RNA, and three- and four-way DNA junctions are central intermediates in homologous genetic recombinations. The ideal (4H) junctions is created by four double helixes that are held together by interchange of strands (Figure 7a). Experimental confirmation of the existence of quadrilateral bonds of cruciform DNA molecules was obtained using electron microscopy, in which it is clearly observed in DNA molecules of X structures obtained from plasmid or bacteriophage DNA. The structure was confirmed by electrophoretic, chemical, and crystallographic studies (Figure 7b). The cruciform structure is formed by base pairing within a strand with an inverted repeating sequence, and their connections are recognized by enzymes that allow the connection. The cruciform shape is another form of quadrilateral bonds found in DNA derived from natural sources [28,29].
The folded X-structure is formed by paired coaxial superposition of spirals forming a right-sided antiparallel cross. The formation of multilayer X-structure from an open square structure involves a decrease in symmetry from four to two times. Electrostatic forces are extremely important in the folding of the quadrilateral DNA junction, as indicated by the critical role of metal ions in the folding process. Formation of a multilayer X-structure requires either bivalent metal ions or very high concentrations of monovalent ions, and the conformer exchange rate strongly depends on the salt concentration [29,30]. A method was developed to track the stereochemical pathway of enzymatic DNA cleavage [25]. In accordance with this method, DNA with a phosphorothioate of known chirality is cleaved by the enzyme in H218O at a scissor bond. Cleavage results in a DNA molecule with a 5′-[16O, 18O, S]-thiophosphoryl group, the chirality of which depends on whether the cleavage reaction proceeds according to a one-step hydrolysis mechanism or a two-step mechanism involving protein-DNA. To determine this chirality, the digested DNA was attached to the oligonucleotide using DNA ligase. Given the strict stereochemistry of the DNA ligase reaction, the initial chirality of the phosphorothioate can be retained or reversed in the product of ligation. In all cases, DNA cleavage occurred with inversion of the configuration, which indicates direct hydrolysis of the phosphodiester bond with water, in contrast to the reaction involving a covalent enzyme-DNA intermediate. Transcription of phosphorothioate-N-substituted RNA by T7 RNA polymerase has the same (RP)-configuration as E. coli RNA-polymerase. Thus, the reaction proceeds with configuration inversion for phosphorus [27]. In the cell, the binding of proteins to specific sequences of the DNA double helix is necessary to control the processes of protein synthesis (at the level of DNA transcription) and cell proliferation (at the level of DNA replication). In the laboratory, the sequence-specific DNA binding/cleaving properties of restriction endonuclease enzymes (secreted by microorganisms to protect them from foreign DNA molecules) have helped revolutionize molecular biology. DNA can take on several conformations, such as A-type, B-type, and others (for example, Z-type), depending on the DNA sequences and environmental conditions, for example, relative humidity, the ionic strength or polarity of the solvent and counterion. In contrast, RNA’s are found in only two related conformations, A and Aʹ, which belong to type A double helical structures. The conformation of A-DNA structure is very similar to the structure of A-RNA. In the B-type double helix, stacking is largely limited by interactions between bases in the same polynucleotide chain. In the A-type double helix, stacking occurs both inside and between the overlapping strands, and bases belonging to two different chains participate in the stack.
In recent years, several successful attempts have been made to use DNA as chiral ligands in transition metal complexes in order to combine the catalytic capabilities of the transition metal with the chiral architecture of the biopolymer [31,32,33,34,35,36,37,38,39,40]. The hybrid catalyst comprises a synthetic catalyst, usually a transition metal complex, which is attached to a chiral biomolecular backbone. For example, salmon testes DNA (stDNA) and calf thymus DNA (ctDNA) are used as ligands. As a result, the catalyzed reaction takes place inside the DNA helix or very close to it, that allows DNA chirality, which is transferred into the reaction, leading to products having an excess one of enantiomers. Two general strategies can be distinguished, which are based either on supramolecular (non-covalent) or covalent attachment of the transition metal complex to DNA (Figure 4). The strategy of non-covalent or supramolecular anchoring is based on the tendency of DNA to bind small molecules using hydrophobic, π-stacking, electrostatic and/or hydrogen interactions leading to intercalation and/or binding of grooves. Supramolecular anchoring can be achieved by incorporating a DNA-binding moiety into a metal center ligand design. Covalent anchoring involves the binding of a transition metal complex through a ligand to DNA using a small spacer fragment. The attachment sites in this case can be modified nitrogenous bases or phosphate esters. The strategy of non-covalent or supramolecular anchoring includes the DNA-binding fragment in the ligand construct (Table 2). Rolfes and Feringa [39] presented a new concept of DNA catalysis, which realized the first DNA-catalyzed formation of symmetric C-C bonds in the Diels-Alder reaction. In the presence of a copper complex, asymmetric catalysis using DNA proceeded especially efficiently as a ligand for transition metal complexes that catalyze Michael addition, Friedel-Crafts alkylation, and others. G-Quadruplex, which induced reasonable levels of enantioselectivity in both Diels-Alder and Friedel-Crafts reactions, confirmed that the G-quadruplex DNA can also act as a suitable chiral template (Figure 8).
The authors have successfully used L-DNA as an enantiomer with the opposite configuration than natural D-DNA. Indeed, L-DNA forms duplexes with identical physical properties in terms of the solubility and thermal duplex stability, resulting in levorotatory double-stranded helices (Table 2).
Erdmann and co-workers recently demonstrated the preferential stabilization of the natural D-configuration in versus the unnatural L-configuration in nucleic acids and have established using X-ray diffraction analysis of crystals grown under microgravity conditions that the D- and L-forms differ not only in their chirality, but also by way of interactions with divalent cations and water molecules (Figure 9) [32,33,34,35,36,37,38].
The chirality of the DNA double helix can be transferred directly to the reaction sites in reactions catalyzed by metal complexes. For example, a complex of DNA with copper catalyzed the Diels-Alder reaction. This was accomplished by placing a non-chiral or racemic catalyst in close contact with DNA and using the chiral information of the DNA double helix to obtain reaction products with an excess of one of the enantiomers. The Table 2 is constructed from different data extracted from article [31].
The catalyst is a complex formed in situ from copper(II) with a DNA ligand that contains three key structural features: A DNA intercalating moiety such as 9-aminoacridine, a spacer component, and a metal-binding group. The ligands were obtained in an efficient and simple way, starting from monoprotected diamines. The Diels-Alder reaction between cyclopentadiene and aza-chalcone was carried out in water in the presence of a copper (II) complex in the presence of salmon testes or calf thymus DNA, both of which are readily available. The reactions were brought to a conversion of >80%. Product of reaction was obtained as a mixture of endo- (major) and exo (minor) isomers, both with significant enantiomeric excess, depending on the ligand used [40]. Conversion of a centrally located phosphate group to an electrically neutral methylphosphonate in a quadrilateral DNA junction can have a large effect on its conformation (Figure 10) [33].
The methodology using DNA catalysts is now at the stage where it can be applied in the process of synthesis. Indeed, the first examples demonstrate that these reactions can be competitive with their “traditional” counterparts, both in terms of practicality and cost. DNA catalysts containing covalently linked catalytic moieties potentially provide greater control over catalyst geometry using a second coordination sphere [40]. Today, using DNA catalysis, an impressive number of chiral compounds with high ee have been obtained, some of which are shown in Figure 11 [33,34,35,36,37,38,39,40].
4. Phosphagens
The important natural compounds of phosphorus are phosphagens, which are energy sources for maintaining vital functions of organisms. Macroergic phosphagens are biological molecules that are capable of storing and transferring energy during a reaction. The hydrolysis of one bond in phosphagen gives usually more than 20 kJ/mol. About 15 representatives of phosphagens are known to support the vital activity of organisms. First of all, adenosine triphosphate (ATP), as well as about 15 phosphagens that contribute to the preservation and restoration of ATP reserves: N-phosphoguanidine, N-phosphocreatine, N-phosphoarginine, N-phosphocreatinine, 1,3-diphosphoglycerate, N-phosphotaurocyamine, 1,3-bisphospoglycerate, phosphoglycocyamine, phosphoagmatine, N-phospholombricine, phosphoopheline, phosphotaurocyamine, phosphothalassemine phosphohypotaurocyamine, phospholombricine (Figure 8) Each phosphagen has its own corresponding phosphagen kinase: arginine kinase, thalassemine kinase, creatine kinase, glycocyamine kinase, taurocyamine kinase, hypotaurocyamine kinase, lombricine kinase and opheline kinase (Figure 12) [41].
Depending on the type of bond being broken, phosphagens release different amounts of energy. For example, during the hydrolysis of phosphoanhydride bond (nucleoside triphosphates and nucleoside diphosphates: ATP, HDF and their analogs), ΔG = 32 kJ/mol is released. The thioether bond gives 42 kJ/mol; acyl phosphate bond (1,3-diphosphoglycerate. 1,3-diphosphoglycerate) ΔG = 46 kJ/mol; and the enol phosphate bond (phosphoenolpyruvate) ΔG = 54 kJ/mol. A universal source of energy in a cell (including muscle) is the free energy of the high-energy phosphate bond of adenosine triphosphate (ATP), released during the hydrolysis of ATP to adenosine di- and adenosine monophosphate (ADP and AMP) and an inorganic phosphorus compound [42,43]. However, the ATP contained in the muscles is sufficient to ensure work for a very short time, therefore, during muscle work, the energy of ATP is used, which is synthesized directly during work using the energy of other high-energy substances contained in the cell, including phosphates (phosphagens). Studying the mechanism of transfer of phosphoryl groups in natural phosphates is important for understanding the main metabolic pathways and for transmitting the cellular signal of fundamental processes in living systems. Monomolecular and bimolecular reactions of nucleophilic substitution at the phosphorus atom SN1(P) and SN2(P) are well known in the chemistry of phosphorus. SN1(P) reactions are involved in the processes of genetic inheritance through nucleic acids and in the generation of chemical energy, which makes it possible to stimulate thermodynamically unfavorable processes necessary for the construction of living cells. There are the following mechanisms of phosphoryl group transfer in substitution reactions at the phosphorus atom: (a) Dissociative mechanism of the SN1 type, which includes the formation of a stable metaphosphate ion (PO3−), which interacts with the nucleophile at the stage of the rate-limiting reaction; (b) an associative, two-step attachment-elimination mechanism through the formation of an intermediate phosphorane (Figure 13) [42,44].
The mechanism of the ATP hydrolysis reaction proceeding through the formation of an intermediate metaphosphate was described by Westheimer [45]. The dissociative method of breaking the P–O–C bond with the formation of an intermediate metaphosphonate is observed in the case of a sterically hindered phosphorus compound, most often in weakly nucleophilic solvents. The pentavalent tri-coordinated metaphosphate is very reactive in aqueous solutions. Consequently, it cannot be registered in aqueous solutions because of the insignificant lifetime under these conditions. However, metaphosphate can be detected in highly polar and weakly nucleophilic nonaqueous media [46,47,48,49,50]. In addition, the metaphosphate anion PO3− exists in a stable form in the gas phase and has been recorded by various physical methods. In the gas phase, metaphosphate has a relatively low reactivity. The enthalpy of deprotonation of HPO3− in the gas phase is only 314 kcal/mol. Metaphosphate anion was recorded in the mass spectra of various pesticides, in the products of ionization cleavage of phosphates, in the mass spectra of adenosine-50-monophosphate, in various phosphate triesters, etc. (Figure 14) [51,52,53,54,55].
The stereochemistry of the SN1(P) reaction proceeding through the formation of an intermediate metaphosphate was studied using the example of phosphates carrying oxygen isotopes. The methanolysis of phenylphosphate monoanion and dinitrophenyl phosphate dianion using [(R)-16O,17O,18O]-phosphocreatine, proceeded with a complete inversion of the configuration was observed at the asymmetric phosphorus center. It was found that the intermediate metaphosphate in protic solvents does not leave the solvation cell in which it was formed, and then enters into a further reaction, which creates conditions for an asymmetric reaction [56,57,58,59]. The metaphosphate ion was detected by X-ray diffraction analysis in some biological molecules in the form of a particle stabilized by coordination bonds, for example, in fructose-1,6-bisphosphatase. The crystals of fructoso-1,6-bisphosphatase were grown up in an equilibrium mixture of substratum and product in almost atomic resolution (1,3 Å) (Figure 15) [60].
Phosphocreatine, as a source of energy for muscle contraction, plays a leading role in the work in the anaerobic alactate power zone, when its reserves in muscle cells limit the duration and intensity of physical activity. The phosphocreatine is a product of reversible metabolic N-phosphorylation of creatine, which, like ATP, is a high-energy compound. However, in unlike ATP, which is hydrolyzed at the pyrophosphate O-P bond, phosphocreatine is hydrolyzed at the phosphamide N-P bond to give a significantly greater energy effect of the reaction. The phosphocreatine is found mainly in muscle and nerve tissues and its biological function is to maintain a constant concentration of ATP due to a reversible rephosphorylation reaction: creatine phosphate + ADP ⇄ creatine + ATP. Thus, the phosphocreatine is an energy depot that provides a rapid synthesis of ATP (Figure 16) [61].
5. Natural Compounds with P-N Bonds
N-Phosphamides. A large number of various organophosphorus compounds with P-N bonds have been isolated from living organisms, which indicates the importance of these compounds in nature. For example, the N-phosphocreatine is one of natural P-N-amidophosphates, which have a significant impact on the formation and development of living organisms, starting from the prebiotic period [62,63]. Many amidophosphates have cytostatic, antitumor, chemotherapeutic properties to be alkylating-type drugs such as cyclophosphamide, diphosphamide, mafosfamide, trophosphamide, perphosphamide and others (Figure 17) [62,63,64,65]. Guanidine compounds containing PN-bonds play a central role in the physiology and biochemistry. For example, up to 20–25% of N-phosphocreatine is converted in vivo, through the intermediate formation of N-phosphocreatinine to creatinine [62,63]. Natural phosphoramides such as N-phosphocreatine and phosphoramides shown in Figure 18 are formed by phosphorylation of histidine groups in histidine kinases.
An analog of N-phosphocreatinine, called dimethyl-N2-creatinyl phosphate, was isolated from a sponge found in Bermuda [64]. Ulosantoin, which is a phosphorylated hydantoin, was isolated from extracts of the Ulosa ruetzleri sponge and exhibited marked insecticidal activity against tobacco hornworm larva and cockroaches. The structure of Ulozantoin was determined by X-ray structural analysis. A structurally related compound, dimethyl N2-creatinyl phosphate, was also isolated from the sponge extracts, but was inactive in the insecticidal screens (Figure 19) [65].
Eschenmoser et al. [66] showed that glycolaldehyde is phosphorylated by AmTP2 in the presence of divalent metals in water at room temperature and at neutral pH, with the formation of glycolaldehyde phosphate (GAP) in quantitative yields. A specific phosphorylation reagent in this process is amidotriphosphate, a compound known to be formed by the ammonolysis of cyclotriphosphate (“metatriphosphate”) in aqueous solution. It is noteworthy that cyclotriphosphate itself is not effective as a phosphorylation agent under the same conditions, its ammonolysis product phosphorylates glycolaldehyde in quantitative yield. The reason for this marked difference in reactivity is the specific ability of amidotriphosphate to reversibly form the addition product of carbonyl A with glycolaldehyde and to phosphorylate the α-hydroxyl group by intramolecular migration of the phosphate group from A to intermediate B, which is then hydrolyzed to GAP (Figure 20) [66].
The stereochemistry of P-N phosphates, isolated from natural sources, is relatively monotonous. Most natural P-N phosphates whose stereochemistry has been established by reliable methods are derivatives of L-amino acids and D-carbohydrates. Therefore, they contain asymmetric centers of the corresponding absolute configurations. For example the reaction shown in the Figure 21. Sutherland et al. [67] investigated the potentially prebiotic synthesis of nucleotides via arabinose-3-phosphate and its cyanamide derivative in an attempt to effect constitutional self-assembly of RNA via a simple pathway to RNA, including nucleotide polymerization. With this aim, the authors investigated several synthetic approaches, including phosphorylation, phosphate migration and 2,3-C-C bond building. In particular, they investigated the phosphorylation of arabinose by diamidophosphate (DAP). An aqueous solution of D-arabinose and DAP was stirred at room temperature for several days, taking aliquots for NMR spectroscopic analysis at regular intervals (Figure 21) [66,67].
An example of a natural compound containing a P-N bond is adenosine-5′-phosphoramide, which was isolated from Chlorella pyrenoidosa cells by high performance liquid chromatography; adenosine-5′-phosphoramide was also synthesized from adenyl 5′-phospho-sulfate and ammonia by adenylyltransferase and SN-enzyme, which is found in various organisms, including bacteria, algae, fungi, and higher plants [68,69]. Therefore, adenosine-5′-phosphoramidate is a normal component of chlorella cells, as is the enzyme that forms it: adenylyl sulfate: ammonia adenylyltransferase. The enzyme that catalyses the formation of adenosine 5′-phosphoramidate from ammonia and adenosine 5′-phosphosulphate was purified to homogeneity from Chlorella by using (NH4)2SO4 precipitation and DEAE-cellulose, Sephadex and Reactive Blue 2-agarose chromatography In a non-enzymatic reaction, both adenosine-5′-phosphoramide and AMP are formed. However in the presence of enzyme adenosine-5′-phosphoramide, which selectively accelerate an already ongoing reaction, the Adenosine 5′-phosphoramidate is formed. Auxenocklorella pyrenoidosa has been identified in cell extracts of many other organisms, including Chlorella pyrenoidosa, Euglena gracilis var. bacillaris, spinach, barley, Dictyostelium discoideum and Escherichia coli (Figure 22) [68,69].
In addition to the identification of individual phosphoramide nucleosides, N-adenylylated proteins were found in the membrane fraction of Dictyostelium discoideum [70,71,72,73]. Eukaryotic cells encode AMP-lysine (AMP-N-epsilon- (methyl N-alpha-acetyl lysinate) -5′-phosphoramide) hydrolases related to the nucleotide binding protein 1 sequence of the rabbit histidine triad (Hint1) [74,75]. Adenosine 5′-phosphoramide (NH2-pA) is a rare naturally occurring nucleotide and its biochemistry and biological functions are poorly recognized (Figure 23).
All organisms have proteins that can be involved in NH2-pA catabolism. They are members of the HIT family of proteins and catalyze the hydrolytic cleavage of NH2-pA into 5′-AMP and ammonia. Hint1 (Histidine triad nucleotide binding protein 1) is a protein encoded by the gene of the same name, located in humans on the short arm of chromosome 5. It is involved in such biological processes as apoptosis, transcription, regulation of transcription. Hint proteins are present in almost all types of living organisms [70,71,72,73,74,75,76,77].
The histidine triad protein binding nucleotide is an enzyme that hydrolyzes lysine-bound AMP and in yeast functions as a positive regulator of the C-terminal domain kinase of RNA polymerase II, Kin28. Rossomando et al. [73,78] reported on protein release and characterization of phosphoamidates formed as a result of a membrane-bound adenylyltransferase reaction. The N-adenylyl-L-lysine derivatives are found, either as covalent enzyme substrate intermediates in DNA ligase EC 6.5.1.2 (i.e., enzyme catalyzing the ligation of DNA strands with 3′-hydroxyl and 5′-phosphate termini) in Esckerickia coli and RNA of wheat germ or as substrates of the evolutionarily conserved family of Hint phosphoramide hydrolases AMP/GMP (Figure 24) [74].
Among the nucleotides containing the P-N bond, nucleotide antibiotics, which are adenosine-containing phospholipids, attract attention. For example, stearic and palmitic acid dinogunellins and their structurally related dinogunellins A-D. A distinctive feature in dynogunellins is the presence of a phosphoramide P-N bond common to many nucleotide antibiotics, for example, fosmidosines, antifungal nucleotide antibiotics from Streptomyces durhameusiz (Figure 25) [79,80,81,82,83,84,85,86,87,88]. The fosmidosine has a specific inhibitory activity against spores of Botrytis cinerea, a worldwide pathogenic fungus that causes gray rot disease in various fruits and vegetables [74,80].
Fosmidosine has a powerful antitumor effect and is able to stop cell growth in the G1 phase of the cell cycle. Another example is Fosmidosine B, where 8-oxoadenosine is attached to proline through a similar phosphoramide bond. This antifungal antibiotic, derived from Streptomyces durhmeusie, also has anti-cancer activity. Both isolated diastereomers of fosmidosine have similar, but about 10 times higher, antitumor activity than fosmidosine B (Table 3 and Table 4) [74]. The structure of phosmidosine was determined by various methods, including mass spectrometry and NMR spectroscopy [81]. Fosmidosine has a chiral center at the phosphorus atom of the N-acylphosphoramide bond and therefore exists in the form of two diastereomers (Figure 26). Demethylated derivative of Phosmidosine, Phosmidosine B, has a achiral phosphorus atom, and thus, has no phosphorus chirality (Table 3). Phosmidosine was also successfully synthesized by use of 5-(3,5-dinitrophenyl)-1H-tetrazole (DNPT) as the powerful activator of an N-prolyl phosphorodiamidite derivative that was condensed with an 8-oxoadenosine derivative.
The phosmidosine derivatives were found to exhibit significant antitumor activities. In connection with these studies, the synthesis and biological properties of aminoacylamido-AMP derivatives having N-P bonds as stable analogues of aminoacyl-AMP were also described (Table 4) [80,81].
The phosmidosine B was successfully synthesized by reaction of N-acetyl-8-oxoadenosine-5′-O-phosphoramidate derivative with N-protected prolinamide in the presence of 5-(3,5-dinitrophenyl)-1H-tetrazole. The synthesis of phosmidosine was achieved through the use of a tert-butoxycarbonyl (Boc) group, which is selectively introduced into the 7-NH-function of 8-oxoadenosine and serves as a pseudoprotective group so that the unmasked 6-amino group is not phosphorylated. The final reaction of coupling the 8-oxoadenosine-5′-phosphoramidite derivative with N-tritylprolinamide and subsequent complete deprotection gave a mixture of phosmidosine diastereoisomers. The 13C NMR spectra of diastereomers suggest that the slowly eluted diastereomer is natural phosmidosine (Figure 27) [74].
A number of phosphoramide nucleotide antibiotics, called phosmidoses, were isolated from the extract of Streptomyces durhameusis (fosmidosine, fosmidosine B and fosmidosin C), as well as two variants of N-methylphosmidosine. Fosmidosines appear to inhibit spore formation of Botrytis ccncrea at a concentration of 0.25 μg/mL. Botrytis cincrea is a common pathogenic fungus responsible for gray mold disease in a variety of commercially important fruits and vegetables. Based on the conducted in vitro studies, it was proposed to use fosmidosines as potential anticancer agents. Phosmidosine analog, having an N-acylsulfamate linkage and possessing potent antitumor activities against cancer cells was synthesized by the sulfamoylation of an 8-oxoadenosine derivative [89,90,91]. Sekine et al. reported the synthesis of a stable biotin–phosmidosine that is required for isolation of biomolecules that bind to phosmidosine (Figure 28) [91].
The selective antibiotic Agrocin 84, which is a member of the adenine nucleotide family, the first member of which is 6-N-AMP (R = H) [83], has attracted close attention. Agrocin 84, which is a 6-N-phosphoramide, was isolated from Agrobacterium radiobacter K84 found in Australia [82,83,84,85,86]. Agrocin 84 is selectively active against several strains of phytopathogenic agrobacteria such as Agrobacterium tumefaciens and Agrobacterium rhizogenes. The toxic effect is achieved by inhibiting the pathogen tRNA synthetase [87,88]. The 1-phosphoglucofuranose sugar fragment at position N-6 is required for proper transport by susceptible bacteria, but it is not essential for the toxicity of agrocin 84. The complete synthesis of the nucleotide antibiotic Agrocin 84, which has two different types of unique phosphoramide bonds, has been described in detail [80]. Agrocin 84 is an active factor in the biological control of coronal gall, a plant cancer caused by certain strains of Agrobacterium. Agrocin 84 neutralizes the pathogenic plant bacterium Agrobacterium tumefaciens (Figure 28).
The activity of agrocin 84 correlates with the pathogenicity and the presence of nopaline (N2-(1,3-dicarboxypropylarginine)) tumor-inducing plasmid. Structural analysis shows that the 5′-phosphoryl bond of the nucleoside core of 9-(3′-deoxy-β-D-threopentofuranosyl) adenine with the amide group of D-threo-2,3-dihydroxy-4-methylpentanamide is necessary for the manifestation of antibiotic activity, but the bactericidal specificity is due to the presence of the D-glucofuranosyloxyphosphoryl substituent at the N6 adenine. Agrocin 84 acts as a molecular Trojan horse, mimicking a substrate derived from a plant tumor, and thus gaining access to the interior of the cell, where it is converted into a toxic moiety (TM84). TM84 is a potent inhibitor of leucyl-tRNA synthetase (LeuRS), an important enzyme that catalyzes the condensation of leucine with its related tRNA and is responsible for the correct translation of the genetic code. It was found that TM84 uses a unique tRNA-dependent mechanism to inhibit leucyl-tRNA synthetase (LeuRS), while the TM84 producer prevents self-poisoning by expressing resistant LeuRS and AgnB2 (Figure 29).
Agrocin 84, when interacting with the pathogen, is cleaved into a toxic fragment of TM84 and a transport fragment. TM84 controls leucyl tRNA synthetase, thereby inhibiting the aminoacylation of tRNALeu. This subsequently leads to the cessation of protein synthesis in the pathogen and leads to cell death. Probably, the stable toxic component of Agrocin 84 can exert an antibiotic effect on bacteria by binding to the catalytic domain of the LeuRS genome-encoded A. tumefaciens as a Leu-AM mimetic (Figure 30) [86].
Microcin C (McC) is a member of the microcin family containing a heptapeptide covalently linked to 3-aminopropyl AMP via an acylphosphoramide bond. McC is a «Trojan horse»-like inhibitor of aspartyl-tRNA synthetase, endowed with strong antibacterial properties, in which the heptapeptide fragment is responsible for the transport of the inhibitory metabolite into the bacterial cell [91,92]. The analogue of aspartyl AMP formed inside the cell carries a chemically more stable phosphoramide bond in comparison with labile aspartyl adenylate and, in addition, is esterified with a 3-aminopropyl group. Biosynthesis and antibacterial activity mechanism [92,93] of McC are well-studied [94,95,96,97,98,99,100,101]. It has been found that some species of bacteria produce antibiotics containing peptide and nucleotide moieties for protection. Escherichia coli, for example, produces the antibiotic Microcin C7 [96]. The peptide moiety of this phosphoramide mediates the transport of the antibiotic to the target bacterial cell, where processing leads to the breaking of the peptide bond between alanine and aspartic acid, releasing adenylphosphoramide aspartic acid, which acts as an inhibitor of protein synthesis in the target bacterium. A similar defense mechanism is used by the eggs of the Japanese fish Stichaeus grigorjewi, which produce the toxin dinogunelin (Figure 31).
Microcins C (McC) consists of a peptide with formylmethionine at the lateral nitrogen and C-terminal asparagine linked to nebularin-5′-monophosphate through a trimethylene bridge. MccC51 is produced by E. coli extract and inhibits the growth of a number of bacteria. The antibiotic is active against gram-negative bacteria of various taxonomic groups, as well as some gram-positive bacteria. As in most ribosomally-synthesized bacterial peptides, the N-terminus of the peptide moiety of McC bears a formyl cap. Various McC derivatives were obtained, that lack either the formyl group (for example, the deformylated McC, 4), the aminopropyl ester moiety (2), or both (3, Figure 31). McC derivatives lacking a formyl group (4, Figure 32) exhibit much lower activity than native McC (1), and a specific deformylase is required to start McC processing. It was shown in vitro that deformylation of the first methionine residue is the limiting step in McC processing [96,97,102]. Chemical structure of different McC derivatives shown in Figure 32: (1) Native McC; (2) obtained from cells harboring a plasmid with either disrupted mccD or double-disrupted mccD and mccE; (3) obtained from mutant cells lacking PepA, B, and N, and harboring a plasmid with disrupted mccD; and (4) deformylated McC, obtained from mutant cells lacking Pep A, B, and N [97].
Microcins (Mc) are peptide antibiotics isolated from gram-negative bacteria that are similar to bacteriocins from various sources. They range from linear unmodified peptides to structures that undergo extensive post-translational modification, resulting in significant chemical diversity. Mc are produced by enterobacteria [97,98,99,100,101], and have potent antibacterial activity against closely related species. Typical gene clusters encoding a microcin precursor, are located either on plasmids or on the chromosome [102]. Unlike most antibiotics of microbial origin, which are synthesized by multimodular enzymes that represent peptide synthetases, Mc are synthesized by means of ribosomes and then undergo enzymatic modification. Mc form a class of potent antibacterial peptides. At the moment, fourteen microcins have been registered, of which seven have been isolated and characterized. Since aminoacyladenylates generally poorly penetrate into bacterial cells, natural compounds that mimic aminoacyl-adenylate intermediates often employ a “Trojan-horse” strategy, wherein the active component is conjugated with a specialized module that facilitates its active transport into bacterial cell. The transport module is subsequently removed, releasing the inhibitor inside the cell. The naturally occurring aminoacyladenylate-based antibiotics include, for example: Aspartyl-adenylate synthesized by aspartyl-tRNA synthetase (AspRS); Leucyl-adenylate synthesized by leucil-tRNA synthetase (LeuRS); Seryl-adenylate synthesized by seryl-tRNA synthetase (SerRS) (See Figure 33) [102].
Antibiotics EM 2487 and 1100-50 were isolated from Streptomyces sp. Mer-2487 and Streptomyces lavendulae SANK 64297, respectively [103,104,105,106,107,108]. Amidophosphate 1100-50 was isolated from the culture of the SANK 64297 strain. The related compound EM 2487 also isolated from Streptomyces lavendulae has anti-HIV activity due to the inhibitory activity of Tat protein biosynthesis at the transcriptional level. This compound has an inhibitory effect on HIV-1 replication in chronically infected cells, as well as in acutely infected cells [103,104,105,106,107,108]. The anti-nematode activity of compound 1100-50 may be due to inhibition of RNA synthesis. Antibiotics 1100-50 and EM2487 differ in their nucleoside substructural composition. EM2487 contains a uridine ring; while 1100-50 includes cytidine. In addition, EM2487 contains an N-methylated hydroxylamine group, while the hydroxylamine group (1100-50) is not N-methylated. Both EM2487 and 1000-50 were tested as potential agents against HIV and non-mitocide, respectively. The third known phosphoramide pyrimidine nucleotide antibiotic HC 62 was isolated from Bacillus sp. HC-62 (Figure 34) [106].
N-phosphorylated proteins and peptides. “Reversible phosphorylation of protein side chains regulates almost all aspects of cell life” (Philip Cohen, 2002 [107]). Phosphorylation and dephosphorylation, catalyzed by protein kinases and protein phosphatases, can alter protein function in almost every possible way. For example, by increasing or decreasing its biological activity [108]. The simplicity, flexibility, and reversibility of phosphorylation, combined with the availability of ATP as a phosphoryl donor, explains its choice as the most general regulatory device adopted by eukaryotic cells. It is believed that 30% of the proteins encoded by the human genome contain covalently bound phosphate, and abnormal phosphorylation is currently recognized as a cause or consequence of many human diseases. Phosphorylation of amino acids is a widespread way for regulating the activity of key cell proteins, including enzymes and proteins of signaling pathways [108,109,110,111]. The protein phosphorylation is understood to mean the attachment of a phosphate group through a phosphoether bond (O-phosphorylation) to the hydroxyl group of the side chain of serine, threonine or tyrosine residue, with ATP being the phosphate donor. In the overwhelming majority of cases, phosphorylation occurs precisely at these three amino acid residues. The introduction of a phosphoric acid residue into a protein molecule, as a rule, changes its properties. This is due to the chemical nature of the phosphate group, which can form hydrogen bonds and electrostatic interactions with the components of the protein molecule. As a result, the spatial structure of the protein can change and, as a result, its activity and ability to bind with other molecules [109,110,111]. Phosphorylation of amino acid residues (Asp, Glu, forming an anhydride P-O bond) and basic (His, Arg, Lys, forming a P–N bond, namely N-phosphorylation) amino acid residues usually generates high-energy molecules that are intermediates for enzymatic catalysis [107]. Arginine is one of the main amino acid residues involving into the formation of protein-DNA interfaces (Figure 35).
N1-phosphohistidine (π-phosphohistidine) and N3-phosphohistidine (τ-phosphohistidine) have been found among prokaryotes, lower eukaryotes, and mammals [112]. Phosphohistidine has been identified as an enzyme intermediate in many biochemical reactions and plays a functional role in many regulatory pathways. In contrast to the phosphoester bond of its analogs—phosphoserine, phosphotreonine and phosphotyrosine, the phosphoramide P-N bond of phosphohistidine is unstable in the presence of acids [113]. Several pathways of origin and co-evolution of nucleic acids and proteins, including amino acids phosphorylated at the N-terminus, have also been proposed as common prebiotic chemical building blocks for peptides and nucleic acids [114,115] (Figure 36).
Several natural phosphorus-sulfur bond-containing (thionophosphates and thiophosphates, respectively) are known, for example, S-phosphocysteine in several proteins and bacterial phosphorothioate modification of DNA. Phosphorus-containing phosphorothione hydrazone was isolated from the Karenia brevis dinoflagellate found off the coast of Florida and identified as an ichthyotoxin [116]. The second known small molecule of natural phosphorothione is a dimerized form of phosphorothione. This compound was isolated from the marine fungus Lignincola laevis and also exhibits cytotoxic properties (Figure 37) [117].
Bacterial clusters of dnd genes have been found to incorporate sulfur into the DNA backbone as a stereospecific modification of phosphorothioate. Phosphorothionation occurs in phylogenetically diverse bacteria with different specific sequences and variable abundance in different bacterial genomes [118]. The dnd gene products have been found to incorporate sulfur into the DNA backbone. Since the initial observation of Streptomyces lividans, the presence of dnd genes has been established in dozens of different bacteria and archaea [119]. Synthetic phosphorothioate internucleotide bonds, in which non-bridging oxygen is replaced by a sulfur atom, have similar physical and chemical properties to phosphodiesters, but give DNA/RNA increased resistance to nucleases. It was found that the modification of phosphorothioates occurs in nature in bacteria selectively in sequence and specific for configuration (RP). The widespread use of dnd systems is thought to be due to extensive horizontal gene transfer (Figure 38) [120,121,122].
6. Natural Organophosphorus Compounds with P-C Bond
Natural phosphonates are represented by low molecular weight metabolites and phosphonylated macromolecules, such as lipids, polysaccharides, and glycoproteins [123]. The widespread use of phosphonates is illustrated by the fact that in some organisms, PC compounds form the overwhelming majority of cellular phosphorus-containing molecules. For example, eggs from the freshwater snail Helisoma sp. contain 95% phosphonoglycans [123,124,125]. Aminophosphonic acids [126,127,128,129] and hydroxyphosphonic acids [130,131] are widely known, many of which have been studied in detail and found practical application [126]. These compounds are analogs of natural amino and hydroxycarboxylic acids, in which the planar carboxyl group is replaced by a tetrahedral phosphonic acid fragment. Due to the wide spectrum of biological activity, these classes of organophosphorus compounds have received considerable attention in recent decades. Some of them have found commercial applications in agriculture and medicine. For example, aminophosphonic acids and their conjugates with peptides have antibacterial, antitumor, antiviral and antifungal effects [126]. Some of their representatives have shown themselves as herbicides and plant growth regulators. The most well-known natural phosphonates are shown in Figure 39: fosfomycin (phosphonomycin) [132], bialaphos [SF-1293) [133], phosphinothricin tripeptide (PTT)] [134], phosalacine [135], trialaphos [136], dehydrophos (A53868) [137], fosmidomycin [138], plumbemycins [139], SF-2312 [140] and fosfonochlorin [141,142]. The most part of compounds shown in Figure 23 were isolated from bacteria, the antibiotic phosphonochlorin was found in fungal strains isolated from soil: Fusarium avenaceum, Fusarium oxysporum, Fusarium tricinctum, and Talaromyces flavus [141]. It is moderately active against some species of gram-negative bacteria, and its synergistic effect with glucose-6-phosphate has been observed on Staphylococcus aureus and Escherichia coli. Cyanophos, which exhibits pesticide properties, was found in Streptomyces regensis [142].
Phosphonic acids of natural origin. Fosfomycin is a structural analogue of phosphoenol pyruvate; it competes with the enzyme N-acetyl-D-glucosamino-3-o-enolpyruvyl transferase. The result is a specific, selective and irreversible inhibition of this enzyme, which ensures the absence of cross-resistance with other classes of antibiotics and the possibility of synergy with other antibiotics. Fosfomycin is used for the treatment of diseases of urinary tract and prostatitis; it is active as an antibiotic against gram-positive and gram-negative bacteria MDR and XDR. Fosmidomycin and FR-900098 have been suggested as drugs against Plasmodium, the causative agent of malaria [138]. The Streptomyces bacteria produces a tripeptide which was called bialaphos. It consists of two alanine residues and a unique amino acid “phosphinothricin”. The phosphinothricin (Gliphosate) was first synthesized as a racemate, which is used as an active ingredient in several commercial herbicidal formulations: L-Phosphinothricin is a broad-spectrum herbicide, which is an analogue of glutamate and works by inhibiting glutamine synthetase [134]. Aminoethylphosphonic acid (ciliatrin) is the most abundant phosphonate present in lower organisms. Ciliatrin and its derivatives have been studied in detail [143,144,145]. The phosphonopyruvate is a key substrate in the synthesis of many naturally occurring phosphonates: ciliatrin, phosphonoalanine, (R)-2-amino-1-hydroxyethylphosphonic acid, phosphonoacetaldehyde, phosphonomethylmalic acid and 2-keto-4-hydroxy-5-phosphonopentanoic acid [146,147]. After the discovery of natural C-P-phosphonates, such as ciliatrin, phosphonotricin, fosfomycin, etc., the mechanism of C-P-bond formation in biological systems aroused considerable interest, and a number of studies were devoted to this problem (Figure 40) [146,147,148].
It was found that the main stage of the reaction resulting in the formation of phosphonate is the rearrangement of phosphoenolpyruvate (PEP) into phosphonopyruvate [148,149,150], catalyzed by the enzymes responsible for the formation of the P-C bond. The synthesis of phosphonate antibiotics starting from phosphonoenolpyruvate is shown in the Figure 25 for the synthesis of phosphonomethylmalic acid and 2-keto-4-hydroxy-5-phosphonopentanoic acid. Following this figure, the fosfomycin is formed as a result of the biocatalytic rearrangement of phosphoenolpyruvate into phosphonopyruvate under the action of PEP-phosphomutase (EC 6.4.2.9), which was isolated from the Tetrahymena pyriformis and Streptomyces hygroscopicus [150]. As a result of intramolecular rearrangement of the acid PEPR then it is decarboxylated with the formation of phosphonoacetaldehyde and, through a series of successive steps, is converted to (1R, 2S)-phosphomycin [148,149]. Using 18O labels, it was proved that the Tetrahymena pyriformis catalyzed transition of the phosphorus group from the oxygen atom to the carbon atom in enolpyruvate. The rearrangement represents an intramolecular process proceeding with the retention of absolute configuration at the phosphorus atom (Figure 41) [151,152].
The formation of the herbicide bialaphos, which is produced by Streptomyces hygroscopicus SF-1293, proceeds by a similar mechanism (Figure 26). Biocatalysis with the carboxyphosphonoenolpyruvate phosphonomutase isolated from Tetrahymena piriformis enzyme, initiated the condensation of phosphonoenolpyruvate with phosphonoformate and the rearrangement of phosphonoenolpyruvate into phosphonopyruvate with the formation of a P-C bond. The reaction catalyzed by carboxyphosphonoenolpyruvate phosphonomutase proceeded with retention of absolute configuration at the phosphorus atom [143] (Figure 42). The biosynthesis of (R)-2-amino-I-hydroxyethyl) phosphinic acid was carried out by enantioselective hydroxylation of 2-aminoethylphosphinic acid by the Acanthamoeba castellanii enzyme (Figure 43) [148].
The biocatalytic epoxidation of (S)-2-hydroxyalkylphosphonic acids to epoxides, analogs of fosfomycin, in the presence of enzyme Streptomyces fradiae was investigated [152]. The replacement of the hydrogen atom at the carbon atom of deuterated 2-hydroxypropylphosphonic acid, catalyzed by Streptomyces fradiae, and the formation of epoxides proceeded stereospecifically [132,153,154,155]. The formation of cis-epoxides proceeded with the inversion of absolute configuration, while trans-epoxides were obtained with the retention of configuration. Deuterated (1S,2S)- and (1R,2S)-2-hydroxy-1-D-propylphosphonic acid were synthesized from (1S,2S)-2-benzyloxy-1-D-propanol, which in turn, was obtained by catalytic reduction of the corresponding aldehyde with horse liver alcohol dehydrogenase (Figure 44) [155,156].
A similar biocatalytic convertion of hydroxyphosphonate to fosfomycin in the presence of (S)-2-hydroxypropylphosphonic acid epoxidase (Ps-HppE) isolated from phytopathogenic bacteria Pseudomonas syringae was reported [157,158]. The Ps-HppE catalyzed the epoxidation of (S)-hydroxypropylphosphonic acid with formation of fosfomycin, while the oxidation of (R)-hydroxyphenylphosphonic acid under the same conditions gave 2-oxopropylphosphonic acid (Figure 45).
Biocatalytic acylation of (S/R)-hydroxyphosphonate with vinyl acetate was carried out in isopropyl ether in the presence of lipase from Pseudomonas syringae. The reaction was complete in 5 days at 30 °C. Chromatographic separation on silica gel, followed by deacetylation afforded optically active (R)-hydroxyphosphonate in yield 46%, which was then converted to a (S,R)-deuterated analogue of fosfomycin. The isotope label experiment showed that the epoxide oxygen atom of (S,R)-fosfomycin is derived from the hydroxyl group of hydroxyphosphonate (Figure 46) [157].
Important from the point of view of practical application are aminophosphonic antibiotics with an amino group at the gamma position relative to the phosphonic functional group, namely fosmidomycin, and its derivatives FR900098, FR-33289 and FR32863, isolated from Streptomyces lavendulae, as well as cyclic SF2312, isolated from Micromonospora sp. [159,160,161]. Glyphosate (Roundup) is a widely used herbicide that inhibits 5-enoylpyruvylshikimate-3-phosphate synthase as a phosphoenolpyruvate substrate mimic. Tenofivir is used as a drug for the treatment of HIV and hepatitis B. Zoledronate and phosphonoformate (Foscarnet) are analogs of pyrophosphate. They are used to treat osteoporosis and herpes. Zoledronate also has antitumor activity due to its ability to target human dimethylallylpyrophosphate farnesylpyrophosphate synthase (Figure 47).
Fosmidomycin and its homologues are potent inhibitors of 1-deoxy-D-xylulose-5-phosphate reductoisomerase, an important enzyme in the nonmevalonate pathway of isoprenoid biosynthesis, active against a wide range of enterobacteria. 2-Acyloxyethylphosphonate, biosurfactant of a lipid nature, was isolated from the aquatic flowers of Aphanizomenon flosaquae [162]. Phosphinotricin is a non-protein amino acid that has been found as a component of the PT-Ala-Ala tripeptide (Bialaphos) in Streptomyces viridochromogenes DSM 40736 and Streptomyces hygroscopicus ATCC 21705 [163,164]. Phosphinothricin is an active inhibitor of glutamine synthetase [164,165,166,167]. The inhibition of the glutamine synthetasein plants determines its herbicidal activity as ammonium salt (ammonium glufosinate). It has been reported that other glutamine synthetase inhibitors are promising for the treatment of tuberculosis and neurological diseases [165]. The D- and L-enantiomers of phosphinothricin (2-amino-4-hydroxymethylphosphinyl-butanoic acid) and its derivative were synthesized by bioenzymatic methods [168]. Acetylated D, L-3-aminopyrrolidin-2-one was converted to acyclic phosphinate by reaction with ethylmethylphosphinate. Sequential enzymatic hydrolysis with phosphodiesterase, acylase, and glutaminase through the formation of intermediates led to phosphinothricin, which after treatment with glutaminase, was converted into an ester of azaphospholidininecarboxylic acid, and then into cyclic phosphonic acid. The last stage also included the formation of an L-phosphinothricin derivative under the action of α-chemotrypsin (Figure 48).
Phosphonotrixin was first isolated from the microorganism Saccharotrix sp. ST-888. This secondary metabolite has attracted interest primarily because of its antibiotic and herbicidal properties, as well as its unique structure containing a C–P bond on the isoprene group. As a result, several methods have been developed for the synthesis of this compound. The formation of phosphonotrixin by rearrangement of phosphoenolpyruvate (PEP) to phosphonopyruvate is shown in Figure 49. The biosynthesis of phosphonates begins with the rearrangement of phosphoenolpyruvate to phosphonopyruvate as a result of a reaction catalyzed by phosphonoenolpyruvate mutase. In this equilibrium process, the formation of phosphonoenolpyruvate is thermodynamically more favorable, at least 500 times. As a result, phosphonopyruvate is converted into metabolically preferred compounds. Therefore, it is a key substrate in the synthesis of phosphonothrixin [130,142].
The total synthesis of phosphonotrixin (PTX) was developed on the basis of commercially available methyl 3-hydroxy-2-methylene butyrate, the reaction of which with diethyl chlorophosphite in the presence of triethylamine gave E-allyl phosphonate in 60% yield. Vicinal dihydroxylation of the diol, accompanied by the oxidation of the resulting secondary alcohol, led to the formation of protected phosphonotrixin in 80% yield. After removal of the protective groups, free RTX was obtained in a total 24% yield (Figure 50) [132,168,169].
Asymmetric epoxidation of alkene phosphonates was used in the synthesis of various hydroxyphosphonates of natural origin. For example, the synthesis of phosphonotrixin was developed starting from bromomethylallyl ketone, which was first converted into allyl phosphonate using the Michaelis-Becker reaction and then allylphosphonate via the vic-asymmetric dihydroxylation with osmium tetroxide according to Sharpless gave the dihydroxyphosphonate. The prepared dihydroxyphosphonate was converted into phosphonothrixine, as shown in the Figure 51 [168,169,170,171].
The enantioselective synthesis of (S)-phosphonothrixin (ee 93%) in nine steps and 12% overall yield was developed starting from 1,3-dihydroxyacetone. The key reaction was the enzymatic desymmetrization of 2-isopropenylpropane-1,2,3-triol, which was resolved with vinyl acetate in the presence of porcine pancreatic lipase with formation of chiral (S)-monoester in good yield (88%) and with high enantiomeric excess 93%. Epoxy-alcohol was transformed into phosphonate, thus completing a formal synthesis of natural (S)-(−)-phosphotrixin. The chemical correlation confirmed the absolute configuration of dibenzyl ether phosphonate to be (R), which was converted into (S)-configuration at the newly created stereogenic centre (Figure 52) [170].
Two unprecedented phosphorus-containing iodinated polyacetylenes, phosphoiodyns A and B (12), were isolated from a Korean marine sponge Placospongia sp.
Plakotilen A showing inhibitory activity against RANKL-induced differentiated osteoclasts at 10 μM was isolated from the marine sponge Placospongia sp. From the same sponge, Placospongia sp. phosphorus derivatives of Plakotilen A and B, phosphoiodines A and B, containing iodinated polyacetylenes were isolated, [172]. Phosphoiodine A was found to exhibit potent agonist activity at the peroxisome proliferator-activated delta receptor (hPPAR8), which is believed to function as an integrator of transcriptional repression and nuclear receptor signaling [164]. This compound, as well as its analogs, demonstrate significant neuroprotective activity in a cell model in vitro, indicating that such phosphonates may be an effective new basis for the development of therapeutic agents for the treatment of neurodegenerative disorders (Figure 53) [173].
Ciliatocholic acid has been found in the bile of the gallbladder in cattle. The structure of ciliatocholic acid was confirmed by independent synthesis from choline and ciliatin [174]. Cholic acid was dissolved in a mixture of tributylamine and dioxane, then treated with chloroethyl formate and a solution of ciliatin in aqueous alkali. The product was recrystallized from ethanol-ethyl acetate. As a result, ciliatocholic acid was obtained in a satisfactory yield. This compound was further purified by preparative thin layer chromatography and confirmed to be ciliatocholic acid by phosphorus content and chromatographic performance. Thus, it was concluded that bovine gallbladder bile contains a small amount of ciliatocholic acid (Figure 54) [174].
Two nucleotide antibiotics, phosphadecin and phosphocytocin, were found in the extract of the Pseudomonas viridiflava PK-5 and Pseudomonas fluorescens PK-52 cultures, respectively. These antibiotics were purified by column chromatography using adsorption, gel filtration and ion exchange resins. These antibiotics were enzymatically and chemically hydrolyzed to form fosfomycin and a new antibiotic, phosphoxacin, which are also formed in culture filtrates. They showed antibacterial activity against gram-positive and gram-negative bacteria (Figure 55) [175].
Phosphonolipids. Lipids containing aminophosphonates are called phosphonolipids [176]. There are three classes of these compounds—Glycerophosphonolipids and Sphingophosphonolipids and Sphingophosphonoglycolipids (typical structures are shown in the Figure 56). Sphingolipids are a class of lipids related to aliphatic amino alcohol derivatives. Glycerophospholipids or phosphoglycerides are glycerol-based phospholipids. Sphingophosphonoglycolipids, a subtype of glycolipids containing the amino alcohol sphingosine, are known and well-studied. The first evidence for the existence of phosphonolipids was obtained in 1963 by Rouser et al. [177] when analyzing the sea anemone Anthropleura elegissima. This new sphingolipid has previously been identified as acyl-sphingosyl-1-(2-aminoethyl) phosphonate (ceramide aminoethyl phosphonate, CAEPn). Subsequent studies have shown that CAEPn is widespread in many species of freshwater and marine bivalve molluscs, snails, cephalopods as well as in various crustaceans, plant seeds and bacteria. Phosphonolipids have been found in fractions of glycocerebrosides (lipids) of many lower marine organisms, bacterial exopolysaccharides. The amount of phosphonolipids in organisms varies depending on the species, tissue, or the location of the cell. For example, vertebrates have sphingophosphonolipids as components of sphingomyelin in neural tissue, whereas at invertebrates often contain high levels of these lipids as components of the outer membrane (Figure 56) [176,177,178].
Phosphonopeptides of natural origin. Phosphonopeptides containing a P–C bond, like phosphopeptides, with a P–O–C bond, are relatively common substances in nature [179]. Currently, both phosphonopeptides isolated from natural sources and a significant amount of synthetic phosphonopeptides with a wide spectrum of biological activity are known. The peptides are thought to enhance cellular uptake through peptide permeases, masking the phosphonate antibiotic in the Trojan horse peptide, which is then hydrolyzed inside the cell by peptidases to release the phosphonate-active fragment. A typical representative is alaphosphin (or alafosfalin—a dipeptide consisting of alanine and 1-aminoethylphosphonic acid) (Figure 57).
Dehydrophos is an antibiotic isolated from Streptomyces luridus, which is effective in vivo against Salmonella-infected chickens snd has a broad spectrum of in vitro activity. Dehydrophos consists of a glycineleucin dipeptide bound to an O-methylated aminovinylphosphonate, its dehydrophosphorylation proceeds analogously to the similar in structure alaphosphine peptide and the Trojan horse peptide dehydrophosphine is imported and cleaved by peptide permeases and peptidases, respectively, to release the alanine racemase inhibitor within the cell (Figure 58) [179,180].
In recent years, a number of P-C antibiotics, possessing antifungal activity: rhizocticins [139,180], phosphonocystoximic acid [181], plumbemycins [139,182], phosphacetamycin [183], have been isolated from Bacillus subtilis and Streptomyces plumbeus. They represent di- and tripeptides containing the C-terminal (Z)-L-2-amino-5-phosphono-3-pentenoic acid, a phosphonotreonine mimetic [184], reminiscent of dehydrophos as a broad-spectrum antibiotic that acts against infection of Salmonella [185]. Rhizocticin A, the main component of the antifungal, hydrophilic phosphono-oligopeptides Bacillus subtilis ATCC 6633, was used for sensitivity testing in experiments with the molecular mechanism of antibiotic action (Figure 59) [182].
Rhizocticins and plumbemycins are natural phosphonate antibiotics produced by the bacterial strains Bacillus subtilis ATCC 6633 and Streptomyces plumbeus, respectively. The non-proteinogenic (S,Z)-2-amino-5-phosphonopent-3-enoic acid [(S,Z)-APPA] were used for the synthesis of Rhizocticin A and Plumbemycin A. The (S,Z)-APPA was synthesized via the Still–Gennari olefination starting from Garner’s aldehyde. The Michaelis-Arbuzov reaction was used to form a P-C bond. Oligopeptides were prepared using liquid phase peptide synthesis (LPPS) and were tested against selected bacteria and fungi [139]. Structurally, plumbemycins A and B, the new antimetabolites from Streptomyces plumbeus that antagonize L-threonine have been assigned to L-alanyl-L-aspartyl-D-2-amino-5-phosphono-3-cis-pentenoic acid and L-alanyl-L-asparaginyl-D-2-amino-5-phosphono-3-cis-pentenoic acid, respectively. Rhizocticins are phosphonate oligopeptide antibiotics containing a C-terminal non-proteinogenic amino acid (APPA). Rhizocticin B was heterologously produced in the non-producing strain Bacillus subtilis. Rhizocticin biosynthesis proceeds on the basis of bioinformatic analysis of Rhi genes. One of the steps in APPA biosynthesis is the aldol reaction between phosphonoacetaldehyde and oxaloacetate, catalyzed by the aldolase homologue RhiG. Recombinant RhiG was obtained and the product of enzymatic transformation in vitro was characterized (Figure 60) [139,182,183,184,185].
One example of compounds isolated from natural sources are phosphazinomycins A and B, which were isolated from Streptomyces lavendofoliae and Streptomyces unzenensis [186,187,188,189]. They contain a hydrazide bond between the peptidylarginine carboxylic acid and phosphonic acid. Phosphonopeptides with an unusual structure include phosphazinomycin, which is a natural product containing a nitrogen-nitrogen (NN) bond, unlike other natural products in which the N–N bond is formed directly at the intermediate stage of biosynthesis during the biosynthesis of phosphazinomycin, kinamycin and related compounds. Phosphazinomycin and kinamycin are natural products that contain nitrogen-nitrogen (NN) bonds, but are otherwise structurally unrelated. Despite their significant structural differences, their biosynthetic gene clusters share a common set of genes that are thought to facilitate N–N bond formation (Figure 61) [189,190,191].
Phosphazinomycins A and B, produced by Streptomyces lavendofoliae, have the same phosphonate fragment with one chiral center of unknown configuration, which was determined by synthesizing both enantiomers of 2-hydroxy-2-phosphonoacetic acid methyl ester. Chiral cyclic phosphite reacted with methylglyoxylate in the Pudovik reaction to form a pair of diastereomeric α-hydroxyphosphonates, which were separated by HPLC. The configurations in C-2 were determined by X-ray diffraction analysis of a single crystal. The removal of protection from these diastereomers gives enantiomeric α-hydroxyphosphonic acids, the (−)-configuration of which has the same sign of optical rotation as the phosphonic acid fragment of two phosphazinomycines (Figure 62) [189,190,191].
Antibiotics, AC549, ATS 1287 (leptomycin) and AM630 (phosphazinomycin), depending on their activity, cause characteristic morphological changes in test organisms. Phosphonopeptides are of limited use in medicine, since they are easily hydrolyzed and release aminophosphonic acids, which are unable to overcome cell barriers and have an antibiotic effect. The genetic approach made it possible to isolate and characterize new peptidomimetics of Streptomyces, such as argolaphos A and B and valinophos [190]. A similar approach was used to isolate phosphonocystoximate and its hydroxylated derivatives: Among these, compounds are argolaphos A and B, a broad-spectrum antibacterial phosphonopeptide, rare amino acid N-(5)-hydroxyarginine; valinophos, N-acetyl-1-Val ester of 2,3-dihydroxypropylphosphonate [190]. Argolaphos has a broad spectrum of antibacterial activity, with the highest efficacy against Salmonella typhimurium, Escherichia coli, Staphylococcus aureu, it also inhibits Mycobacterium smegmatis. In addition valinophos, N-acetyl-L-Val ester of 2,3-dihydroxypropylphosphonate; and phosphonocystoximate, an unusual molecule containing sulfide group, representing a new chemotype of sulfur-containing phosphonate natural products. The absolute configuration of phosphonocystoximic acid was defined as (1S,6S)- (Figure 63) [181].
Antibiotic K-26 was isolated from the extract of the actiomycete K-26, which is a water-soluble acidic peptide composed of equal moles of L-isoleucine, L-tyrosine and (R)-1-amino-2-(4-hydroxyphenyl)ethylphosphonic acid. The IC50 of K-26 for ACE inhibition was 6.7 ng/mL when hippuril-L-histidyl-L-leucine was used as an ACE substrate. K-26 has hypotensive activity in vivo (Figure 64) [190,191].
The family of bacterial secondary metabolites of tripeptides K-26, K4, and I5B2 is a special group of phosphonic peptidomimetics [190]. They are produced by three different actinomycetes and act as potent inhibitors of the human angiotensin I, converting enzyme, selectively targeting the eukaryotic family of this enzyme. K-26 was isolated from the extract of the actiomycete K-26 [192,193]. The activity of the peptide in inhibiting ACE (angiotensin converting enzyme) in vitro is comparable to that of captopril. The absolute configuration of this connection has been determined. The conformation of the molecule is relatively rigid due to intramolecular restrictions, and the relative arrangement of heteroatoms necessary for its biological activity (Figure 20) [194,195,196,197]. Shibuya et al. [195] carried out a stereoscopic synthesis of hydroxyphosphonate tyrosine analogs by stereoselective reaction of 4-formyl-L-phenylalanine derivatives in the presence of a chiral heterobimetallic catalyst (Figure 65].
The tripeptide of α-hydroxy-phosphonate, which is highly effective inhibitor of renin was prepared by stereoselective phosphadol reaction of dialkyl phosphites with chiral aldehyde [195]. The reaction with Titanium(IV) isopropoxide as a catalyst gave a mixture of diastereomers in a ratio of ~1:1, from which the (1S,2S)-diastereomer was isolated by column chromatography on silica gel (Figure 66) [198].
The phosphonic tripeptides Bialaphos and Phosalacine, as well as the tetrapeptide Trialaphos, are biological herbicides isolated from soil actinomycetes that attracted close attention. The Bialaphos tripeptide was isolated from the filtrates of Streptomyces viridochromogenes and Streptomyces hygroscopicus [199,200,201]. The antibacterial activity of Bialaphos is a result of active transport of the peptide across the bacterial membrane, followed by hydrolysis of the peptide and the release of the terminal phosphonate, phosphinotricin, which inhibits glutamine synthetase. Further studies of Bialaphos led to the isolation of the tetrapeptide Trialaphos [136] and Phosalacine [135] from the Streptomyces hygroscopicus strain with a similar mechanism of action. Trialaphos (L-alanyl-L-alanyl-L-alanyl-phosphinothricin) is a naturally occurring tetrapeptide phosphinate composed of the combination of the three amino acids Alanine and Glufosinate (Figure 67).
Phosalacine was isolated from the fermentation broth of actinomycete strain KA-338 during the screening of antimetabolites competing with L-glutamine as structural analogs of the catalytic intermediate glutamine synthetase. Glufosinate actively inhibits this enzyme, but has low activity as a free amino acid, probably due to poor absorption. However, the “Trojan horse” peptides derived from glufosinate are readily imported into cells, where the peptidases release the active inhibitor [202,203]. Plant death due to inhibition of plant GS has made these substances successful herbicides, which are usually applied in the form of an ammonium salt. Phosalacin, a herbicidal antibiotic containing phosphinothricin, was isolated from a culture filtrate of Kitasatosporia phosalacinea KA-338 soil extract. Bialaphos and phosphalacin, which are produced by soil actinomycetes, are tripeptides and contain phosphinothricin as the N-terminal amino acid. Phosphinothricin is released from both Bialaphos and Phosalacine as an active substances by peptidases. The difference between the two peptides is due to the type of C-terminal amino acid, which is L-alanine for bialaphos and L-Leucine for Phosalacine. Bialaphos, Trialaphos and Phosalacine are pro-herbicides and are active only after their metabolic hydrolysis in the plant. Only the (S)–enantiomer of phosphinothricin exhibits herbicidal activity. The mechanism of action is shown in Figure 68.
It is assumed that phosalacin transported across cell membranes, incorporated into microbial or plant cells and, after hydrolysis decomposes with the formation of phosphinothricin, which exhibits antimicrobial and herbicidal effect by inhibiting vital parasite enzymes.1-Phosphinothricin is a phosphinic acid that mimics (S)-glutamic acid, a natural substrate for glutamine synthetase. This target enzyme provides a reaction that incorporates inorganic nitrogen into organic forms (such as amino acids and nitrogenous bases) by releasing glutamine to detoxify ammonia. Inhibition of glutamine synthetase leads to the accumulation of ammonia in plant cells to toxic levels, and leads to the suppression of photorespiration due to a decrease in the level of amino acid donors [136,204,205,206].
7. Conclusions
This literature review has emphasized and described the importance of natural organophosphorus derivatives, which are a wide class of chemical compounds containing organic moieties usually bonded directly to phosphorus or bonded through a heteroatom, such as sulfur, oxygen or nitrogen. In recent years, significant advances have been made in understanding the influence of phosphates on the chemistry of prebiotics, among which two important aspects stand out. First, the phosphorylation reaction of prebiotics; second, the non-structural roles that phosphates may play in prebiotic chemistry. Several prebiotic pathways for the synthesis of nucleotides, amino acids and basic metabolites have been described, which are controlled by phosphates acting as acid-base catalysts, pH buffers or chemical buffers. These simple and subtle mechanisms of using phosphates to organize selective, robust prebiotic chemistry, combined with the central and universal role of phosphates in biochemistry, are increasingly making it clear that phosphate chemistry may be a key element in the origin of life on Earth. Phosphorous acid is adapted to play its role in nucleic acids because it can bind two nucleotides and be ionized. The resulting negative charge serves to both stabilize the diesters from hydrolysis and keep the molecules inside the lipid membrane. For the resulting material to remain inside the cell membrane, it must be charged, and therefore the binding element must have a third, ionizable group. The bond is usually carried out by ester groups, but for the ester to be hydrolytically stable, this charge must be negative and must be physically close to the ester groups. Phosphoric acid meets all these conditions, and there is no alternative to it. The properties and role of phosphorus in maintaining life processes, combined with the conditions created by stereochemistry, homochirality, racemization and deracemization, open the way to solving one of the most important problems facing the science of the 21st century—to answer the question “How did Life on Earth arise?” In addition to the great problems of fundamental science, the study of the chemistry of natural phosphorus compounds is of great utilitarian importance [207]. Natural phosphonates can be considered as simple analogs of phosphoric acid esters and/or carboxylic acids. The stability of the C-P bond leads to the fact that phosphonates can act as inhibitors of enzymes and, therefore, can be considered as drugs or agrochemicals [208]. The development of modern physicochemical analysis and genomics, supplemented by biochemical research, has led to the development of new detection technologies that significantly accelerate the discovery of new natural phosphonates, the identification of their metabolic pathways, and their use as starting compounds for the creation of new promising drugs. Aminophosphonic and hydroxyphosphonic acids, phosphagens, phosphonopeptides, phosphonolipids, phosphorus antibiotics, agrochemicals, etc., confirm the importance of natural phosphorus compounds for science and economics [209].
Author Contributions
O.I.K. wrote the paper. The author has read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are openly available.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
Ala—Alanine; AMP—Adenosine monophosphate, Arg—Argenine; ATF—Adenosyn triphosphate; Gly—Glycine; Asp—Asparagine; Boc—tert-Butoxycarbonyl; de—Diastereomeric excess; DIBAL-H—Diisobutylaluminum hydride; DMSO—Dimethylsulfoxide; DiPT—Diisopropyl tartrate; DiPE—Diisopropyl ether; ee—Enantiomeric excess; Gly—Glycine; GTP—Guanosine-5′-triphosphate; Hint- Histidine triad nucleotide binding protein; HppE—2-Hydroxypropylphosphonic acid epoxidase; i-Pr—iso-Propyl; Leu—Leucine; McC—Microcin C; Mes—Mesyl; Mnt—(1R,2S,5R)-Menthyl; Mops—3-Morpholinopropanesulfonic acid; MTBE—Methyl tert-butyl ether; PEP–Phosphoenolpyruvate; rac—Racemate; TBDMS—tert-Butyldimethylsilyl; THF—Tetrahydrofurane; TFA—Trifluoroacetic acid; TMS—Trimethylsilyl; Tol—Tolyl; Ts—toluenesulfonyl (tosyl); Val—Valine.
References
- Fernández-García, C.; Coggins, A.J.; Powner, M.W. A Chemist’s Perspective on the Role of Phosphorus at the Origins of Life. Life 2017, 7, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Abdillah, A.; Palma, J.; Churchill, D.G. Introductory Chapter: Phosphorus—Nature’s Versatile Pentavalent and Tetrahedral Covalent Building Block and Agent for Energy, Disease and Health. In Contemporary Topics about Phosphorus in Biology and Materials; Intech Open: London, UK, 2020; pp. 1–17. [Google Scholar] [CrossRef]
- Lazcano, A.; Miller, S.L. The origin and early evolution of life: Prebiotic chemistry, the pre-RNA world, and time. Cell 1996, 85, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Butusov, M.; Jerneliiv, A. Chapter 1. The Role of Phosphorus in the Origin of Life and in Evolution Phosphorus. In An Element that Could Have Been Called Lucifer; Springer Briefs in Environmental Science: New York, NY, USA, 2013; Volume 9, pp. 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Rossi, J.-C.; Pascal, R. How Prebiotic Chemistry and Early Life Chose Phosphate. Life 2019, 9, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westheimer, F. Why nature chose phosphates. Science 1987, 235, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Altstein, A.D. Origin of Biological Nucleic Acids and the First Genetic System (The Progene Hypothesis). Transcriptomics 2017, 5, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Goldford, J.E.; Hartman, H.; Smith, T.F.; Segrè, D. Remnants of an ancient metabolism without phosphate. Cell 2017, 168, 1126–1134.e9. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, F.; Chen, K.; Mojica, M.; Conley, M.; Napoline, J.; Butch, C.; Pollet, P.; Krishnamurthy, R.; Liotta, C. A plausible prebiotic origin of glyoxylate: Nonenzymatic transamination reactions of glycine with formaldehyde. Synlett 2016, 28, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Bean, H.D.; Anet, F.A.L.; Gould, I.R.; Hud, N.V. Glyoxylate as a backbone linkage for a prebiotic ancestor of RNA. Orig. Life Evol. Biol. 2006, 36, 39–63. [Google Scholar] [CrossRef]
- Nelson, K.E.; Levy, M.; Miller, S.L. Peptide nucleic acids rather than RNA may have been the first genetic molecule. Proc. Natl. Acad. Sci. USA 2000, 97, 3868–3871. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, A.W. Phosphorus in prebiotic chemistry. Philos. Trans. R. Soc. B. 2006, 361, 1743–1749. [Google Scholar] [CrossRef] [Green Version]
- Dass, A.V.; Jaber, M.; Brack, A.; Foucher, F.; Kee, T.P.; Georgelin, T.; Westall, F. Potential Role of Inorganic Confined Environments in Prebiotic Phosphorylation. Life 2018, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Pasek, M.A.; Lauretta, D.S. Aqueous corrosion of phosphide minerals from iron meteorites: A highly reactive source of prebiotic phosphorus on the surface of the early earth. Astrobiology 2005, 5, 515–535. [Google Scholar] [CrossRef]
- Bryant, D.E.; Kee, T.P. Direct evidence for the availability of reactive, water soluble phosphorus on the early Earth. H-Phosphinic acid from the Nantan meteorite. Chem. Commun. 2006, 22, 2344–2346. [Google Scholar] [CrossRef]
- Pasek, M.A. Schreibersite on the early earth: Scenarios for prebiotic Phosphorylation. Geosci. Front. 2017, 8, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Kolodiazhnyi, O.I. Stereochemistry of electrophilic and nucleophilic substitutions at phosphorus. Pure Appl. Chem. 2019, 91, 43–57. [Google Scholar] [CrossRef]
- Pasek, M.A.; Kee, T.P.; Bryant, D.E.; Pavlov, A.A.; Lunine, J.I. Production of potentially prebiotic condensed phosphates by phosphorus redox chemistry. Angew. Chem. Int. Ed. 2008, 47, 7918–7920. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Liu, Y.; Gao, X.; Xu, P. The international background of the origin of life. In Phosphorus Chemistry; Walter de Gruyter GmbH: Berlin, Germany, 2019; pp. 1–18. [Google Scholar] [CrossRef]
- Hall, B.G.; Zuzel, T. Evolution of a new enzymatic function by recombination within a gene. Proc. Natl. Acad. Sci. USA 1980, 77, 3529–3533. [Google Scholar] [CrossRef] [Green Version]
- Eldredge, N. Life Pulse Episodes from the Story of the Fossil Record; Pelican Edition: London, UK; New York, NY, USA, 1987. [Google Scholar]
- Skulachev, V.P.; Bogachev, A.V.; Kasparinsky, F.O. Principles of Bioenergetics; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; p. 252. ISBN 978-3-642-33430-6. [Google Scholar]
- Bustamante, C.; Bryant, Z.; Smith, S.B. Ten years of tension: Single-molecule DNA mechanics. Nature 2003, 421, 423–427. [Google Scholar] [CrossRef]
- Bruce, A.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walters, P. Molecular Biology of the Cell, 4th ed.; Alberts, B., Ed.; Garland Science: New York, NY, USA, 2002; p. 1400, ISBN-13: 978-081533218, ISBN-10: 0815332181. [Google Scholar]
- Ussery, D.W. DNA Structure: A-, B- and Z-DNA Helix Families. In Encyclopedia of Life Sciences; Published Online; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2002; pp. 1–7. [Google Scholar] [CrossRef]
- Tomassi, S.; Montalban, F.F.; Russo, R.; Novellino, E.; Messere, A.; Di Maro, S. Investigation of the Stereochemical-Dependent DNA and RNA Binding of Arginine-Based Nucleopeptides. Symmetry 2019, 11, 567. [Google Scholar] [CrossRef] [Green Version]
- Hickman, A.B.; Dyda, F. DNA Transposition at Work. Chem. Rev. 2016, 116, 12758–12784. [Google Scholar] [CrossRef]
- Liu, J.; Declais, A.-C.; McKinney, S.A.; Ha, T.; Norman, D.G.; Lilley, D.M.J. Stereospecific Effects Determine the Structure of a Four-Way DNA Junction. Chem. Biol. 2005, 12, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilley, D.M.J. Structures of helical junctions in nucleic acids. Q. Rev. Biophys. 2000, 33, 109–159. [Google Scholar] [CrossRef] [PubMed]
- Askari, M.S.; Lachance-Brais, C.; Rizzuto, F.J.; Toader, V.; Sleiman, H. Remote control of charge transport and chiral induction along a DNA-metallohelicate. Nanoscale 2019, 11, 11879–11884. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Benedetti, E.; Bethge, L.; Vonhoff, S.; Klussmann, S.; Vasseur, J.-J.; Cossy, J.; Smietana, M.; Arseniyadis, S. DNA vs. Mirror Image DNA: A Universal Approach to Tune the Absolute Configuration in DNA Based Asymmetric Catalysis. Angew. Chem. Int. Ed. 2013, 52, 11546–11549. [Google Scholar] [CrossRef]
- Roelfes, G.; Feringa, B.L. DNA-Based Asymmetric Catalysis. Angew. Chem. Int. Ed. 2005, 44, 3230–3232. [Google Scholar] [CrossRef]
- Coquire, D.; Feringa, B.L.; Roelfes, G. DNA-Based Catalytic Enantioselective Michael Reactions in Water. Angew. Chem. Int. Ed. Engl. 2007, 46, 9308–9311. [Google Scholar] [CrossRef]
- Li, Y.; Wang, C.; Jia, G.; Lu, S.; Li, C. Enantioselective Michael addition reactions in water using a DNA-based catalyst. Tetrahedron 2013, 69, 6585–6590. [Google Scholar] [CrossRef]
- Qiu, H.; Gilroya, J.B.; Manners, I. DNA-induced chirality in water-soluble poly (cobaltoceniumethylene). Chem. Commun. 2013, 49, 42–44. [Google Scholar] [CrossRef]
- Boersma, A.J.; Feringa, B.L.; Roelfes, G. Enantioselective Friedel–Crafts Reactions in Water Using a DNA Based Catalyst. Angew. Chem. Int. Ed. Engl. 2009, 48, 3346–3348. [Google Scholar] [CrossRef]
- Park, S.; Ikehata, K.; Watabe, R.; Hidaka, Y.; Rajendran, A.; Sugiyama, H. Deciphering DNA-based asymmetric catalysis through intramolecular Friedel–Crafts alkylations. Chem. Commun. 2012, 48, 10398–10400. [Google Scholar] [CrossRef] [Green Version]
- Shibata, N.; Yasui, H.; Nakamura, S.; Toru, T. DNA-Mediated Enantioselective Carbon–Fluorine Bond Formation. Synlett 2007, 7, 1153–1157. [Google Scholar] [CrossRef]
- Boersma, A.J.; Coquiere, D.; Geerdink, D.; Rosati, F.; Feringa, B.L.; Roelfes, G. Catalytic enantioselective syn hydration of enones in water using a DNA-based catalyst. Nat. Chem. 2010, 2, 991–995. [Google Scholar] [CrossRef] [Green Version]
- Boersma, A.J.; Megens, R.P.; Feringa, B.L.; Roelfes, G. DNA-based asymmetric catalysis. Chem. Soc. Rev. 2010, 39, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Ellington, W.R. Evolution and Physiological Roles of Phosphagen Systems. Annu. Rev. Physiol. 2001, 63, 289–325. [Google Scholar] [CrossRef]
- Webster, C.E. High-Energy Intermediate or Stable Transition State Analogue: Theoretical Perspective of the Active Site and Mechanism of β-Phosphoglucomutase. J. Am. Chem. Soc. 2004, 126, 6840–6841. [Google Scholar] [CrossRef]
- Yamamoto, T. Preferred dissociative mechanism of phosphate monoester hydrolysis in low dielectric environments. Chem. Phys. Lett. 2010, 500, 263–266. [Google Scholar] [CrossRef]
- Lassila, J.K.; Zalatan, J.G.; Herschlag, D. Biological phosphoryl-transfer reactions: Understanding mechanism and catalysis. Annu. Rev. Biochem. 2011, 80, 669–702. [Google Scholar] [CrossRef] [Green Version]
- Westheimer, F.H. Monomeric metaphosphates. Chem. Rev. 1981, 81, 313–326. [Google Scholar] [CrossRef]
- Keglevich, G.; Kovács, T.; Csatlós, F. The Deoxygenation of Phosphine Oxides under Green Chemical Conditions. Heteroat. Chem. 2015, 26, 199–205. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I.; Kolodiazhna, A.O. Nucleophilic substitution at phosphorus: Stereochemistry and Mechanisms. Tetrahedron Asymmetry 2017, 28, 1651–1674. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Asymmetric Electrophilic Reactions in Phosphorus Chemistry. Symmetry 2020, 12, 108. [Google Scholar] [CrossRef] [Green Version]
- Duarte, F.; Barrozo, A.; Åqvist, J.; Williams, N.H.; Kamerlin, S.C.L. The Competing Mechanisms of Phosphate Monoester Dianion Hydrolysis. J. Am. Chem. Soc. 2016, 138, 10664–10673. [Google Scholar] [CrossRef] [Green Version]
- Duarte, F.; Åqvist, J.; Williams, N.H.; Kamerlin, S.C.L. Resolving Apparent Conflicts between Theoretical and Experimental Models of Phosphate Monoester Hydrolysis. J. Am. Chem. Soc. 2015, 137, 1081–1093. [Google Scholar] [CrossRef]
- Bhengu, T.T.; Wood, T.P.; Shumane, M.; Tafesse, F. Detection of metaphosphate intermediates in reaction solutions of phosphate esters: Chromatographic and spectroscopic studies. Phosph. Sulf. Silicon Relat. Elem. 2017, 192, 1144–1152. [Google Scholar] [CrossRef]
- Henchman, M.; Viggiano, A.A.; Paulson, J.F.; Freedman, A.; Wormhoudt, J. Thermodynamic and kinetic properties of the metaphosphate anion, PO3-, in the gas phase. J. Am. Chem. Soc. 1985, 107, 1453–1455. [Google Scholar] [CrossRef]
- Range, K.; McGrath, M.J.; Lopez, X.; York, D.M. The Structure and Stability of Biological Metaphosphate, Phosphate, and Phosphorane Compounds in the Gas Phase and in Solution. J. Am. Chem. Soc. 2004, 126, 1654–1665. [Google Scholar] [CrossRef]
- Lahiri, S.D.; Zhang, G.; Dunaway-Mariano, D.; Allen, K.N. The Pentacovalent Phosphorus Intermediate of a Phosphory Transfer Reaction. Science 2003, 299, 2067–2071. [Google Scholar] [CrossRef] [Green Version]
- Buchwald, S.L.; Friedman, J.M.; Knowles, J.R. Stereochemistry of Nucleophilic Displacement on Two Phosphoric Monoesters and a Phosphoguanidine: The Role of Metaphosphate. J. Am. Chem. Soc. 1984, 106, 4911–4916. [Google Scholar] [CrossRef]
- Catrina, I.E.; Czyryca, P.G.; Hengge, A.C. Isotope effects on enzymatic and nonenzymatic reactions of phosphorothioates. Nukleonika 2002, 47, S17–S23. [Google Scholar]
- Cullis, P.M.; Nicholls, D. The existence of monomeric metaphosphate in hydroxylic solvent: A positional isotope exchange study. J. Chem. Soc. Chem. Commun. 1987, 10, 783–785. [Google Scholar] [CrossRef]
- Kiani, F.A.; Fischer, S. Catalytic strategy used by the myosin motor to hydrolyze ATP. Proc. Natl. Acad. Sci. USA 2014, 111, E2947–E2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.M.; Freeman, S.; Knowles, J.R. The quest for free metaphosphate in solution. Racemization at phosphorus in the transfer of the phospho group from aryl phosphate monoesters to tert-butyl alcohol in acetonitrile or in tert-butyl alcohol. J. Am. Chem. Soc. 1988, 110, 1268–1275. [Google Scholar] [CrossRef]
- Choe, J.Y.; Iancu, C.V.; Fromm, H.J.; Honzatko, R. Metaphosphate in the active site of fructose-1,6-bisphosphatase B. J. Biol. Chem. 2003, 278, 16015–16020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular System Bioenergetics: Energy for Life; Saks, V. (Ed.) Wiley-VCH: Weinheim, Germany, 2007; p. 633. ISBN 978-3-527-31787-5. [Google Scholar]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and Creatinine Metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef]
- Iyengar, M.R.; Coleman, D.W.; Butler, T.M. Phosphocreatinine, a high-energy phosphate in muscle, spontaneously forms phosphocreatine and creatinine under physiological conditions. J. Biol. Chem. 1985, 260, 7562–7567. [Google Scholar] [CrossRef]
- VanWagenen, B.C.; Larsen, R.; Cardellina, J.H.; Randazzo, D.; Lidert, Z.C.; Swithenbank, C. Ulosantoin, a potent insecticide from the sponge Ulosa ruetzleri. J. Org. Chem. 1993, 58, 335–337. [Google Scholar] [CrossRef]
- Karki, M.; Gibard, C.; Bhowmik, S.; Krishnamurthy, R. Nitrogenous Derivatives of Phosphorus and the Origins of Life: Plausible Prebiotic Phosphorylating Agents in Water. Life 2017, 7, 32. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, R.; Guntha, S.; Eschenmoser, A. Regioselective ex-phosphorylation of aldoses in aqueous solution. Angew. Chem. Int. Ed. Engl. 2000, 39, 2281–2285. [Google Scholar] [CrossRef]
- Anastasi, C.; Buchet, F.F.; Crowe, M.A.; Helliwell, M.; Raftery, J.; Sutherland, J.D. The Search for a Potentially Prebiotic Synthesis of Nucleotides via Arabinose-3-phosphate and Its Cyanamide Derivative. Chem. Eur. J. 2008, 14, 2375–2388. [Google Scholar] [CrossRef]
- Fankhauser, H.; Schiff, J.A.; Garber, L.J. Purification and properties of adenylyl sulphate:ammonia adenylyltransferase from Chlorella catalysing the formation of adenosine 50-phosphoramidate from adenosine 50-phosphosulphate and ammonia. Biochem. J. 1981, 195, 545–560. [Google Scholar] [CrossRef] [Green Version]
- Fankhauser, H.; Berkowitz, G.A.; Schiff, J.A. A nucleotide with the properties of adenosine 50 phosphoramidate from Chlorella cells. Biochem. Biophys. Res. Commun. 1981, 101, 524–532. [Google Scholar] [CrossRef]
- Wojdyla-Mamon, A.M.; Guranowski, A. Adenylylsulfate-ammonia adenylyltransferase activity is another inherent property of Fhit proteins. Biosci. Rep. 2015, 35, e00235–e00243. [Google Scholar] [CrossRef]
- Guranowski, A.; Wojdyla, A.M.; Rydzik, A.M.; Stepinski, J.; Jemielity, J. Plant nucleoside 5’-phosphoramidate hydrolase; simple purification from yellow lupin (Lupinus luteus) seeds and properties of homogeneous enzyme. Acta Biochim. Pol. 2011, 58, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Bretes, E.; Wojdyla-Mamon, A.M.; Kowalska, J.; Jemielity, J.; Kaczmarek, R.; Baraniak, J.; Guranowski, A. Hint2, the mitochondrial nucleoside 50-phosphoramidate hydrolase; properties of the homogeneous protein from sheep (Ovis aries) liver. Acta Biochim. Pol. 2013, 60, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Rossomando, E.F.; Hadjimichael, J. Characterization and cAMP inhibition of a lysyl-(N-ε-5′-phospho) adenosyl phosphoamidase in Dictyostelium discoideum. Int. J. Biochem. 1986, 18, 481–484. [Google Scholar] [CrossRef]
- Moriguchi, T.; Asai, N.; Okada, K.; Seio, K.; Sasaki, T.; Sekine, M. First synthesis and anticancer activity of phosmidosine and its related compounds. J. Org. Chem. 2002, 67, 3290–3300. [Google Scholar] [CrossRef]
- Ozga, M.; Dolot, R.; Janicka, M.; Kaczmarek, R.; Krakowiak, A. Histidine Triad Nucleotide-binding Protein 1 (HINT-1) Phosphoramidase Transforms Nucleoside 5′-O-Phosphorothioates to Nucleoside 5′-O-Phosphates. J. Biol. Chem. 2010, 285, 40809–40818. [Google Scholar] [CrossRef] [Green Version]
- Brenner, C. Hint, Fhit, GalT: Function, structure, evolution, and mechanism of three branches of the histidine triad superfamily of nucleotide hydrolases and transferases. Biochemistry 2002, 41, 9003–9014. [Google Scholar] [CrossRef] [Green Version]
- Bieganowski, P.; Garrison, P.N.; Hodawadekar, S.C.; Faye, G.; Barnes, L.D.; Brenner, C. Adenosine monophosphoramidase activity of Hint and Hnt1 supports function of Kin28, Ccl1, and Tfb3. J. Biol. Chem. 2002, 277, 10852–10860. [Google Scholar] [CrossRef] [Green Version]
- Hadjimichael, J.; Rossomando, E.F. Isolation and characterization of the protein phosphoamidates formed by a membrane bound adenylyl transferase reaction in Dictyostelium discoideum. Int. J. Biochem. 1991, 23, 535–539. [Google Scholar] [CrossRef]
- Hatano, M.; Hashimoto, Y. Properties of a toxic phospholipid in the northern blenny roe. Toxicon 1974, 12, 231–236. [Google Scholar] [CrossRef]
- Sekine, M.; Moriguchi, T.; Wada, T.; Seio, K. Total Synthesis of Agrocin 84 and Phosmidosine as Naturally Occurring Nucleotidic Antibiotics Having P-N Bond Linkages. J. Synth. Org. Chem. Jpn. 2001, 59, 1109–1120. [Google Scholar] [CrossRef] [Green Version]
- Phillips, D.R.; Uramoto, M.; Isono, K.; McCloskey, J.A. Structure of the antifungal nucleotide antibiotic phosmidosine. J. Org. Chem. 1993, 58, 854–859. [Google Scholar] [CrossRef]
- Roberts, W.P.; Tate, M.E.; Kerr, A. Agrocin 84 is a 6-N-phosphoramidate of an adenine nucleotide analogue. Nature 1977, 265, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-G.; Park, B.K.; Kim, S.-U.; Choi, D.; Nahm, B.H.; Moon, J.S. Bases of biocontrol: Sequence predicts synthesis and mode of action of agrocin 84, the Trojan Horse antibiotic that controls crown gall. Proc. Natl. Acad. Sci. USA 2006, 103, 8846–8851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertusati, F.; McGuigana, C. Diastereoselective synthesis of P-chirogenic phosphoramidate prodrugs of nucleoside analogues (ProTides) via copper catalysed reaction. Chem. Commun. 2015, 51, 8070–8073. [Google Scholar] [CrossRef] [Green Version]
- Tate, M.E.; Murphy, P.J.; Roberts, W.P.; Kerr, A. Adenine N6-substituent of agrocin 84 determines its bacteriocin-like specificity. Nature 1979, 280, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Reader, J.S.; Ordoukhanian, P.T.; Kim, J.-G.; de Crecy-Lagard, V.; Hwang, I.; Farrand, S.; Schimmel, P. Major Biocontrol of Plant Tumors Targets tRNA Synthetase. Science 2005, 309, 1533 LP–1553 LP. [Google Scholar] [CrossRef] [Green Version]
- Chopra, S.; Palencia, A.; Virus, C.; Tripathy, A.; Temple, B.R.; Velazquez-Campoy, A.; Cusack, S.; Reader, J.S. Plant tumour biocontrol agent employs a tRNA-dependent mechanism to inhibit leucyl-tRNA synthetase. Nat. Commun. 2013, 4, 1417–1430. [Google Scholar] [CrossRef] [Green Version]
- Chopra, S.; Palencia, A.; Virus, C.; Schulwitz, S.; Temple, B.R.; Cusack, S.; Reader, J. Structural characterization of antibiotic self-immunity tRNA synthetase in plant tumour biocontrol agent. Nat. Commun. 2016, 7, 12928–12941. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, H.; Ohkubo, A.; Sekine, M.; Seio, K.; Kakeya, H.; Osada, H. Synthesis and Biological Properties of New Phosmidosine Analogs Having an N-Acylsulfamate. Nucleosides Nucleotides Nucleic Acids 2006, 25, 647–654. [Google Scholar] [CrossRef]
- Sekine, M.; Okada, K.; Seio, K.; Kakeya, H.; Osada, H.; Obata, T.; Sasaki, T. Synthesis of Chemically Stabilized Phosmidosine Analogues and the Structure-Activity Relationship of Phosmidosine. J. Org. Chem. 2004, 69, 314–326. [Google Scholar] [CrossRef]
- Sekine, M.; Okada, K.; Seio, K.; Obata, T.; Sasaki, T.; Kakeya, H.; Osada, H. Synthesis of a biotin-conjugate of phosmidosine O-ethyl ester as a G1 arrest antitumor drug. Bioorganic. Med. Chem. 2004, 12, 6343–6349. [Google Scholar] [CrossRef]
- Paz-Yepes, J.; Brahamsha, B.; Palenik, B. Role of a microcin-C-like biosynthetic gene cluster in allelopathic interactions in marine Synechococcus. Proc. Natl. Acad. Sci. USA 2013, 110, 12030–12035. [Google Scholar] [CrossRef] [Green Version]
- Hengralph, N.C.K.; Jack, W. Chapter 13—Microcins. In Handbook of Biologically Active Peptides; Kastin, A.J., Ed.; Academic Press: Cambridge, MA, USA, 2006; pp. 75–81. [Google Scholar] [CrossRef]
- Rebuffat, S. Chapter 20—Microcins. In Handbook of Biologically Active Peptides: Bacterial/Antibiotic Peptides, 2nd ed.; Kastin, A., Ed.; Academic Press: Boston, MA, USA, 2013; pp. 129–137. ISBN 978-0-12-385095-9. [Google Scholar]
- Kazakov, T.; Vondenhoff, G.H.; Datsenko, K.A.; Novikova, M.; Metlitskaya, A.; Wanner, B.L.; Severinov, K. Escherichia coli Peptidase A, B, or N Can Process Translation Inhibitor Microcin, C. J. Bacteriol. 2008, 190, 2607–2610. [Google Scholar] [CrossRef] [Green Version]
- Baquero, F.; Lanza, V.F.; Baquero, M.-R.; del Campo1, R.; Bravo-Vázquez, D.A. Microcins in Enterobacteriaceae: Peptide Antimicrobials in the Eco-Active Intestinal Chemosphere. Front. Microbiol. 2019, 10, 2261. [Google Scholar] [CrossRef]
- Vondenhoff, G.H.M.; Van Aerschot, A. Microcin C: Biosynthesis, Mode of Action, and Potential as a Lead in cAntibiotics Development. Nucleosides Nucleotides Nucleic Acids 2011, 30, 465–474. [Google Scholar] [CrossRef]
- Kulikovsky, A.; Tsibulskaya, D.; Piskunova, J.; Severinov, K.; Serebryakva, M.; Dubiley, S. Microcin C-like peptide-adenylate antibiotic from Rubidus massiliensis. In FEBS Open Bio; WILEY: Hoboken, NJ, USA, 2018; Volume 8, p. 280. [Google Scholar]
- Tsibulskaya, D.; Mokina, O.; Kulikovsky, A.; Piskunova, J.; Severinov, K.; Serebryakova, M.; Dubiley, S. The Product of Yersinia pseudotuberculosis mcc Operon Is a Peptide-Cytidine Antibiotic Activated Inside Producing Cells by the TldD/E Protease. J. Am. Chem. Soc. 2017, 139, 16178–16187. [Google Scholar] [CrossRef]
- Duquesne, S.; Destoumieux-Garzón, D.; Peduzzi, J.; Rebuffat, S. Microcins, gene-encoded antibacterial peptides from enterobacteria. Nat. Prod. Rep. 2007, 24, 708–734. [Google Scholar] [CrossRef]
- Serebryakova, M.; Tsibulskaya, D.; Mokina, O.; Kulikovsky, A.; Nautiyal, M.; Van Aerschot, A.; Severinov, K.; Dubiley, S. A Trojan-Horse Peptide-Carboxymethyl-Cytidine Antibiotic from Bacillus amyloliquefaciens. J. Am. Chem. Soc. 2016, 138, 15690–15698. [Google Scholar] [CrossRef]
- Severinov, K.; Nair, S.K. Microcin C: Biosynthesis and mechanisms of bacterial resistance. Future Microbiol. 2012, 7, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takatsu, T.; Horiuchi, N.; Ishikawa, M.; Wanibuchi, K.; Moriguchi, T.; Takahashi, S. 1100-50, a novel nematocide from Streptomyces lavendulae SANK 64297. J. Antibiot. 2003, 56, 306–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, M.; Okamoto, M.; Takeuchi, H. Inhibition of human immunodeficiency virus type 1 replication in acutely and chronically infected cells by EM2487, a novel substance produced by a Streptomyces species. Antimicrob. Agents Chemother. 1999, 43, 2350–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, H.; Asai, N.; Tanabe, K.; Kozaki, T.; Fujita, M.; Sakai, T.; Okuda, A.; Naruse, N.; Yamamoto, S.; Sameshima, T. EM2487, a Novel Anti-HIV-1 Antibiotic, Produced by Streptomyces sp. Mer-2487. J. Antibiot. 1999, 52, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Liu, Y.; Gao, X.; Xu, P. Chemistry, Biochemistry, Organic Chemistry, Industrial Chemistry, Biochemical Engineering, Life Sciences, Evolutionary Biology; Walter de Gruyter GmbH: Berlin, Germany; Munich, Germany; Boston, MA, USA, 2018; ISBN 9783110562552. [Google Scholar]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef]
- Besant, P.G.; Attwood, P.V.; Piggott, M.J. Focus on phosphoarginine and phospholysine. Curr. Protein Pept. Sci. 2009, 10, 536–550. [Google Scholar] [CrossRef]
- Vlastaridis, P.; Kyriakidou, P.; Chaliotis, A.; de Peer, Y.V.; Oliver, S.G.; Amoutzias, G.D. Estimating the total number of phosphoproteins and phosphorylation sites in eukaryotic proteomes. Gigascience 2017, 6, 1–11. [Google Scholar] [CrossRef]
- Guerra-Castellano, A.; Baños-Jaime, B.; Díaz-Quintana, A.; González-Arzola, K.; Ángel De la Rosa, M.; Díaz-Moreno, I. Exploring protein phosphorylation by combining computational approaches and biochemical methods. Comput. Struct. Biotechnol. J. 2020, 18, 1852–1863. [Google Scholar] [CrossRef]
- Piggott, M.J.; Attwood, P.V. Focus on O-phosphohydroxylysine, O-phosphohydroxyproline, N 1-phosphotryptophan and S-phosphocysteine. Amino Acids 2017, 49, 1309–1323. [Google Scholar] [CrossRef]
- Gonzalez-Sanchez, M.B.; Lanucara, F.; Helm, M.; Eyers, C.E. Attempting to rewrite History: Challenges with the analysis of histidine-phosphorylated peptides. Biochem. Soc. Trans. 2013, 41, 1089–1095. [Google Scholar] [CrossRef] [Green Version]
- Attwood, P.V.; Piggott, M.J.; Zu, X.L.; Besant, P.G. Focus on phosphohistidine. Amino Acids 2007, 32, 145–156. [Google Scholar] [CrossRef]
- Cheng, C.M.; Liu, X.H.; Li, Y.M.; Ma, Y.; Tan, B.; Wan, R.; Zhao, Y.F. N-Phosphoryl Amino Acids and Biomolecular Origins. Review Paper in Honor of the 50th Anniversary of the Publication of “A Production of Amino Acids under Possible Primitive Earth Conditions” (Miller, 1953). Orig. Life Evol. Biosph. 2004, 34, 455–464. [Google Scholar] [CrossRef]
- Guillaume, H.A.; Perich, J.W.; Johns, R.B.; Tregear, G.W. Synthesis of N(1)-phosphorylated tryptophan derivatives. J. Org. Chem. 1989, 54, 1664–1668. [Google Scholar] [CrossRef]
- Alam, M.; Sanduja, R.; Hossain, M.B.; Van der Helm, D. Gymnodinium breve toxins. 1. Isolation and X-ray structure of O,O-dipropyl (E)-2-(1-methyl-2-oxopropylidene) phosphorohydrazidothioate (E)-oxime from the red tide dinoflagellate Gymnodinium breve. J. Am. Chem. Soc. 1982, 104, 5232–5234. [Google Scholar] [CrossRef]
- Abraham, S.P.; Hoang, T.D.; Alam, M.; Jones, E.B.G. Chemistry of the cytotoxic principles of the ma rine fungus Lignincola laevis. Pure Appl. Chem. 1994, 66, 2391–2394. [Google Scholar] [CrossRef]
- Tong, T.; Chen, S.; Wang, L.; Tang, Y.; Ryu, J.Y.; Jiang, S.; Wu, X. Occurrence, evolution, and functions of DNA phosphorothioate epigenetics in bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, E2988–E2996. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, S.; Vergin, K.L.; Giovannoni, S.J.; Chan, S.W.; DeMott, M.S.; Taghizadeh, K.; Cordero, O.X.; Cutler, M.; Timberlake, S. DNA phosphorothioation is widespread and quantized in bacterial genomes. Proc. Natl. Acad. Sci. USA 2011, 108, 2963–2968. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Seth, P.P.; Vickers, T.A.; Liang, X.-H. The Interaction of Phosphorothioate-Containing RNA Targeted Drugs with Proteins Is a Critical Determinant of the Therapeutic Effects of These Agents. J. Am. Chem. Soc. 2020, 142, 14754–14771. [Google Scholar] [CrossRef]
- Wang, L.; Chen, S.; Xu, T.; Taghizadeh, K.; Wishnok, J.S. Phosphorothioation of DNA in bacteria by dnd genes. Nat. Chem. Biol. 2007, 3, 709–710. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, S.; Deng, Z.; Dedon, P.C.; Chen, S. DNA phosphorothioate modification—A new multi-functional epigenetic system in bacteria. FEMS Microbiol. Rev. 2019, 43, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Horsman, G.P.; Zechel, D.L. Phosphonate Biochemistry. Chem. Rev. 2017, 117, 5704–5783. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, W.W.; van der Donk, W.A. Biosynthesis of phosphonic and phosphinic acid natural products. Annu. Rev. Biochem. 2009, 78, 65–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarreal-Chiu, J.F.; Quinn, J.P.; McGrath, J.W. The genes and enzymes of phosphonate metabolism by bacteria, and their distribution in the marine environment. Front. Microbiol. 2012, 3, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Hudson, H.; Kukhar, V.P. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; John Wiley: New York, NY, USA, 2000; p. 660. ISBN 978-0-471-89149-9. [Google Scholar]
- Mikołajczyk, M. Phosphonate reagents and building blocks in the synthesis of bioactive compounds, natural products and medicines. Pure Appl. Chem. 2019, 91, 811–838. [Google Scholar] [CrossRef]
- Ordonez, M.; Viveros-Ceballos, J.L.; Cativiela, C.; Sayago, F.J. An update on the stereoselective synthesis of α-aminophosphonic acids and derivatives. Tetrahedron 2015, 71, 1745–1784. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Łyżwa, P.; Drabowicz, J. Asymmetric addition of dialkyl phosphite and diamido phosphite anions to chiral, enantiopure sulfinimines: A new, convenient route to enantiomeric α-aminophosphonic acids. Tetrahedron Asym. 1997, 8, 3991–3994. [Google Scholar] [CrossRef]
- Kafarski, P. Phosphonates: Their Natural Occurrence and Physiological Role. In Contemporary Topics about Phosphorus in Biology and Materials; Churchill, D.G., Čolović, B., Milhofer, H.F., Eds.; Intechopen Publ.: London, UK, 2020; pp. 1–19. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Asymmetric synthesis of hydroxyphosphonates. Tetrahedron Asymm. 2005, 16, 3295–3341. [Google Scholar] [CrossRef]
- Falagas, M.E.; Vouloumanou, K.; Samonis, G.; Vardakas, K.Z. Fosfomycin. Clin. Microbiol. Rev. 2016, 29, 321–347. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Anzai, H.; Imai, S.; Satoh, A.; Nagaoka, K.; Thompson, C.J. The bialaphos biosynthetic genes of Streptomyces hygroscopicus: Molecular cloning and characterization of the gene cluster. MGG Mol. Gen. Genet. 1986, 205, 42–50. [Google Scholar] [CrossRef]
- Wu, G.; Yuan, M.; Wei, L.; Zhang, Y.; Lin, Y.; Zhang, L.; Liu, Z. Characterization of a Novel Cold-Adapted Phosphinothricin N-Acetyltransferase From the Marine Bacterium Rhodococcus Sp. Strain YM12. J. Mol. Catal. B Enzym. 2014, 104, 23–28. [Google Scholar] [CrossRef]
- Omura, S.; Murata, M.; Hanaki, H.; Hinotozawa, K.; Oiwa, R.; Tanaka, H. Phosalacine, a new herbicidal antibiotic containing phosphinothricin. Fermentation, isolation, biological activity and mechanism of action. J. Antibiot. 1984, 37, 829–835. [Google Scholar] [CrossRef]
- Kato, H.; Nagayama, K.; Abe, H.; Kobayashi, R.; Ishihara, E. Isolation, Structure and Biological Activity of Trialaphos. Agr. Biol. Chem. 1991, 55, 1133–1134. [Google Scholar] [CrossRef]
- Bougioukou, D.J.; Mukherjee, S.; van der Donk, W.A. Revisiting the biosynthesis of dehydrophos reveals a tRNA-dependent pathway. Proc. Natl. Acad. Sci. USA 2013, 110, 10952–10957. [Google Scholar] [CrossRef] [Green Version]
- Lell, B.; Ruangweerayut, R.; Wiesner, J.; Missinou, M.A.; Schindler, A.; Baranek, T.; Hintz, M.; Hutchinson, D.; Jomaa, H.; Kremsner, P.G. Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrob. Agents Chemother 2003, 47, 735–738. [Google Scholar] [CrossRef] [Green Version]
- Gahungu, M.; Arguelles-Arias, A.; Fickers, P.; Zervosen, A.; Joris, B.; Damblon, C. Synthesis and biological evaluation of potential threonine synthase inhibitors: Rhizocticin A and Plumbemycin, A. Bioorg. Med. Chem. 2013, 21, 4958–4967. [Google Scholar] [CrossRef]
- Leonard, P.G.; Satani, N.; Maxwell, D.; Lin, Y.-H.; Hammoudi, N.; Peng, Z.F. SF2312 is a natural phosphonate inhibitor of enolase. Nat. Chem. Biol. 2016, 12, 1053–1058. [Google Scholar] [CrossRef]
- Takeuchi, M.; Nakajima, M.; Ogita, T.; Inukai, M.; Kodama, K.; Furuya, K.; Nagaki, H.; Haneishi, T. Fosfonochlorin, a new antibiotic with spheroplast forming activity. J Antibiot 1989, 42, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Cioni, J.P.; Doroghazi, J.R.; Kou-San, J.; Yu, X.; Evans, B.S.; Lee, J.; Metcalf, W.W. Cyanohydrin Phosphonate Natural Product from Streptomyces regensis. J. Nat. Prod. 2014, 77, 243–249. [Google Scholar] [CrossRef]
- Takahashi, E.; Kimura, T.; Nakamura, K.; Arahira, M.; Iida, M. Phosphonothrixin, a novel herbicidal antibiotic produced by Saccharothrix sp. ST-888. I. Taxonomy, fermentation, Hitakaisolation and biological properties. J. Antibiot. 1995, 48, 1124–1129. [Google Scholar] [CrossRef]
- Hidaka, T.; Seto, H.; Imai, S. Biosynthesis mechanism of carbon-phosphorus bond formation. Isolation of carboxyphosphonoenolpyruvate and its conversion to phosphinopyruvate. J. Am. Chem. Soc. 1989, 111, 8012–8013. [Google Scholar] [CrossRef]
- Hammerschmidt, F. Biosynthese von Naturstoffen mit einer P—C-Bindung, IV. Synthese der (R)- und (S)-(2-Amino[2-D1]ethyl)phosphonsäure und Hydroxylierung zu (2-Amino-1-hydroxyethyl)phosphonsäure in Acanthamoeba castellanii (Neff). Liebigs Ann. Chem. 1988, 961–964. [Google Scholar] [CrossRef]
- Harnrnerschrnidt, F. Synthese der (R)- und (S)-(2-Amino[2-D1]ethy1)phosphonsaure und Hydroxylierung zu (2-Amino-1-hydroxyethy1)phosphonsaure in Acanthamoeba castellanii (Neff). Lieb Ann. Chem. 1988, 1988, 961–964. [Google Scholar] [CrossRef]
- Peck, S.C.; van der Donk, W. Phosphonate biosynthesis and catabolism: A treasure trove for unusual enzymology. Curr. Opin. Chem. Biol. 2013, 17, 580–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C.T. The Chemical Biology of Phosphorus; RSC: Cambridge, UK, 2020; p. 546. ISBN 139781839162022. [Google Scholar]
- Blodgett, J.A.V.; Thomas, P.M.; Li, G.; Velasquez, J.E.; van der Donk, W.A. Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide. Nat. Chem. Biol. 2007, 3, 480–485. [Google Scholar] [CrossRef] [Green Version]
- McQueney, M.S.; Lee, S.L.; Swartz, W.H.; Ammon, H.L.; Mariano, P.S.; Dunaway-Mariano, D. Evidence for an intramolecular, stepwise reaction pathway for PEP phosphomutase catalyzed phosphorus-carbon bond formation. J. Org. Chem. 1991, 56, 7121–7130. [Google Scholar] [CrossRef]
- McQueney, M.S.; Lee, S.L.; Bowman, E.; Mariano, P.S.; Dunaway-Mariano, D. A remarkable pericyclic mechanism for enzyme-catalyzed phosphorus-carbon bond formation. J. Am. Chem. Soc. 1989, 111, 6885–6887. [Google Scholar] [CrossRef]
- Freeman, S.; Seidel, H.M.; Schwalbe, C.H.; Knowles, J.R. Phosphonate biosynthesis: The stereochemical course of phosphoenolpyruvate phosphomutase. J. Am. Chem. Soc. 1989, 111, 9233–9234. [Google Scholar] [CrossRef]
- Hammerschmidt, F. Incorporation of ~-Methyl-*H,. methionine and 2-Hydroxy-’80. hydroxyethylphosphonic Acid into Fosfomycin in Streptomyces frudiue-An Unusual Methyl Transfer. Angew. Chem. Intern. Ed. Engl. 1994, 106, 334–342. [Google Scholar] [CrossRef]
- Behrman, E.J.; Gopalan, V. Phosphoenolpyruvate. Biochem. Mol. Biol. 2008, 36, 323–324. [Google Scholar] [CrossRef]
- Petkowski, J.J.; Bains, W.; Seager, S. Natural Products Containing ‘Rare’ Organophosphorus Functional Groups. Molecules 2019, 24, 866. [Google Scholar] [CrossRef] [Green Version]
- Kolodiazhnyi, O.I. Enzymatic synthesis of organophosphorus compounds. Russ. Chem. Rev. 2011, 80, 883–910. [Google Scholar] [CrossRef]
- Simov., B.P.; Wuggenig., F.; Lammerhofer, M.; Lindner, W.; Zarbl, E.; Hammerschmidt, F. Indirect Evidence for the Biosynthesis of (1S,2S)-1,2-Epoxypropylphosphonic Acid as a Co-Metabolite of Fosfomycin (1R,2S)-1,2-Epoxypropylphosphonic Acid. by Streptomyces fradiae. Eur. J. Org. Chem. 2002, 1139–1142. [Google Scholar] [CrossRef]
- Munos, J.W.; Moon, S.-J.; Mansoorabadi, S.O.; Chang, W.; Hong, L.; Yan, F.; Liu, A.; Liu, H.-W. Purification and Characterization of the Epoxidase Catalyzing the Formation of Fosfomycin from Pseudomonas syringae. Biochemistry 2008, 47, 8726–8735. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Moon, S.-J.; Liu, P.; Zhao, Z.; Lipscomb, J.D.; Liu, A.; Liu, H.-W. Determination of the Substrate Binding Mode to the Active Site Iron of (S)-2-Hydroxypropylphosphonic Acid Epoxidase Using 17O-Enriched Substrates and Substrate Analogues. Biochemistry 2007, 46, 12628–23638. [Google Scholar] [CrossRef] [Green Version]
- Kramer, G.J.; Mohd, A.; Schwager, S.L.U.; Masuyer, G.; Ravi Acharya, K.; Sturrock, E.D.; Bachmann, B.O. Interkingdom Pharmacology of Angiotensin-I Converting Enzyme Inhibitor Phosphonates Produced by Actinomycetes. ACS Med. Chem. Lett 2014, 5, 346–351. [Google Scholar] [CrossRef]
- Ohba, K.; Sato, Y.; Sasaki, T.; Sezaki, M. Studies on a new phosphonic acid antibiotic SF2312.II isolation, physicochemical properties and structure. Sci. Rep. 1986, 25, 18–22. [Google Scholar]
- Okuhara, M.; Kuroda, Y.; Goto, T.; Okamoto, M.; Terano, H.; Kohsaka, M. Studies on a new phosphonic acid antibiotic III. Isolation and characterisation of FRFR-31564, FR-32863 and FR-33289. J. Antibiot. 1980, 33, 24–28. [Google Scholar] [CrossRef] [Green Version]
- Kaya, K.; Morrison, L.F.; Codd, G.A.; Metcalf, J.S.; Sano, T.; Takagi, H. A novel biosurfactant,2-Acyloxyethylphosphonate, isolated from Waterblooms of Aphanizomenon flos-aquae. Molecules 2006, 11, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Bayer, E.; Gugel, K.H.; Hägele, K.; Hagenmaier, H.; Jessipow, S.; König, W.A.; Zähner, H. Metabolic Products of Microorganisms.98. Phosphinothricin and Phosphinothricyl-Alanyl-Alanine. Helv. Chim. Acta 1972, 55, 224–239. [Google Scholar] [CrossRef]
- Donn, G.; Köcher, H. Inhibitors of Glutamine Synthetase. In Herbicide Classes in Development; Springer: Berlin/Heidelberg, Germany, 2002; pp. 87–101. [Google Scholar]
- Mowbray, S.L.; Kathiravan, M.K.; Pandey, A.A.; Odell, L.R. Inhibition of Glutamine Synthetase: A Potential Drug Target in Mycobacterium Tuberculosis. Molecules 2014, 19, 13161–13176. [Google Scholar] [CrossRef] [Green Version]
- Natchev, I.A. Enzymatic synthesis of D,D,L, and Lphosphinothricin and their cyclic analogs. Bull. Chem. Soc. Jpn. 1988, 61, 3699–3704. [Google Scholar] [CrossRef] [Green Version]
- Kolodiazhnyi, O.I. Asymmetric Synthesis in Organophosphorus Chemistry; John Wiley: Weinheim, Germany, 2016; p. 400. [Google Scholar]
- Nakamura, K.; Yamamura, S. Enantioseleetive Synthesis of Phosphonothrixin and Its Absolute Stereochemistry. Tetrahedron Lett. 1997, 38, 437–438. [Google Scholar] [CrossRef]
- Chênevert, R.; Simard, M.; Bergeron, J.; Dasser, M. Chemoenzymatic formal synthesis of (S)-(−)-phosphonotrixin. Tetrahedron Asymmetry 2004, 15, 1889–1892. [Google Scholar] [CrossRef]
- Field, S.C. Total synthesis of (±)-Phosphonothrixin. Tetrahedron Lett. 1998, 39, 6621–6624. [Google Scholar] [CrossRef]
- Kim, H.; Chin, J.; Choi, H.; Baek, K.; Lee, T.-G. Phosphoiodyns A and B, Unique Phosphorus-Containing Iodinated Polyacetylenes from a Korean Sponge Placospongia sp. Org. Lett. 2013, 15, 100–103. [Google Scholar] [CrossRef]
- Kinarivala, N.; Suh, J.H.; Botros, M.; Webb, P.; Trippier, P.C. Pharmacophore elucidation of phosphoiodyn A—Potent and selective peroxisome proliferator-activated receptor β/δ agonists with neuroprotective activity. Bioorg. Med. Chem. Lett. 2016, 26, 1889–1893. [Google Scholar] [CrossRef]
- Tamari, M.; Ogawa, M.; Kametaka, M. A New Bile Acid Conjugate, Ciliatocholic Acid, from Bovine Gall Bladder Bile. J. Biochem. 1976, 80, 371–377. [Google Scholar] [CrossRef]
- Katayama, N.; Tsubotani, S.; Nozaki, Y.; Harada, S.; Ono, H. Fosfadecin and fosfocytocin, new nucleotide antibiotics produced by bacteria Affiliations expand PMID: 2182591. J. Antibiot. 1990, 43, 238–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschidis, M.C. Phosphonolipids. Prog. Lipid Res. 1984, 23, 223–246. [Google Scholar] [CrossRef]
- Rouser, G.; Kritchevsky, G.; Heeler, D.; Lieaer, E. Lipid composition of beef brain, beef liver, and the sea anemone: Two approaches to quantitative fractionation of complex lipid mixture. J. Am. Oil Chem. Soc. 1963, 40, 425–454. [Google Scholar] [CrossRef]
- Mukhamedova, K.S.; Glushenkova, A.I. Natural phosphonolipids. Chem. Nat. Compd. 2000, 36, 329–341. [Google Scholar] [CrossRef]
- Marrs, E.C.L.; Varadi, L.; Bedernjak, A.F.; Day, K.M.; Gray, M.; Jones, A.L.; Cummings; Anderson, S.P.; Anderson, R.J.; Perry, J.D. Phosphonopeptides Revisited, in an Era of Increasing Antimicrobial Resistance. Molecules 2020, 25, 1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Kwo-Kwang; Wang, A.; van der Donk, W.A. New insights into the biosynthesis of fosfazinomycin. Chem. Sci. 2016, 7, 5219–5223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goettge, M.N.; Cioni, J.P.; Ju, K.-S.; Pallitsch, K.; Metcalf, W.W. PcxL and HpxL are flavin-dependent, oxime-forming N-oxidases in phosphonocystoximic acid biosynthesis in Streptomyces. J. Biol. Chem. 2018, 293, 6859–6868; [Google Scholar] [CrossRef] [Green Version]
- Rapp, C.; Jung, G.; Kugler, M.; Loeffler, W. Rhizocticins—New phosphono- oligopeptides with antifungal activity. Eur. J. Org. Chem. 1988, 7, 655–661. [Google Scholar] [CrossRef]
- Pallitsch, K.; Happl, B.; Stieger, C. Determination of the Absolute Configuration of (−)-Hydroxynitrilaphos and Related Biosynthetic Questions. Chem. Eur. J. 2017, 23, 15655–15665. [Google Scholar] [CrossRef]
- Borisova, S.A.; Circello, B.T.; Zhang, J.K.; van der Donk, W.W.; Metcalf, W.W. Biosynthesis of rhizocticins, antifungal phosphonate oligopeptides produced by Bacillus subtilis ATCC6633. Chem. Biol. 2010, 17, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.S.; Zhao, C.; Gao, J.; Evans, C.M.; Ju, K.-S.; Doroghazi, J.R.; Metcalf, W.W. Discovery of the antibiotic phosacetamycin via a new mass spectrometry-based method for phosphonic acid detection. ACS Contemporary Topics about Phosphorus in Biology and Materials. Chem. Biol. 2013, 8, 908–913. [Google Scholar] [CrossRef] [Green Version]
- Kugler, M.; Loeffler, W.; Rapp, C.; Kern, A.; Jung, G. Rhizocticin A, an antifungal phosphono-oligopeptide of Bacillus subtilis ATCC 6633: Biological properties. Archiv. Microbiol. 1990, 153, 276–281. [Google Scholar] [CrossRef]
- Johnson, R.D.; Kastner, R.M.; Larsen, S.H.; Ose, E.E. Antibiotic A53868 and Process for Production Thereof. U.S. Patent 4,482,488, 13 November 1988. [Google Scholar]
- Gunji, S.; Arima, K.; Beppu, T. Screening of antifungal antibiotics according to activities inducing morphological abnormalities. Agric. Biol. Chem. 1983, 41, 2016–2069. [Google Scholar] [CrossRef]
- Gao, J.; Ju, K.-S.; Yu, X.; Velásques, J.E.; Mukherjee, S.; Lee, J.; Van Der Donk, W.A. Use of phosphonate methyltransferase in the identification of the fosfazinomycin biosynthetic gene cluster. Angew. Chem. 2014, 53, 1334–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneemann, I.; Nagel, K.; Labes, A.; Wiese, J.; Imhoff, J.F. Comprehensivei of marine Actinobacteria associated with the sponge Halichondria panacea. Appl. Environ. Microbiol. 2010, 76, 3702–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.-K.A.; Ng, T.L.; Wang, P.; Huang, Z.; Balskus, E.P.; van der Donk, W.A. Glutamic acid is a carrier for hydrazine during the biosyntheses of fosfazinomycin and kanamycin. Nat. Commun. 2018, 9, 3687–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamato, M.; Koguchi, T.; Okachi, R.; Yamada, K.; Nakayama, K.; Kase, H.; Karasawa, A.; Shuto, K. K-26, a novel inhibitor of angiotensin I converting enzyme produced by an actinomycete K-26. J. Antibiot. 1986, 39, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Ju, K.-S.; Gao, J.; Doroghazi, J.R.; Wang, K.-K.A.; Thibodeaux, C.J. Discovery of phosphonic acid natural products by mining the genomes of 10,000 actinomycetes. Proc. Natl. Acad. Sci. USA 2015, 112, 12175–12180. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.A.; Wang, K.-K.; Lee, J.; van der Donk, W. A Biosynthesis of fosfazinomycin is a convergent process. Chem. Sci. 2015, 6, 1282–1287. [Google Scholar] [CrossRef] [Green Version]
- Yokomatsu, T.; Yamagishi, T.; Matsumoto, K.; Shibuya, S. Stereocontrolled synthesis of hydroxymethylene phosphonate analogues of phosphorylated tyrosine and their conversion to monofluoromethylene phosphonate analogues. Tetrahedron 1996, 52, 11725–11738. [Google Scholar] [CrossRef]
- Masuyer, G.; Cozier, G.E.; Kramer, G.J.; Bachmann, B.O.; Acharya, K.R. Crystal structure of a peptidyl-dipeptidase K-26-DCP from Actinomycete in complex with its natural inhibitor. FEBS J. 2016, 283, 4357–4369. [Google Scholar] [CrossRef] [Green Version]
- Kido, Y.; Hamakado, T.; Anno, M.; Miyagawa, E.; Motoki, Y.; Wakamiya, T. Isolation and characterization of I5B2, a new phosphorus containing inhibitor of angiotensin i converting enzyme produced by actinomadura sp. J. Antibiot. 1984, 37, 965–969. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.V.; Rielly-Gauvin, K.; Ryono, D.E.; Free, C.A.; Lynn Rogers, W. Hydroxy Phosphinyl-Based Inhibitors of Human Renin. J. Med. Chem. 1995, 38, 4557–4569. [Google Scholar] [CrossRef]
- Allen, J.G.; Atherton, F.R.; Hall, M.J.; Hassall, C.H.; Holmes, S.W.; Lambert, R.W.; Nisbet, L.J.; Ringrose, P.S. Phosphonopeptides, a new class of synthetic antibacterial agents. Nature 1978, 272, 56–58. [Google Scholar] [CrossRef]
- Kafarski, P. Phosphonopeptides containing free phosphonic groups: Recent advances. RSC Adv. 2020, 10, 25898–25910. [Google Scholar] [CrossRef]
- Lamberth, C. Naturally occurring amino acid derivatives with herbicidal, fungicidal or insecticidal activity. Amino Acids 2016, 48, 929–940. [Google Scholar] [CrossRef]
- Hoagland, R.E. Naturally Occurring Carbon—Phosphorus Compounds as Herbicides. Biol. Act. Nat. Prod. 1988, 13, 182–210. [Google Scholar] [CrossRef]
- Blodgett, J.A.V.; Zhang, J.K.; Yu, X.; Metcalf, W.W. Conserved biosynthetic pathways for phosalacine, bialaphos and newly discovered phosphonic acid natural products. J. Antibiot. 2016, 69, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Hunt, A.; Elzey, T. Revised structure of A53868A. J. Antibiot. 1988, 41, 802. [Google Scholar] [CrossRef] [Green Version]
- Dragicevic, M.; Platiša, J.; Nikolic, R.; Todorovic, S.; Bogdanovic, M.; Mitic, N.; Simonovic, A. Herbicide phosphinothricin causes direct stimulation hormesis Formerly Nonlinearity in Biology, Toxicology, and Medicine. Dose-Response 2013, 11, 344–360. [Google Scholar] [CrossRef]
- Schwartz, D.; Berger, S.; Heinzelmann, E.; Muschko, K.; Welzel, K.; Wohlleben, W. Biosynthetic Gene Cluster of the Herbicide Phosphinothricin Tripeptide from Streptomyces viridochromogenes Tu494. Appl. Environ. Microbiol. 2004, 70, 7093–7102. [Google Scholar] [CrossRef] [Green Version]
- Timsit, Y. DNA Self-Assembly: From Chirality to Evolution. Int. J. Mol. Sci. 2013, 14, 8252–8270. [Google Scholar] [CrossRef] [Green Version]
- Tajti, Á.; Keglevich, G. 3. The Importance of Organophosphorus Compounds as Biologically Active Agents. In Organophosphorus Chemistry; Walter de Gruyter GmbH: Boston, Germany; Boston, MA, USA, 2018; ISBN 978-3-11-053453-5. [Google Scholar] [CrossRef]
- Mikulík, K.; Melčová, M.; Zídková, J. Antibacterial peptides from thermophilic bacteria. Int. J. Eng. Res. Sci. 2017, 3, 46–57. [Google Scholar] [CrossRef]
Figure 1.
Natural organophosphotus compounds.

Figure 2.
Biologically important phosphates.

Figure 3.
An example of the substrate-level phosphorylation.

Figure 4.
Structure of nucleic acids.

Figure 5.
A-, B and Z-forms of DNA.

Figure 6.
Nucleopeptides.

Figure 7.
Cross-shaped structure of DNA: (a)—Schematic representation; (b)—Drawing from a photograph obtained using an electron micrograph of a four-way DNA junction (permission on reproduction of Cambridge University Press and Copyright Clearance Center, Order Number: 5065840479765 from 11 May 2021) [29].
Figure 7.
Cross-shaped structure of DNA: (a)—Schematic representation; (b)—Drawing from a photograph obtained using an electron micrograph of a four-way DNA junction (permission on reproduction of Cambridge University Press and Copyright Clearance Center, Order Number: 5065840479765 from 11 May 2021) [29].

Figure 8.
Schematic representation of DNA-based asymmetric catalysis using the supramolecular anchoring strategy.
Figure 8.
Schematic representation of DNA-based asymmetric catalysis using the supramolecular anchoring strategy.

Figure 9.
L- and D-Enantiomers of DNA: L-DNA (left) and natural D-DNA (right).

Figure 10.
The asymmetric Michael addition reaction catalyzed by complexes formed between copper(II) ions and achiral ligands in the presence of DNA.
Figure 10.
The asymmetric Michael addition reaction catalyzed by complexes formed between copper(II) ions and achiral ligands in the presence of DNA.

Figure 11.
Some chiral compounds obtained by asymmetric DNA catalysis.

Figure 12.
Some of the most important phosphagens.

Figure 13.
Mechanisms of phosphoryl group transfer in substitution reactions at phosphorus atom. (*) is a well-known designation for chirality, the absolute configuration of which is not defined.
Figure 13.
Mechanisms of phosphoryl group transfer in substitution reactions at phosphorus atom. (*) is a well-known designation for chirality, the absolute configuration of which is not defined.

Figure 14.
ATP hydrolysis by ATPase with formation of ADP.

Figure 15.
(a) Mechanism of solvolysis and racemization of dinitrophenyl phosphorous acid dianion; (b) The metaphosphate-anion in a crystal lattice of fructoso-1,6-bisphosphatase found by X-ray crystallographic analysis.
Figure 15.
(a) Mechanism of solvolysis and racemization of dinitrophenyl phosphorous acid dianion; (b) The metaphosphate-anion in a crystal lattice of fructoso-1,6-bisphosphatase found by X-ray crystallographic analysis.

Figure 16.
Interconversions of phosphocreatine/creatine kinase: the system of ATP + creatine is in equilibrium with ADP + phosphocreatine.
Figure 16.
Interconversions of phosphocreatine/creatine kinase: the system of ATP + creatine is in equilibrium with ADP + phosphocreatine.

Figure 17.
Examples of N-phosphamides.

Figure 18.
Examples of Phosphaguanidines.

Figure 19.
Analogs of N-phosphocreatinine.

Figure 20.
Regioselective intramolecular phosphorylation of glycolaldehyde (R = H) and D-glyceraldehyde (R = CH2OH) by amidotriphosphate.
Figure 20.
Regioselective intramolecular phosphorylation of glycolaldehyde (R = H) and D-glyceraldehyde (R = CH2OH) by amidotriphosphate.

Figure 21.
Intramolecular phosphorylation by transient tethering in the arabino series.

Figure 22.
Formation of adenosine-5′-phosphoramide from ammonia and adenosine-5′-phosphosulfate.

Figure 23.
Structures of the nucleotidyl derivatives Hint 2.

Figure 24.
Nucleotides that bind protein of the histidine triad.

Figure 25.
Natural products containing N-acylphosphoramide bonds.

Figure 26.
Synthesis of phosmidosine diasteremers. Reagents and conditions: (I) MeOH-Et3N (9:1, v/v), rt, 1 h; (II) diisopropylammonium tetrazolide, CH2Cl2, rt, 1 h; (III) 17, 5-(3,5-dinitrophenyl)-1H-tetrazole, CH2Cl2-MeCN (1:1, v/v), rt, 10 min; tert-BuOOH, MeCN, rt, 5 min; I2, pyridine-H2O (9:1, v/v), rt, 30 min; (IV) 80% HCOOH, rt, 12 h. (*) is a well-known designation for chirality, the absolute configuration of which is not defined.
Figure 26.
Synthesis of phosmidosine diasteremers. Reagents and conditions: (I) MeOH-Et3N (9:1, v/v), rt, 1 h; (II) diisopropylammonium tetrazolide, CH2Cl2, rt, 1 h; (III) 17, 5-(3,5-dinitrophenyl)-1H-tetrazole, CH2Cl2-MeCN (1:1, v/v), rt, 10 min; tert-BuOOH, MeCN, rt, 5 min; I2, pyridine-H2O (9:1, v/v), rt, 30 min; (IV) 80% HCOOH, rt, 12 h. (*) is a well-known designation for chirality, the absolute configuration of which is not defined.

Figure 27.
The synthesis of nucleotide antibiotic phosmidosine B.

Figure 28.
Agrocine 84 and Biotin-Phosmidosine.

Figure 29.
Metabolites of Agrocin 84.

Figure 30.
Conversion of agrocin 84 into a toxic fragment of TM84.

Figure 31.
Antibiotics of the McC family.

Figure 32.
McC derivatives.

Figure 33.
Examples of naturally occurring aminoacyladenylate-based antibiotics.

Figure 34.
Phosphoramide antibiotics.

Figure 35.
Protein-DNA intermediates.

Figure 36.
Examples of N-phosphohistidine.

Figure 37.
Natural phosphorus-sulfur bond containing phosphates.

Figure 38.
Phosphorothioate modificated oligonucleotides.

Figure 39.
The most famous natural phosphonates.

Figure 40.
Overview of P-C phosphonate biosynthesis.

Figure 41.
Synthesis of (2S,1R)-Fosfomycin starting from phosphonoenolpyruvate.

Figure 42.
Formation of herbicide Bialaphos.

Figure 43.
Biosynthesis of (2-amino-I-hydroxyethyl) phosphinic acid.

Figure 44.
Biocatalytic epoxidation of (S)-2-hydroxyalkylphosphonic acids to epoxides.

Figure 45.
Biocatalytic conversion of hydroxyphosphonate to fosfomycin.

Figure 46.
Synthesis of a deuterated analog of (S,R)-phosphomycin.

Figure 47.
Biologically active phosphonates.

Figure 48.
Enzymatic synthesis of D- and L-stereomers of phosphinotricin.

Figure 49.
The enzymatic synthesis of Phosphonothrixin.

Figure 50.
Synthesis of phosphonothrixin.

Figure 51.
Asymmetric synthesis of phosphonothrixin.

Figure 52.
Determination of (S)-phosphonoth rixin configuration by chemical correlation.

Figure 53.
Phosphorus derivatives of Plakotilen A and B.

Figure 54.
Reaction system for ciliatocholic acid synthesis.

Figure 55.
Nucleotide antibiotics—phosphadecin and phosphocytocin.

Figure 56.
Representatives of phosphonolipids.

Figure 57.
Hydrolytic treatment of alaphosphin peptides to obtain an alanine analog.

Figure 58.
Hydrolytic treatment of dehydrophos peptides to release pyruvate analog.

Figure 59.
Antibiotic phosphonopeptides.

Figure 60.
Seven step synthesis of (S,Z)-APPA starting from (R)-Garner’s aldehyde. Reagents and conditions: (a) (CF3CH2O)2P(O)CH2CO2Me, 18-Crown-6, NaH, THF, 78 °C; (b) DIBAL-H 1 M sol. in hexanes, toluene, 78 °C; (c) CBr4, PPh3, CH2Cl2, 0 °C; (d) P(OMe)3, 80–100 °C; (e) Dowex 50WX4 H+, MeOH/H2O:(9/1); (f) (i) H5IO6/CrO3, CH3CN/H2O:(99/1), 0 °C; (ii) CH3I, K2CO3, CH3CN; (g) (i) 10% (CH3)3SiBr in CH2Cl2, and then MeOH/H2O:(9/1); (ii) 1 N LiOH, THF/H2O:(4/1).
Figure 60.
Seven step synthesis of (S,Z)-APPA starting from (R)-Garner’s aldehyde. Reagents and conditions: (a) (CF3CH2O)2P(O)CH2CO2Me, 18-Crown-6, NaH, THF, 78 °C; (b) DIBAL-H 1 M sol. in hexanes, toluene, 78 °C; (c) CBr4, PPh3, CH2Cl2, 0 °C; (d) P(OMe)3, 80–100 °C; (e) Dowex 50WX4 H+, MeOH/H2O:(9/1); (f) (i) H5IO6/CrO3, CH3CN/H2O:(99/1), 0 °C; (ii) CH3I, K2CO3, CH3CN; (g) (i) 10% (CH3)3SiBr in CH2Cl2, and then MeOH/H2O:(9/1); (ii) 1 N LiOH, THF/H2O:(4/1).

Figure 61.
Phosphonopeptides containing a nitrogen-nitrogen (NN) bond.

Figure 62.
Determination of absolute configuration of phosphazinomycin by chemical correlation. (*)—center of chirality with an undefined absolute configuration.
Figure 62.
Determination of absolute configuration of phosphazinomycin by chemical correlation. (*)—center of chirality with an undefined absolute configuration.

Figure 63.
Some representatives of phosphonopeptides.

Figure 64.
Representatives of peptide antibiotics.

Figure 65.
Stereocontrolled synthesis of the tyrosine hydroxyphosphonate analog.

Figure 66.
Stereoselective synthesis of tripeptide renin inhibitors.

Figure 67.
Phosphonic tripeptides and tetrapeptide.

Figure 68.
Mechanism of Trialaphos and Phosalacine transformation inside of cell.


{kind=link}
{kind=link}
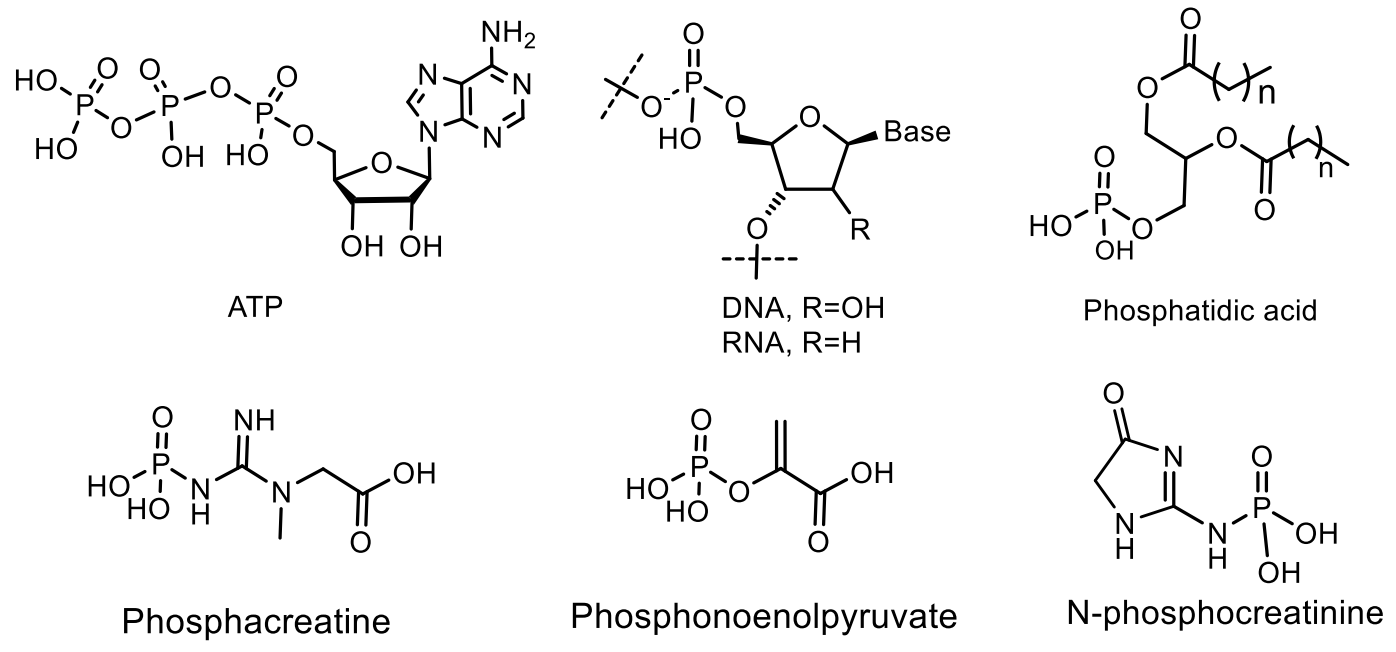{kind=link}
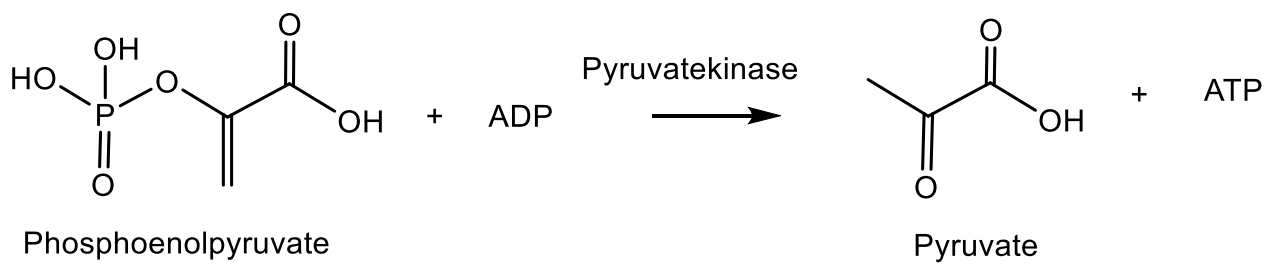{kind=link}
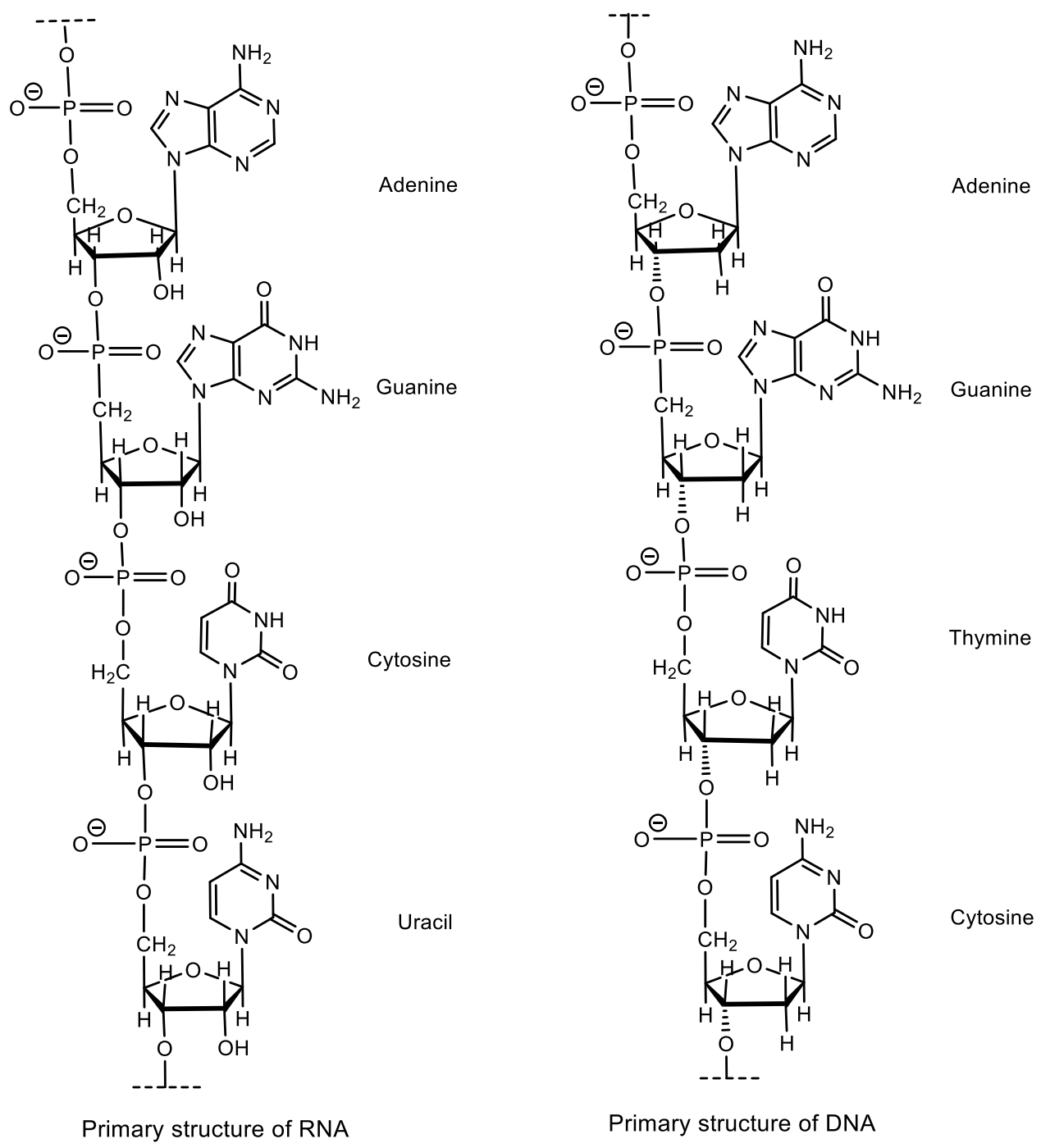{kind=link}
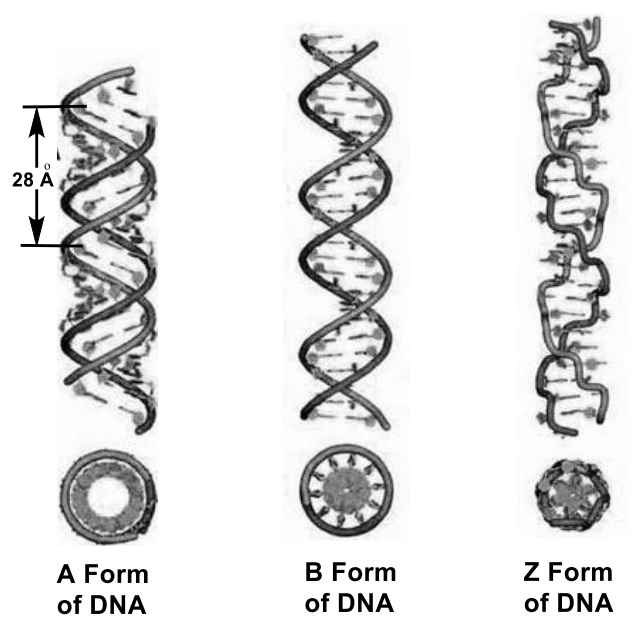{kind=link}
{kind=link}
{kind=link}
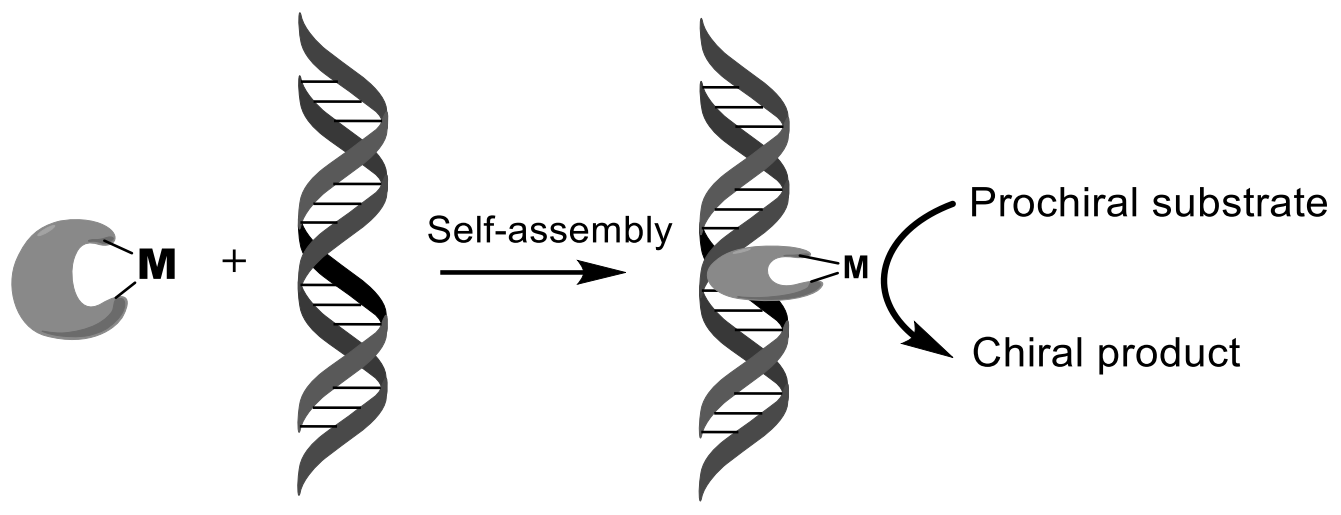{kind=link}
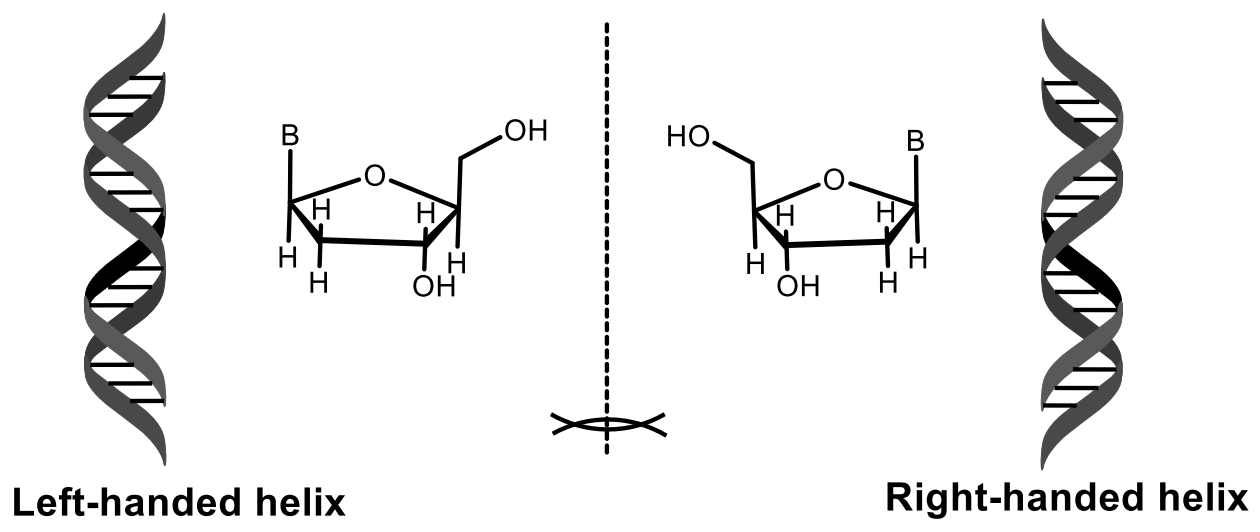{kind=link}
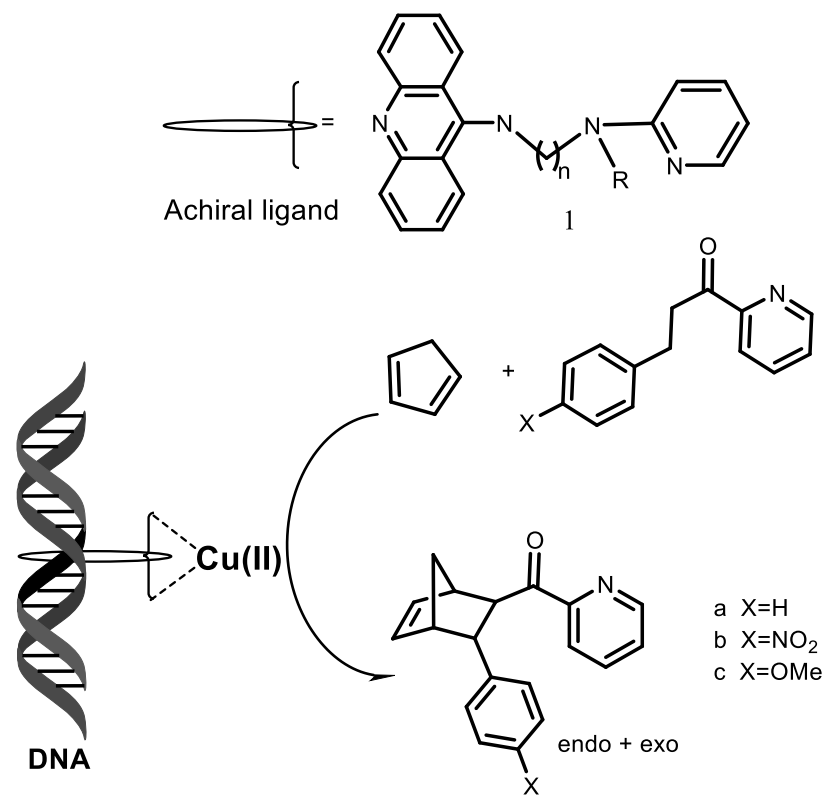{kind=link}
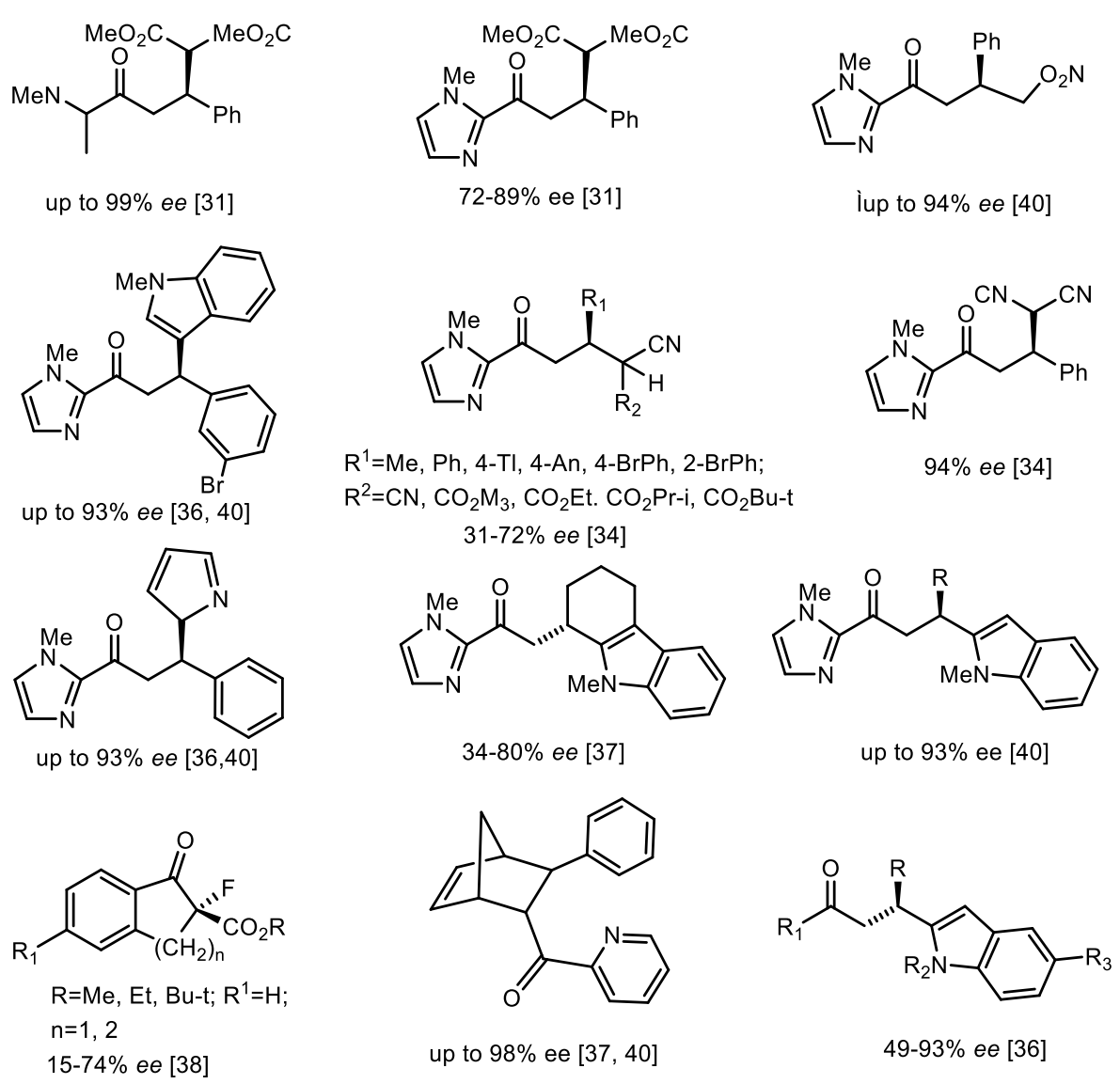{kind=link}
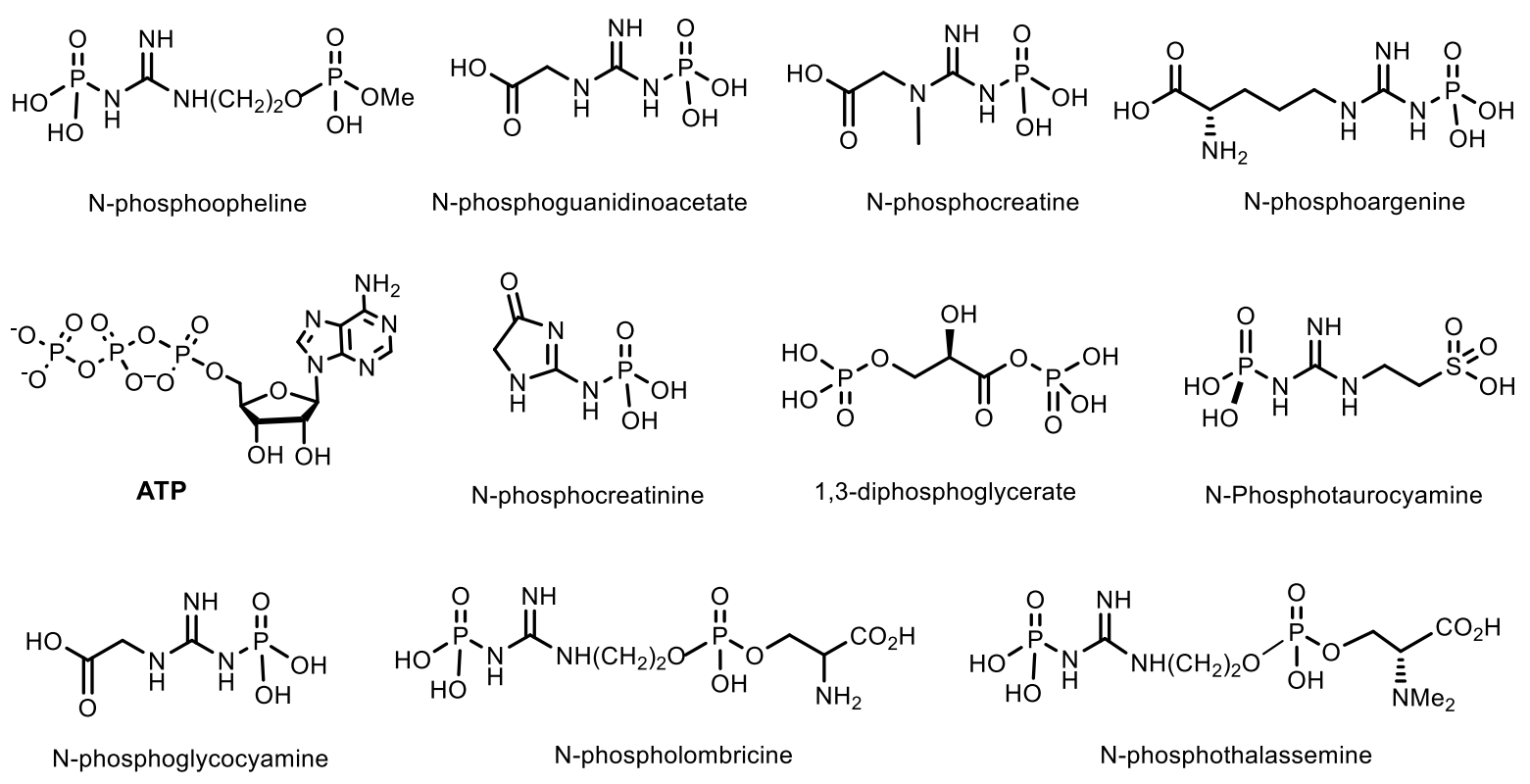{kind=link}
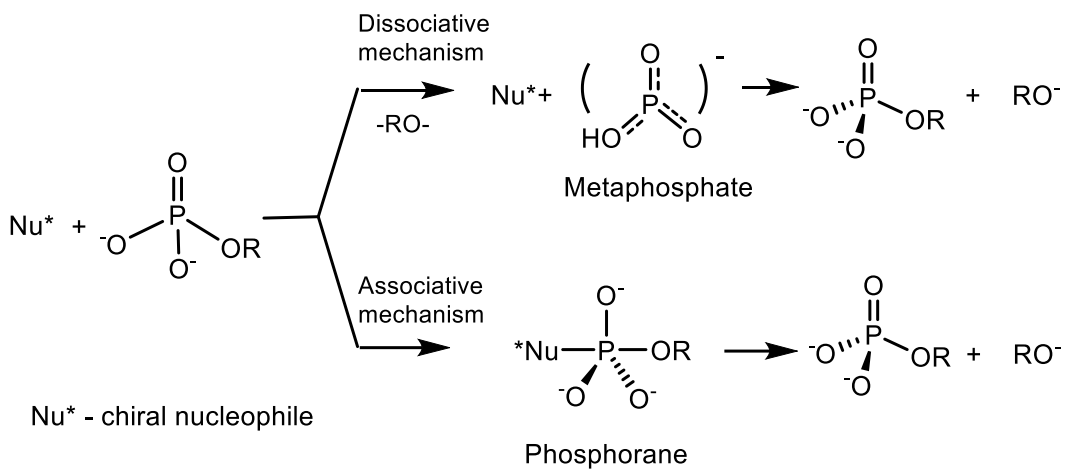{kind=link}
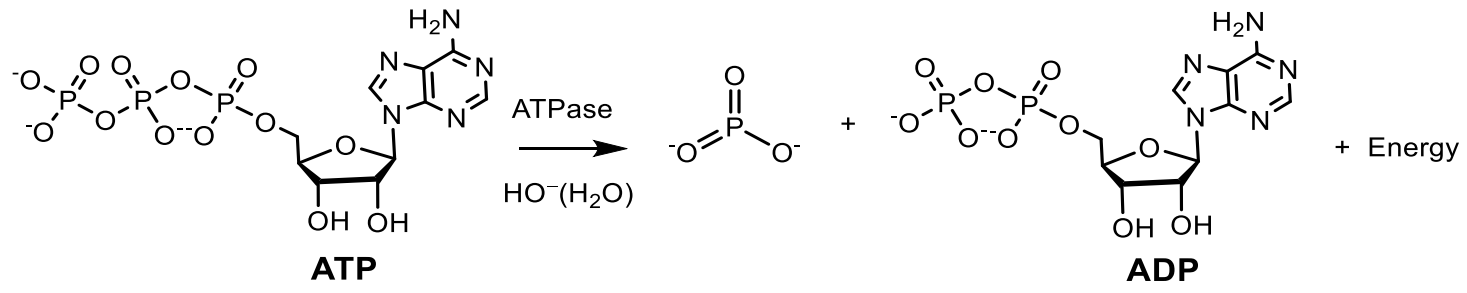{kind=link}
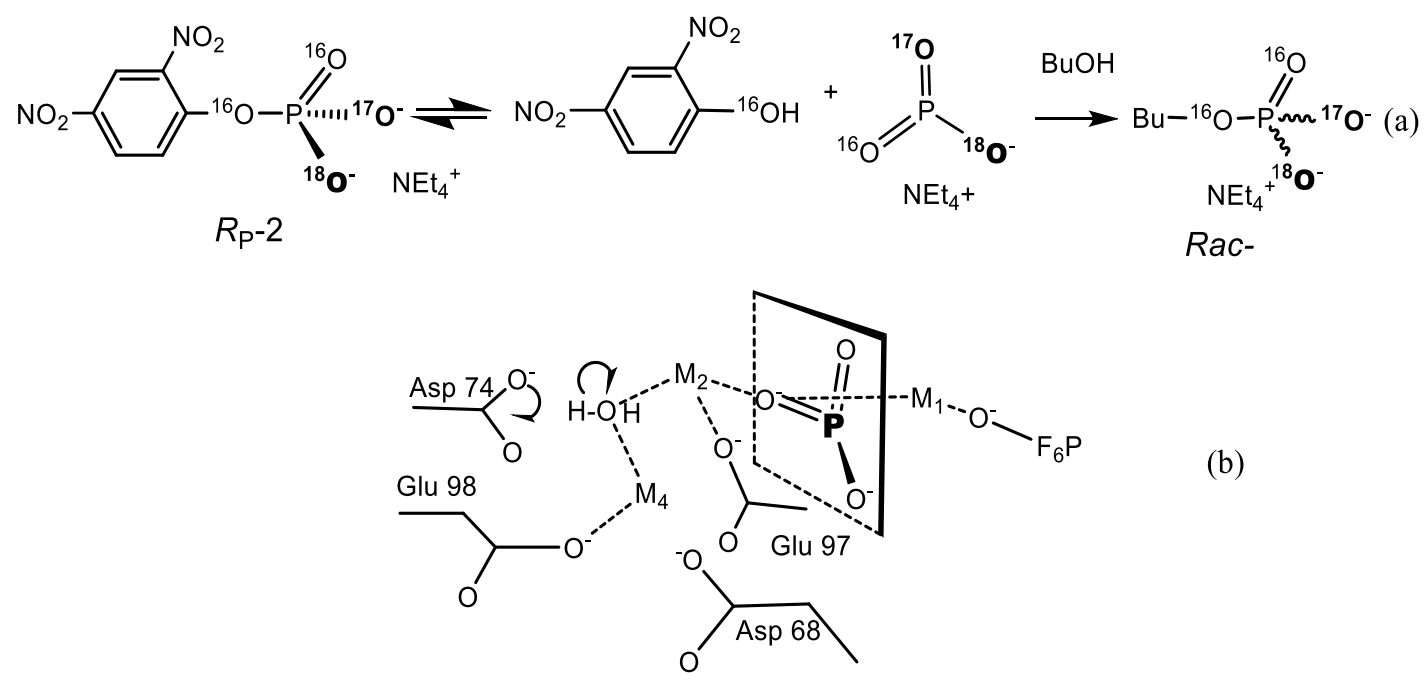{kind=link}
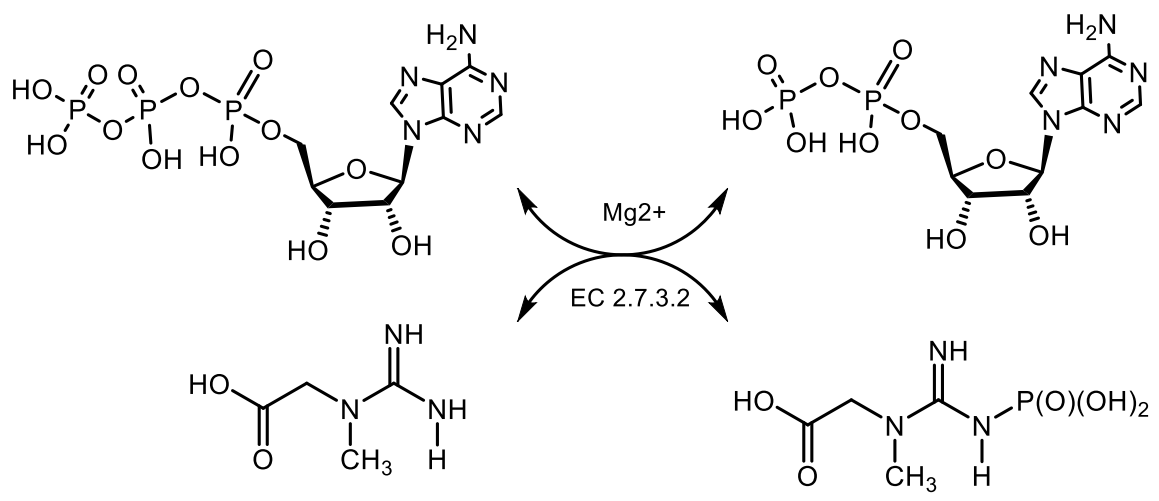{kind=link}
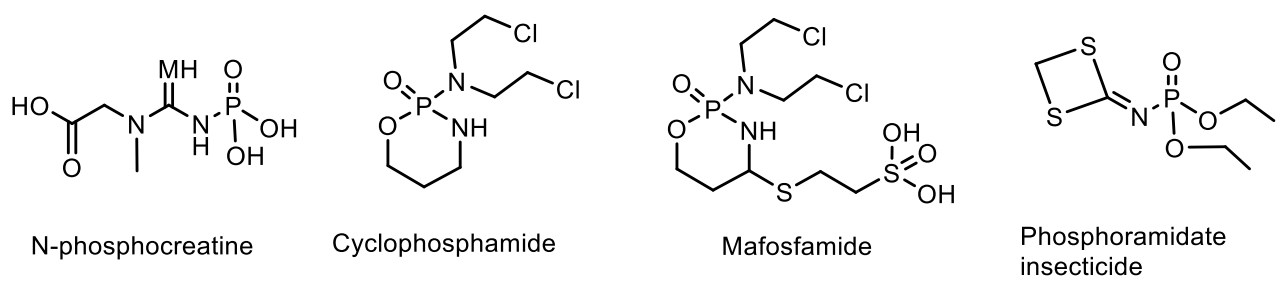{kind=link}
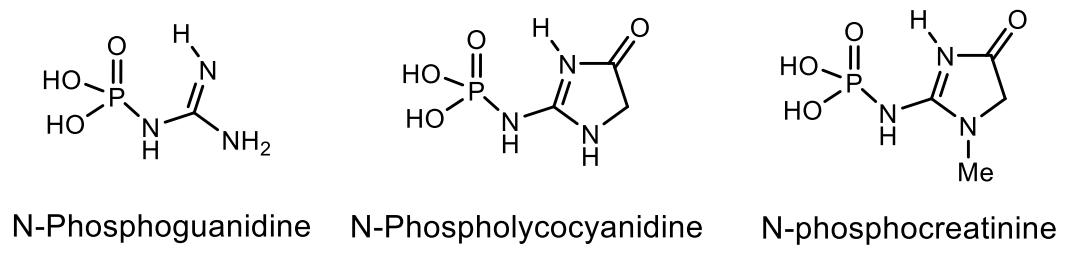{kind=link}
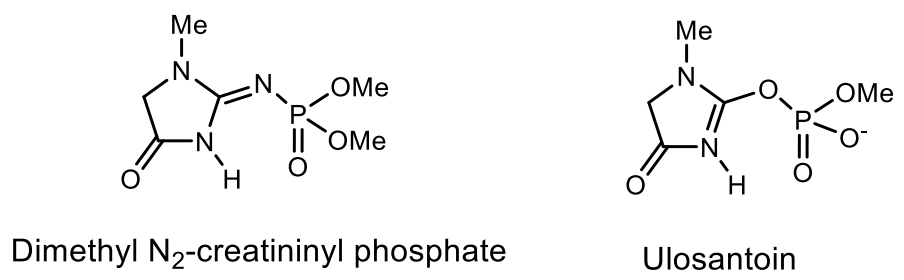{kind=link}
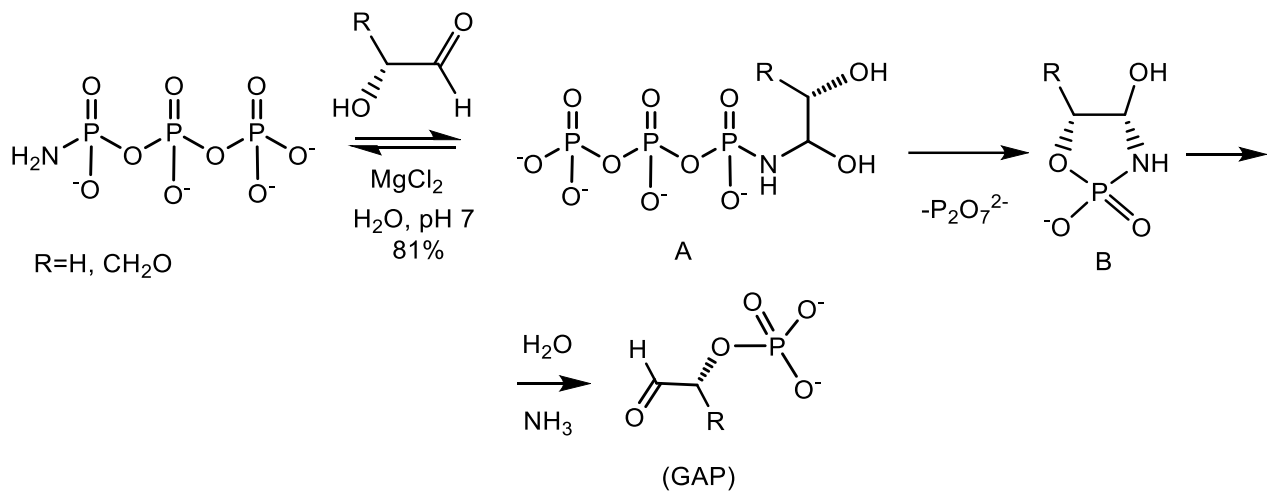{kind=link}
{kind=link}
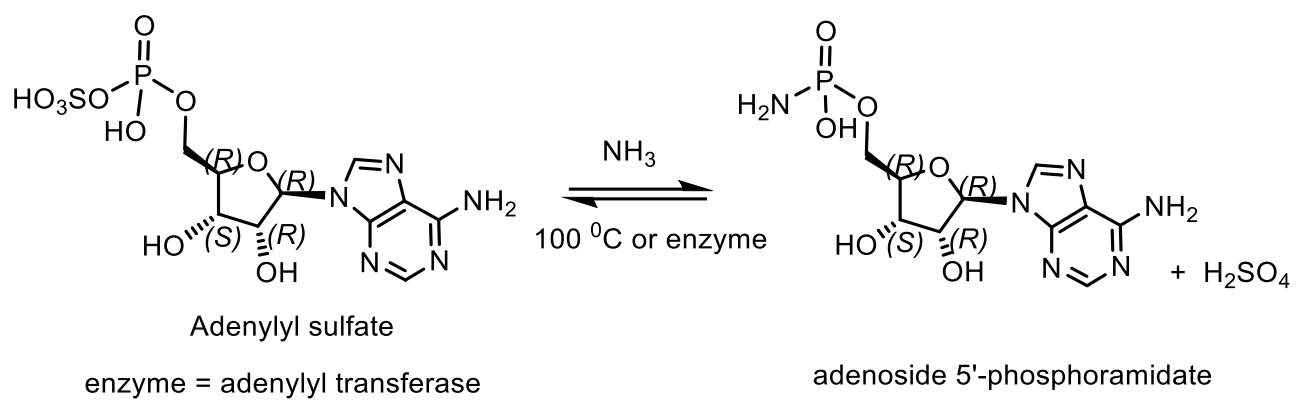{kind=link}
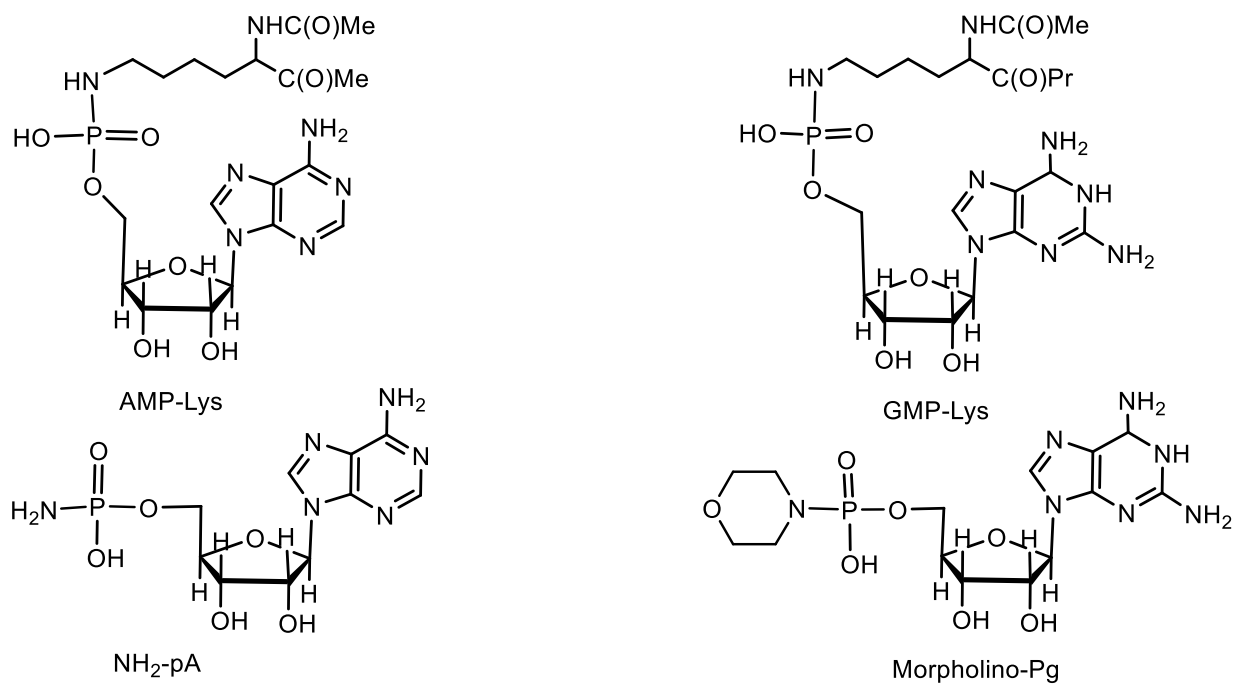{kind=link}
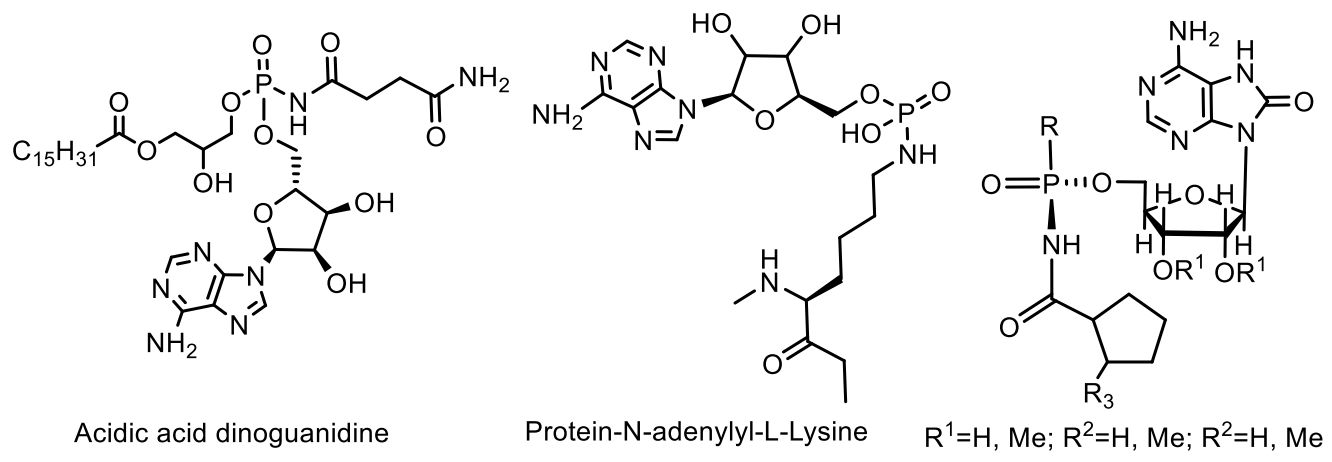{kind=link}
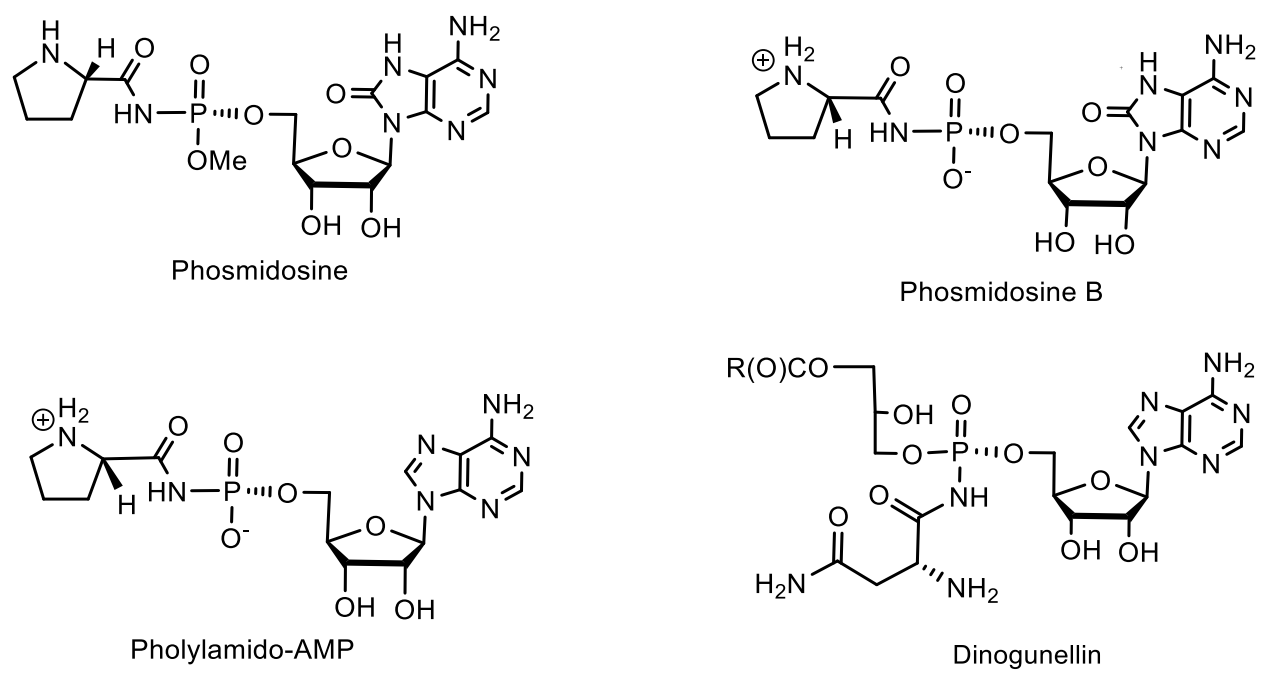{kind=link}
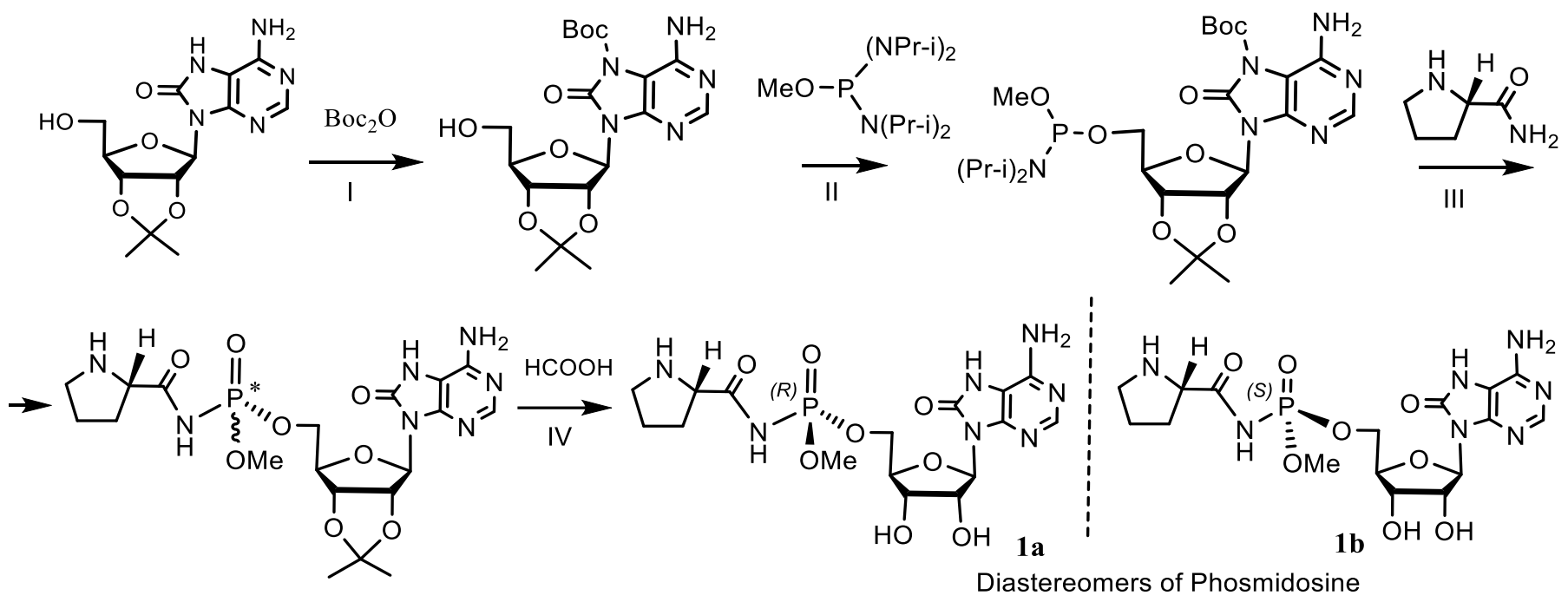{kind=link}
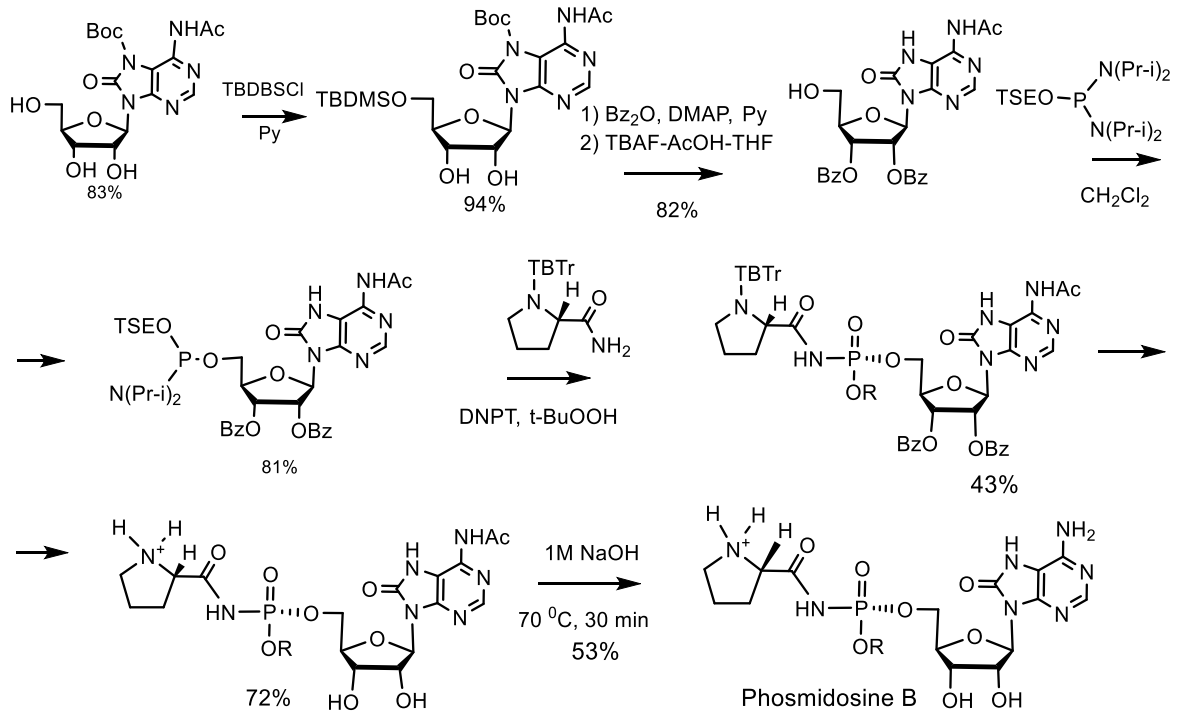{kind=link}
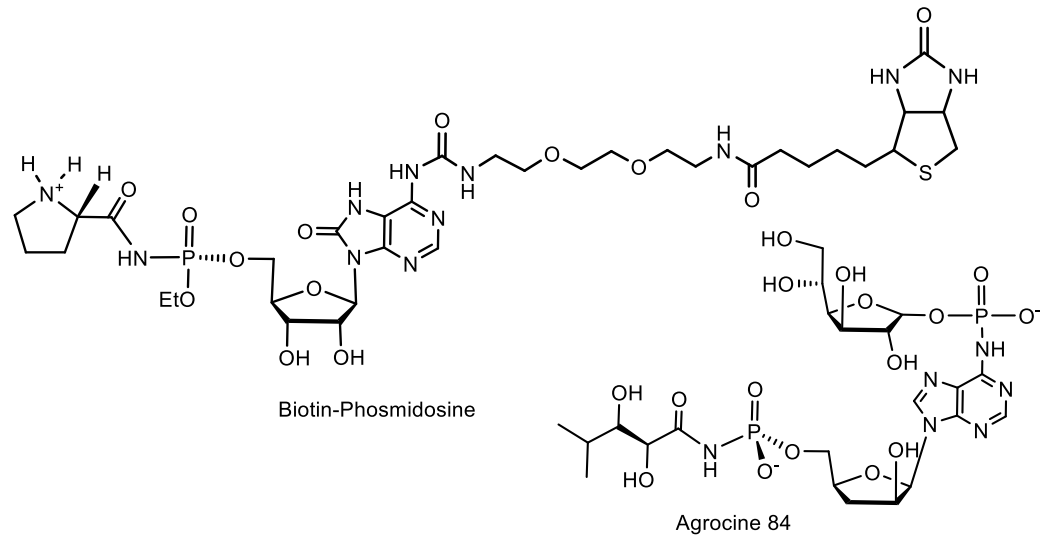{kind=link}
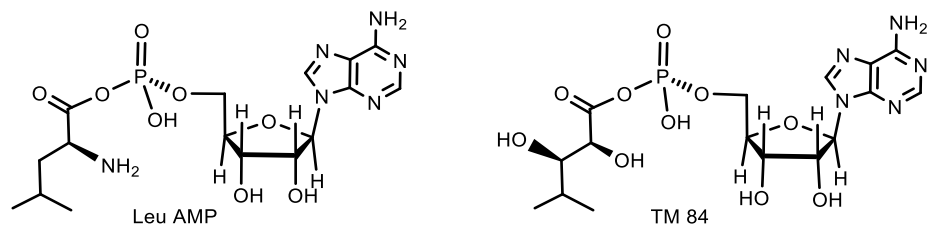{kind=link}
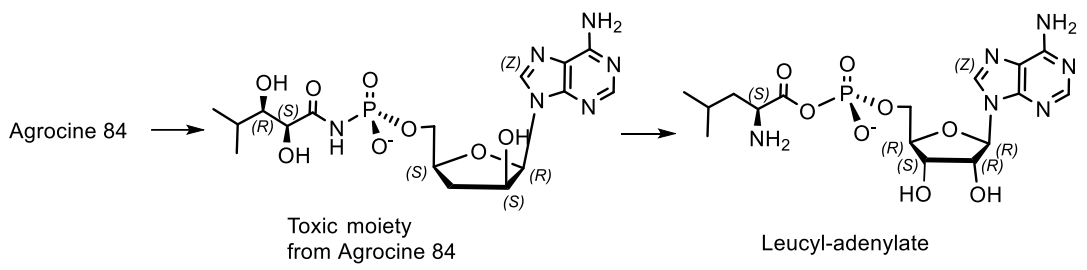{kind=link}
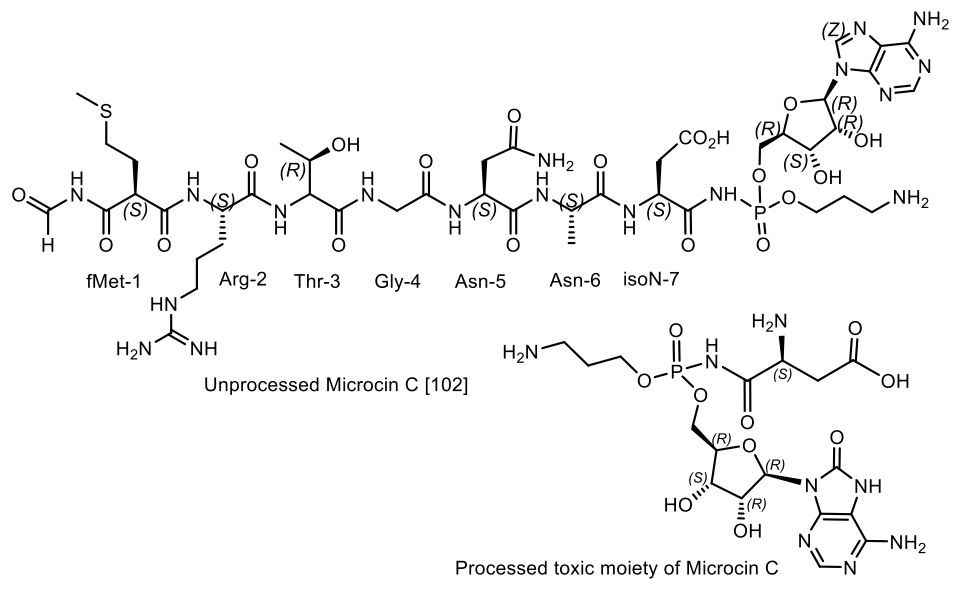{kind=link}
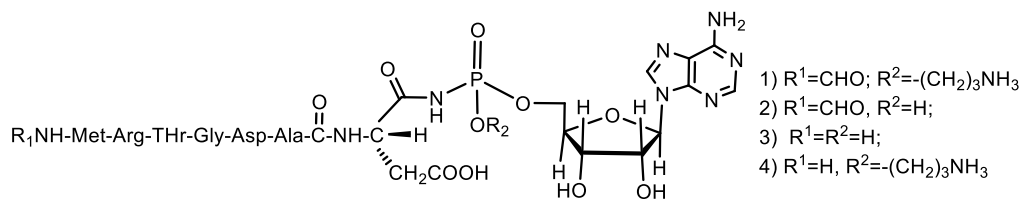{kind=link}
{kind=link}
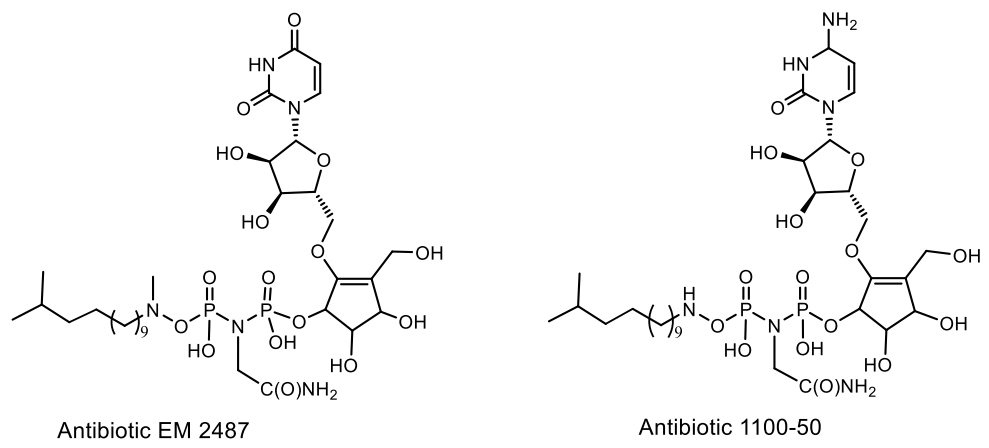{kind=link}
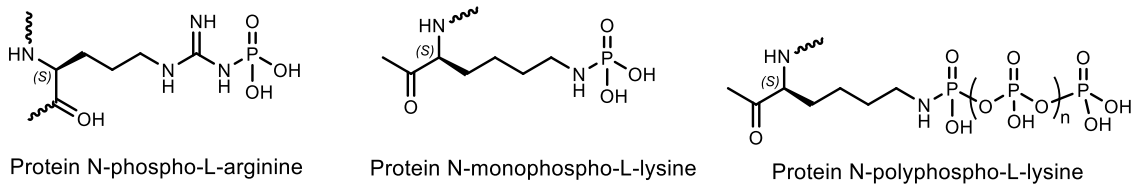{kind=link}
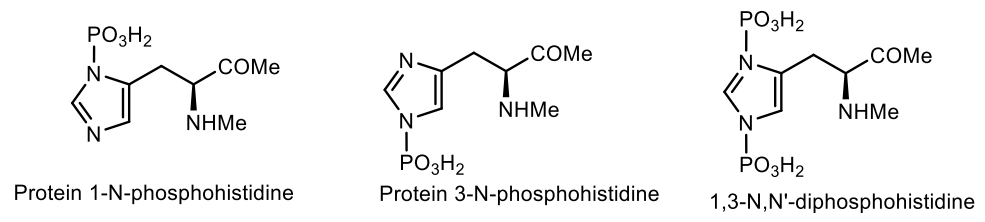{kind=link}
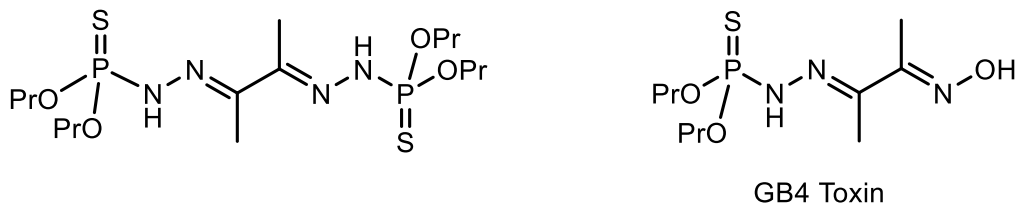{kind=link}
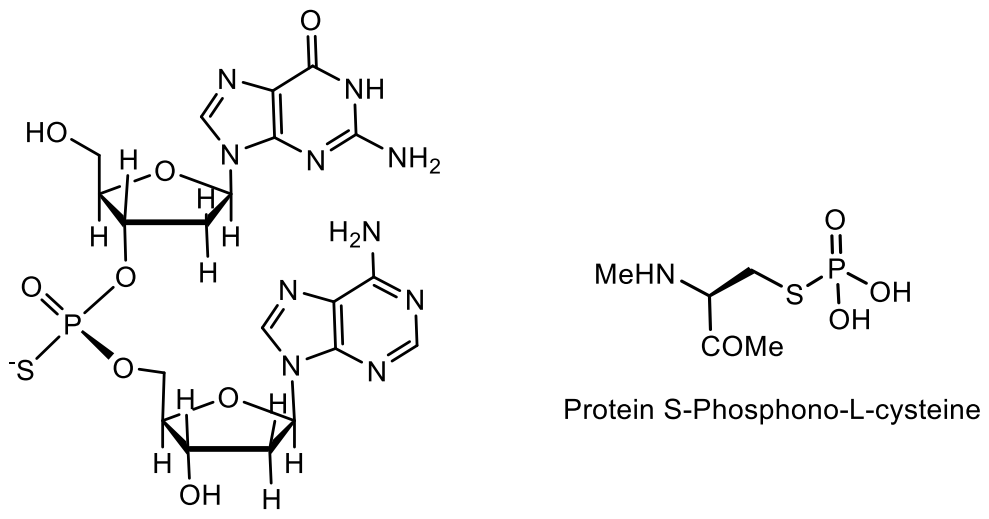{kind=link}
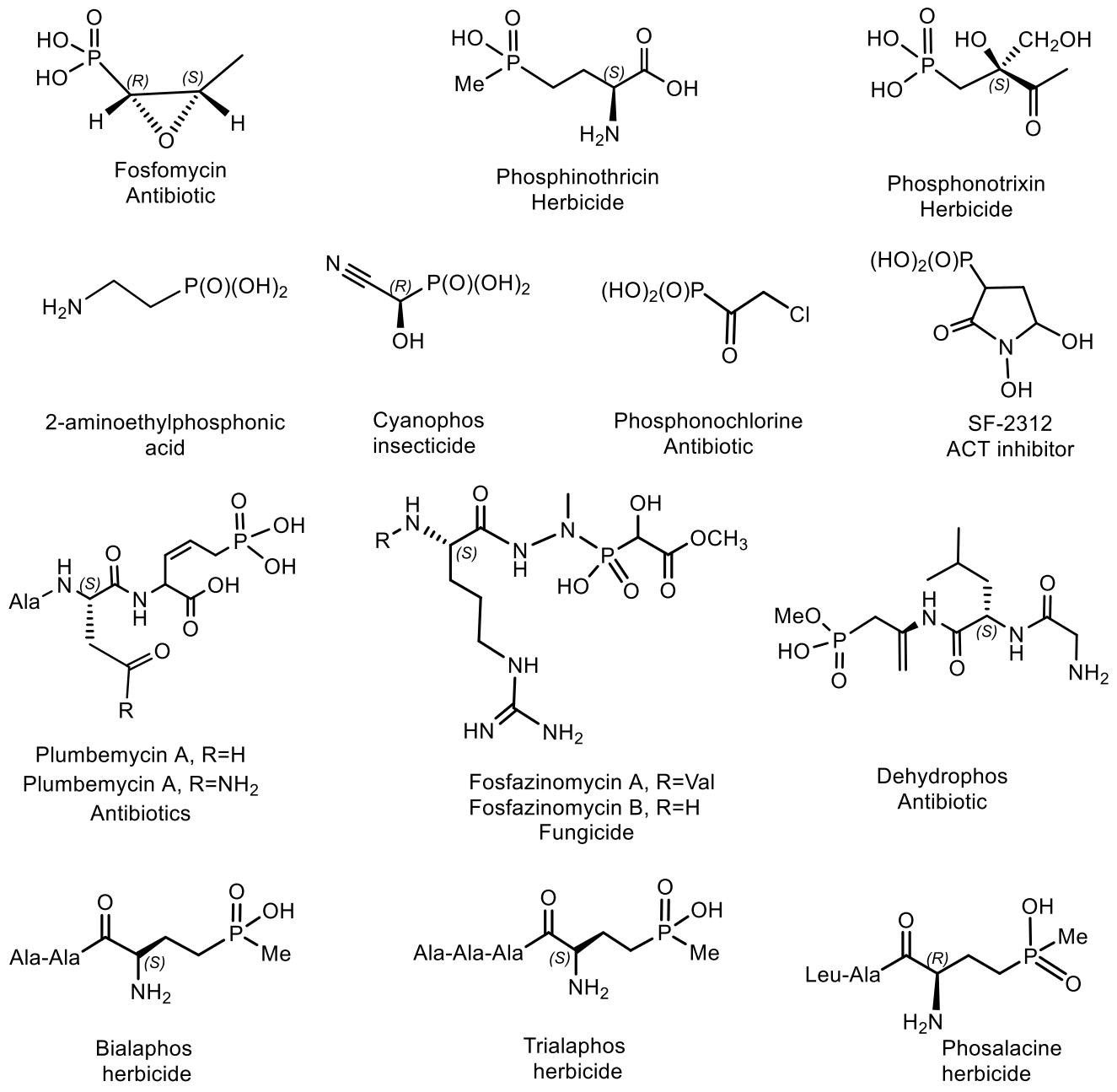{kind=link}
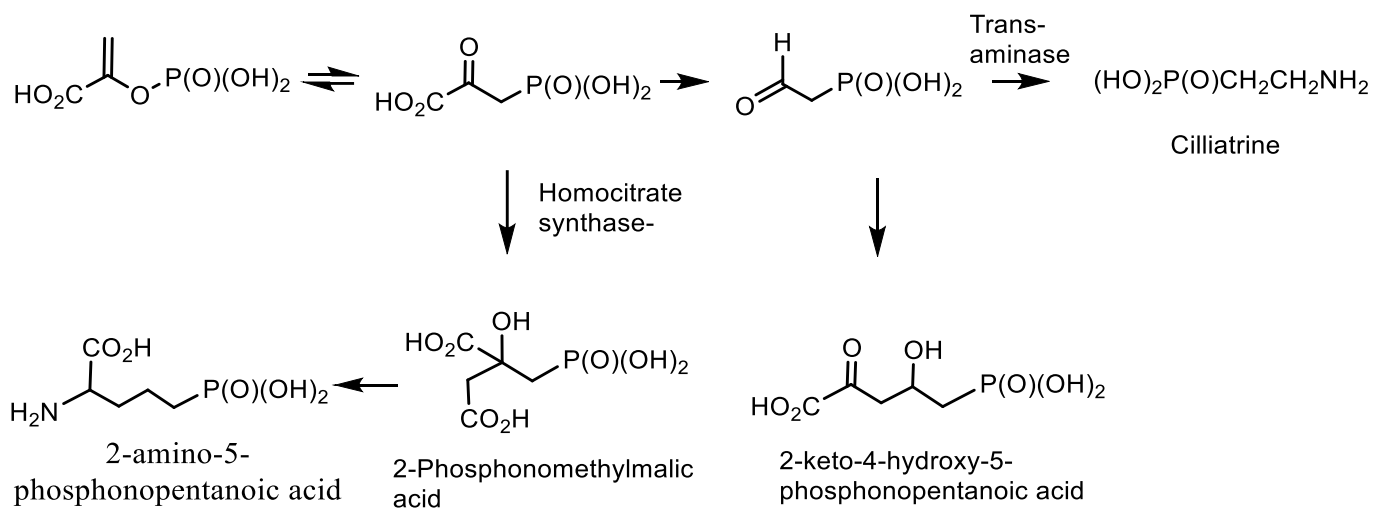{kind=link}
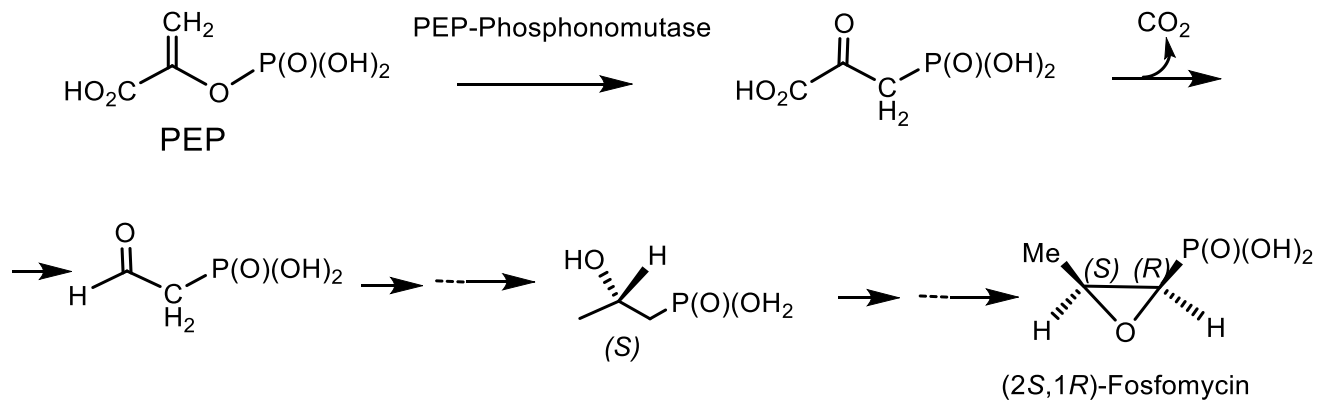{kind=link}
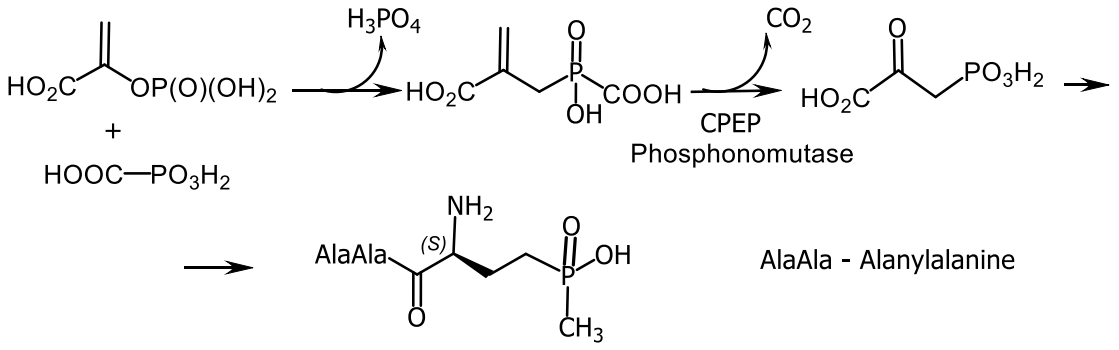{kind=link}
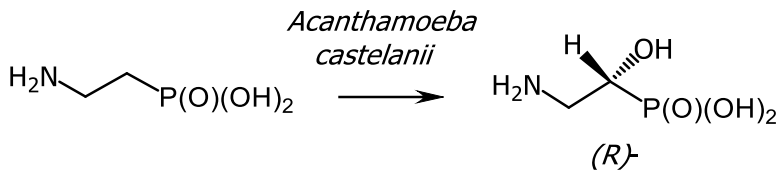{kind=link}
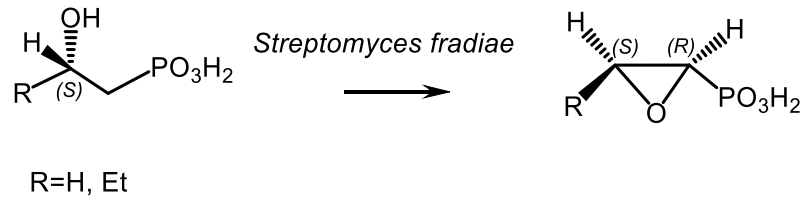{kind=link}
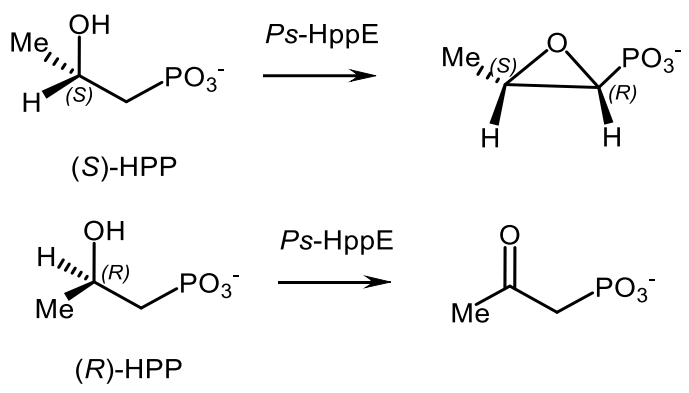{kind=link}
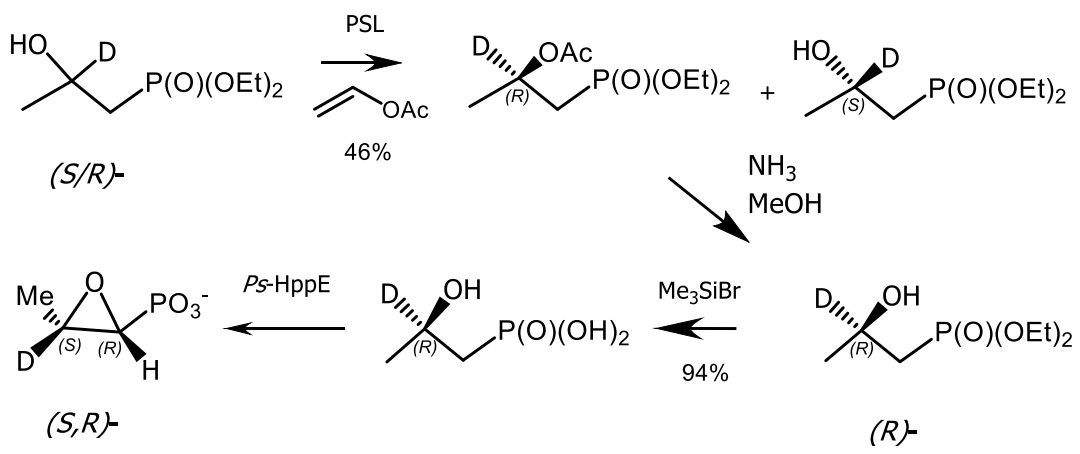{kind=link}
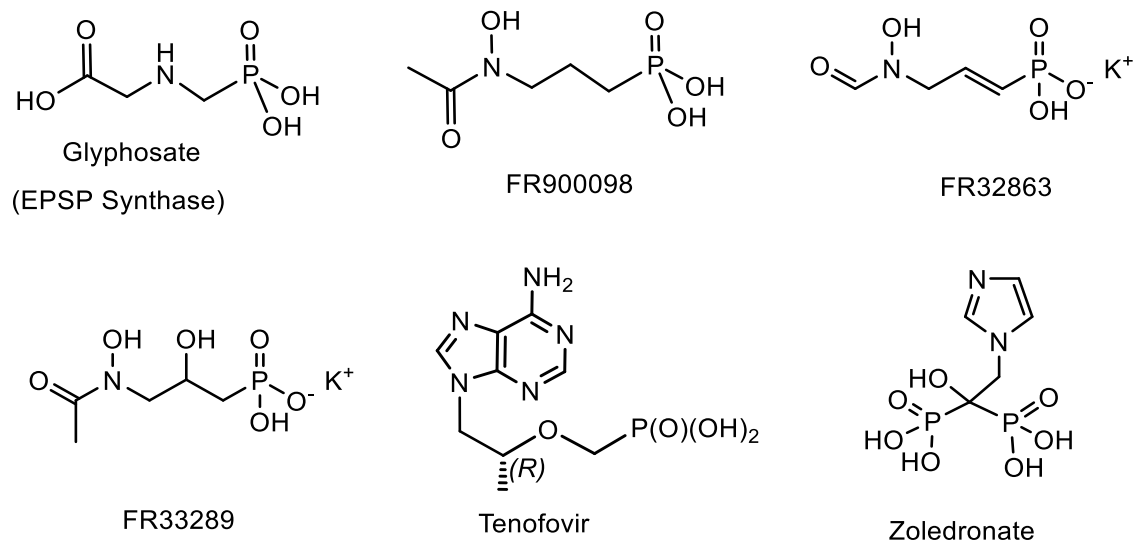{kind=link}
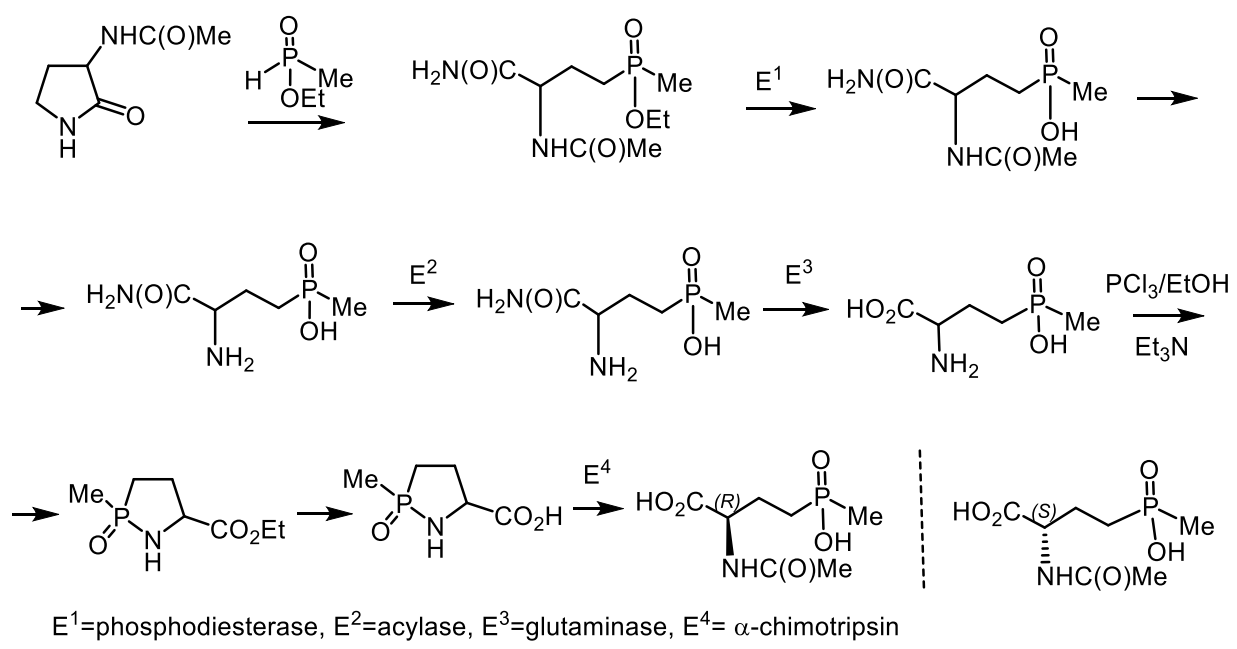{kind=link}
{kind=link}
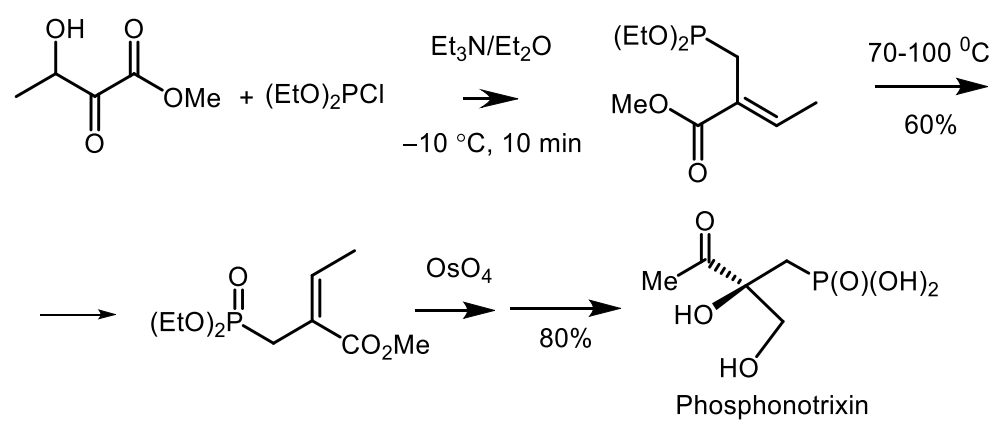{kind=link}
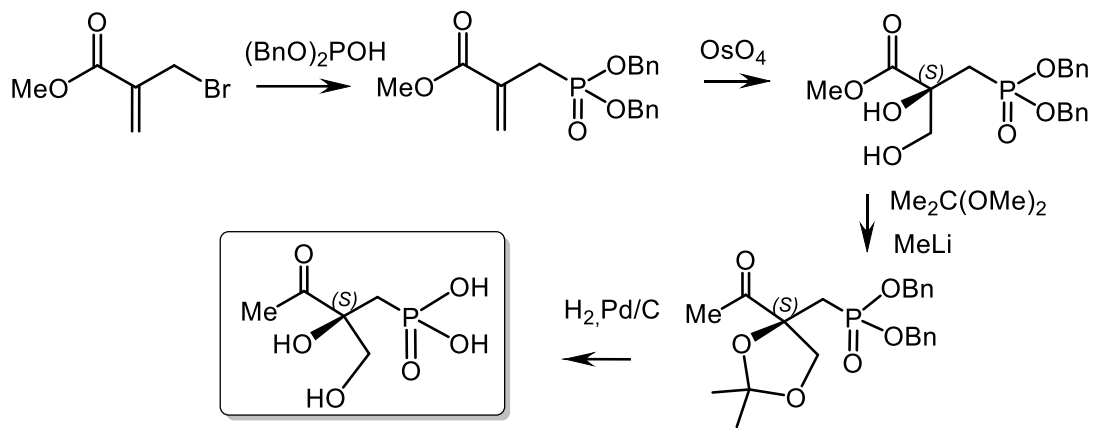{kind=link}
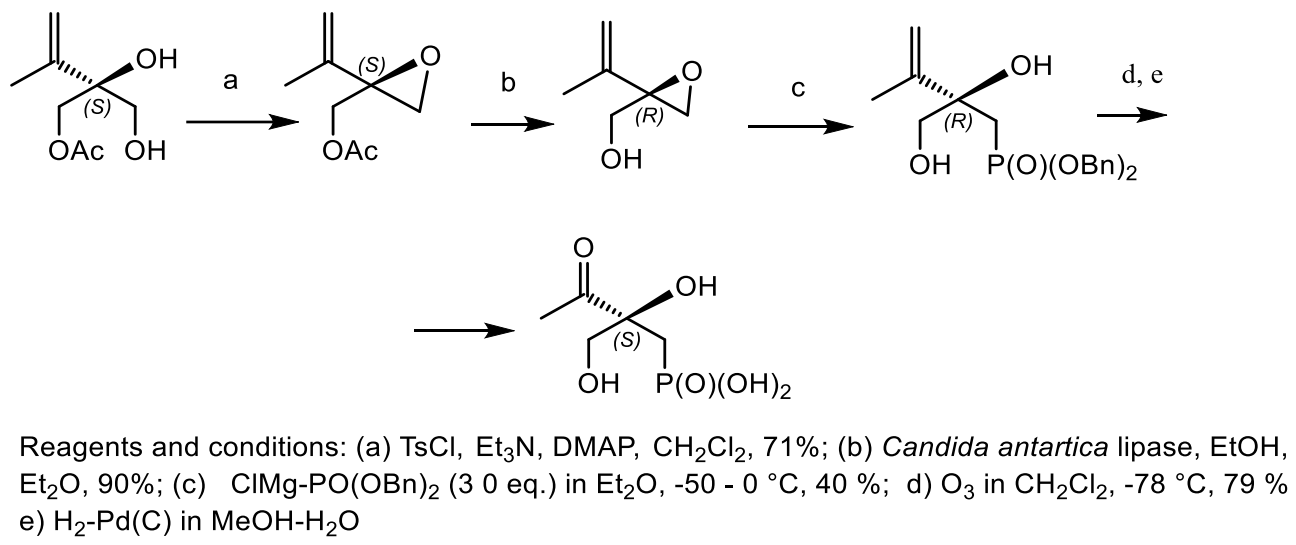{kind=link}
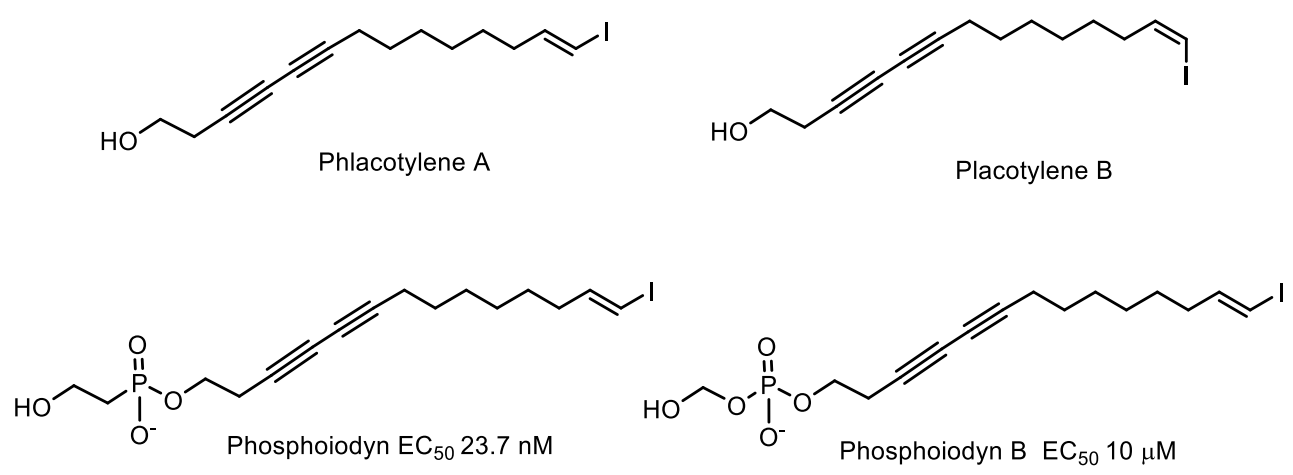{kind=link}
{kind=link}
{kind=link}
{kind=link}
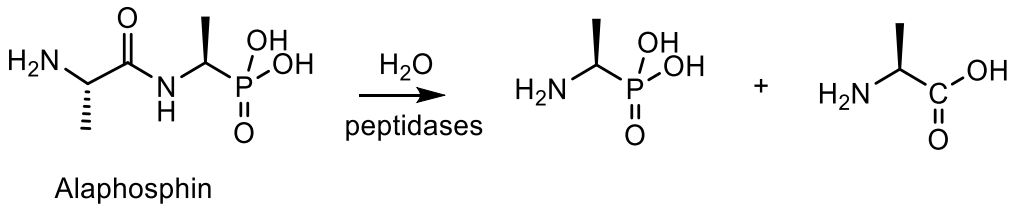{kind=link}
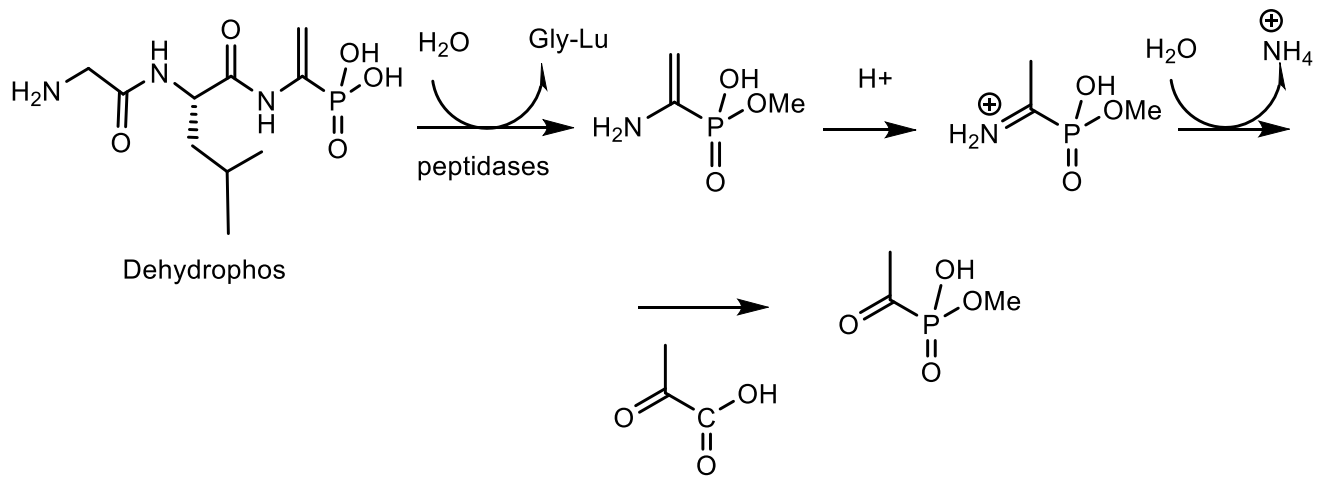{kind=link}
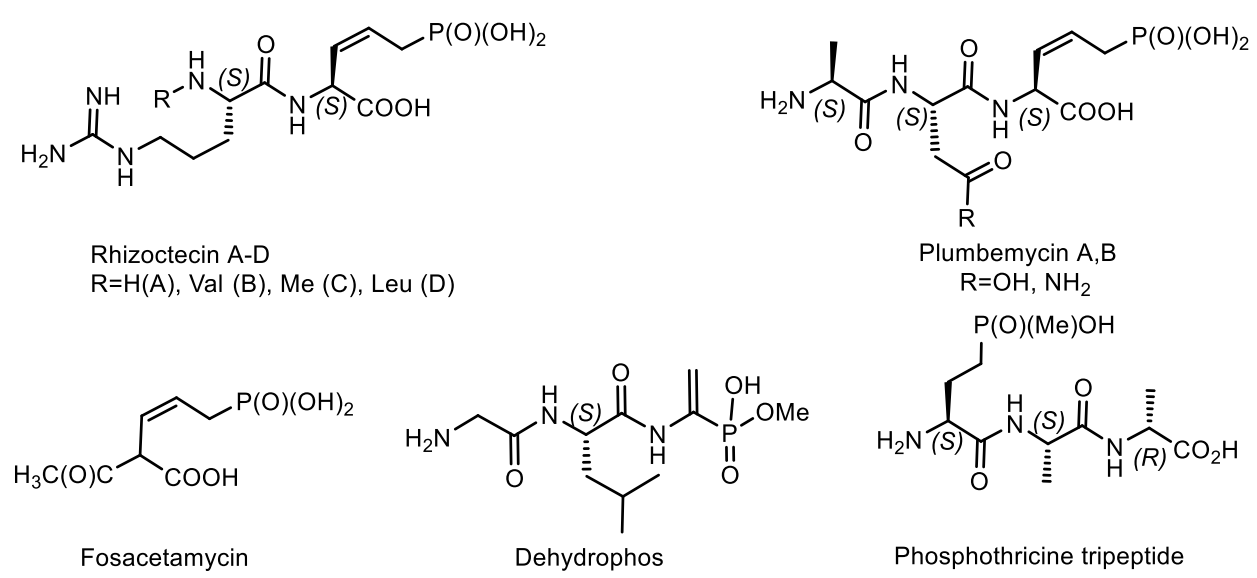{kind=link}
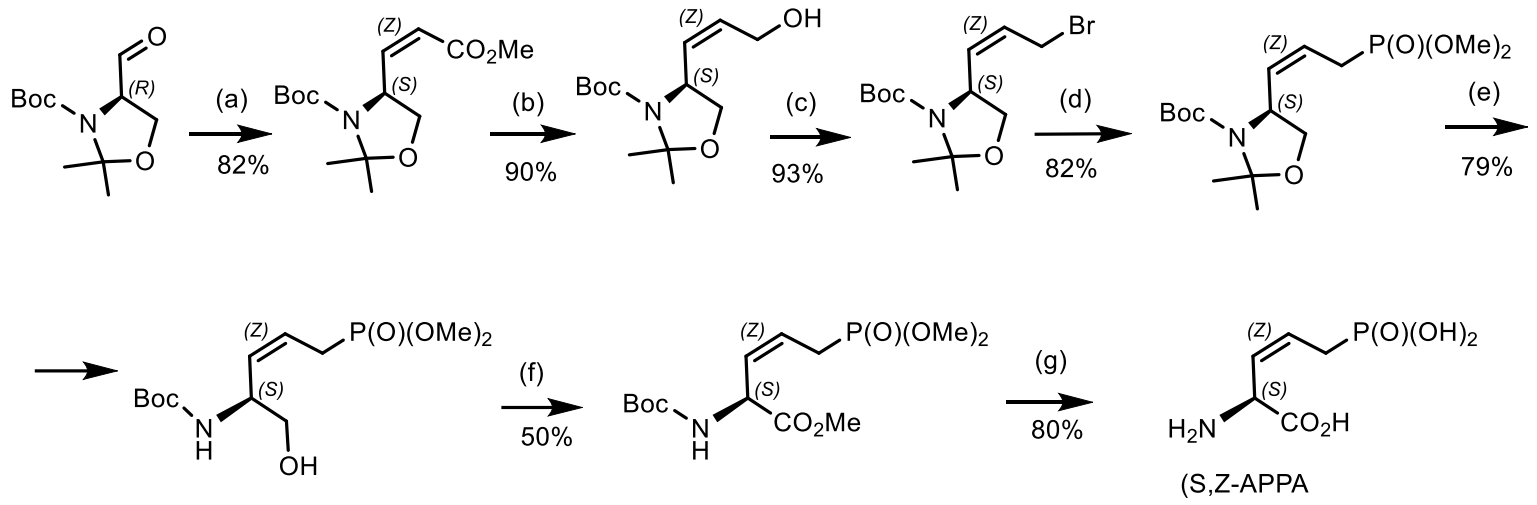{kind=link}
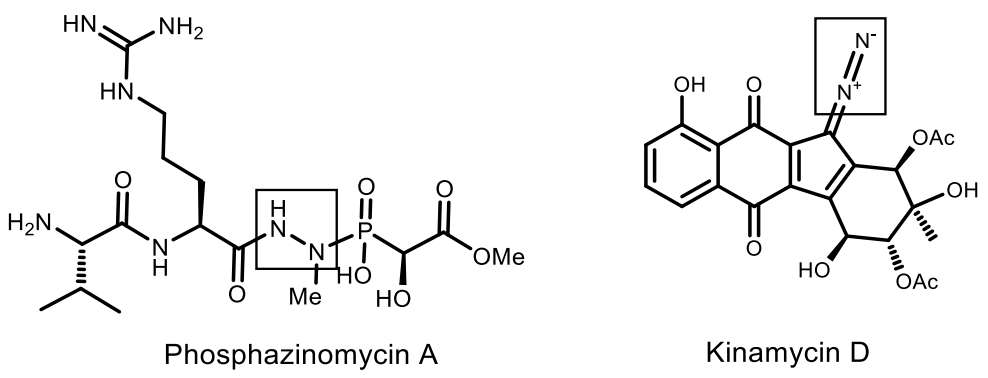{kind=link}
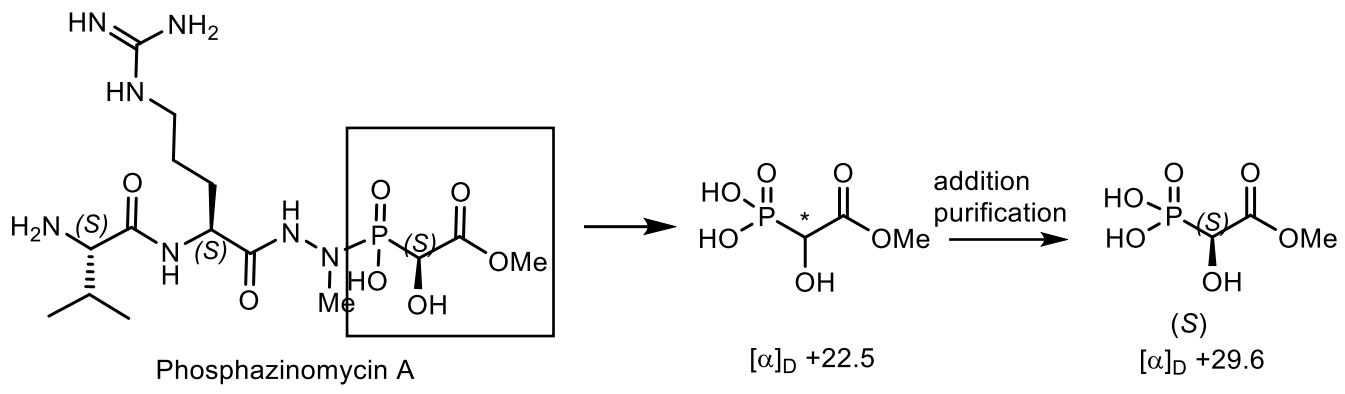{kind=link}
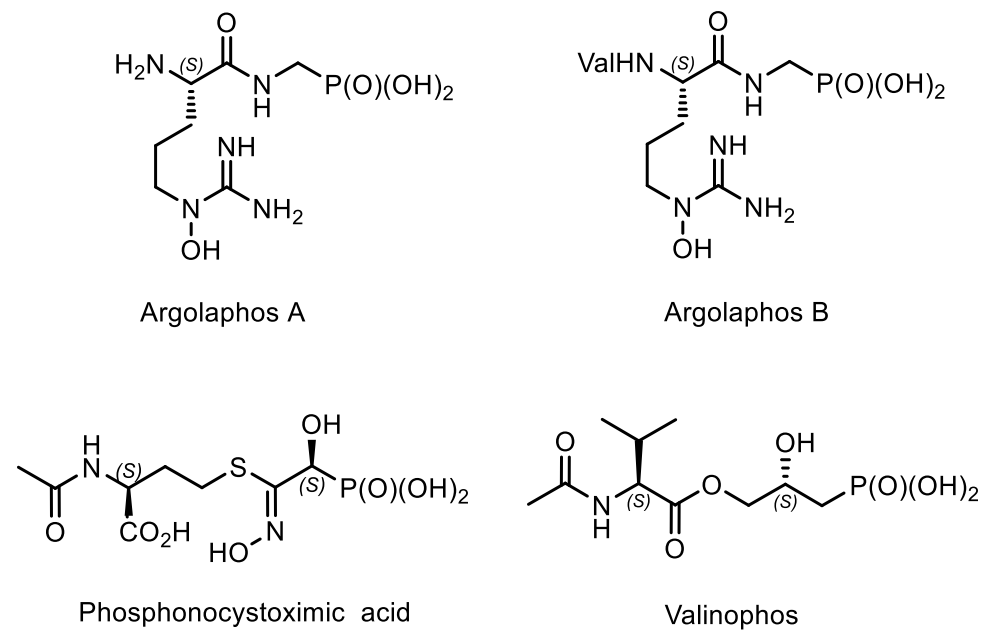{kind=link}
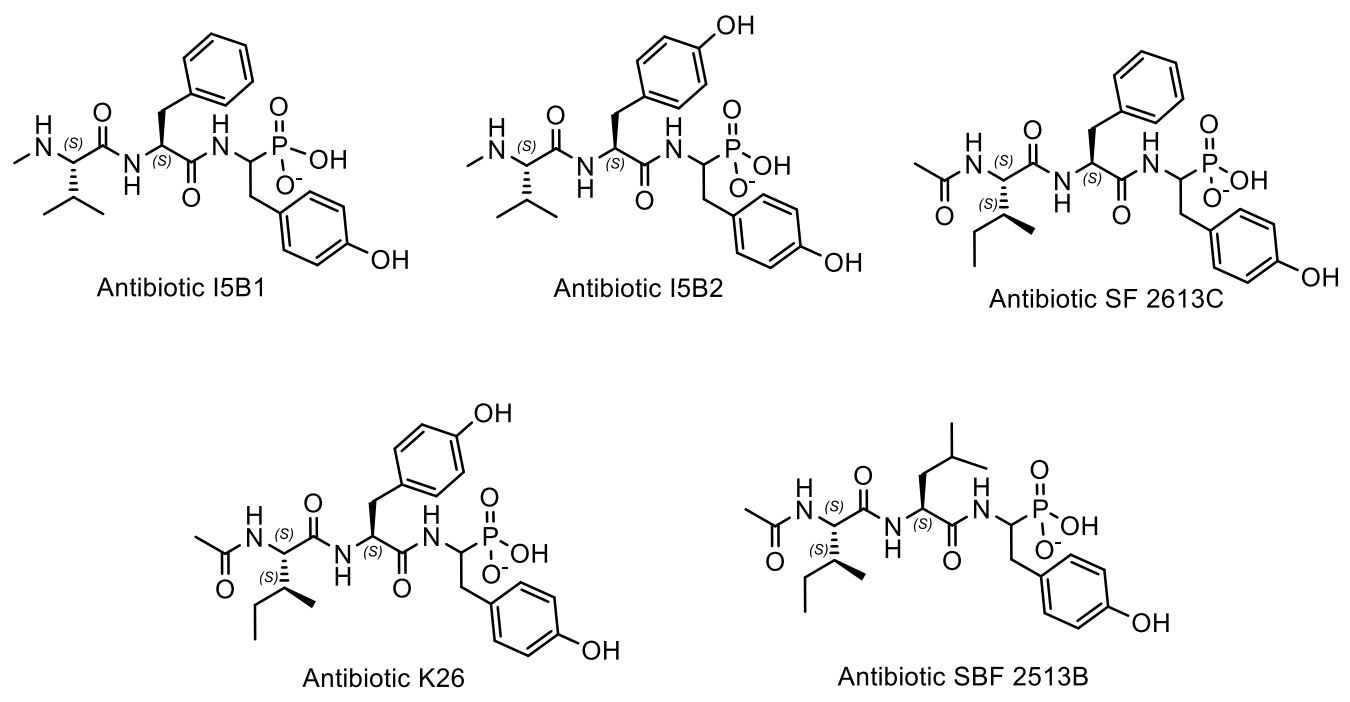{kind=link}
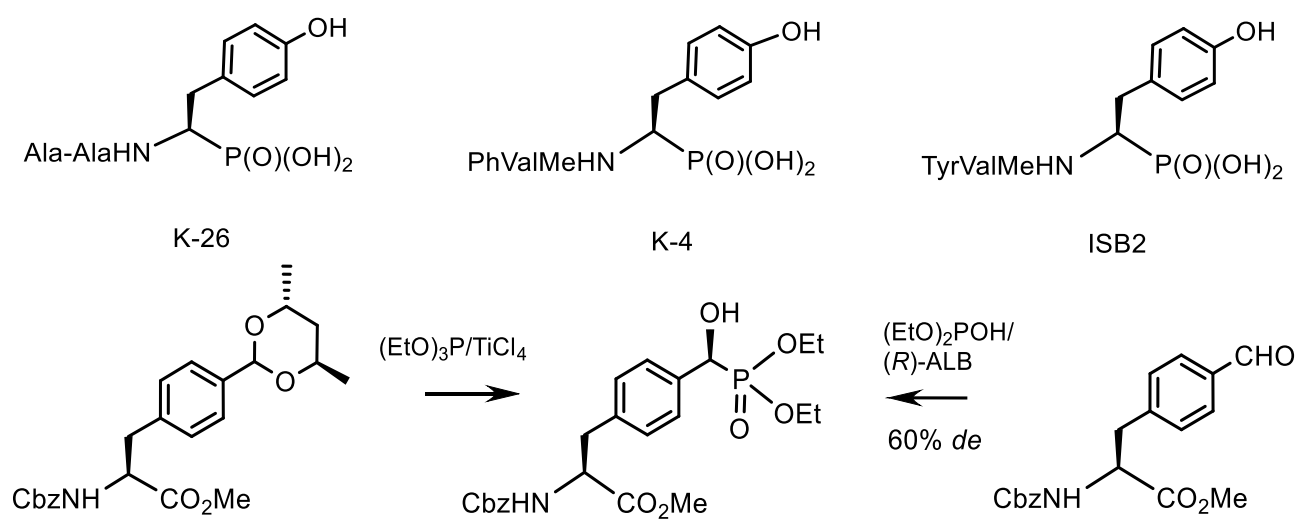{kind=link}
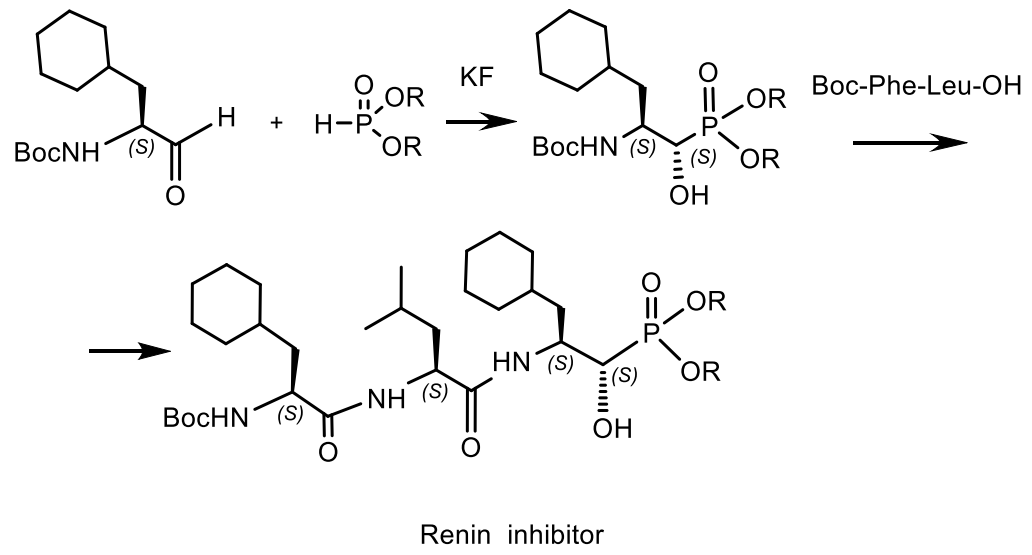{kind=link}
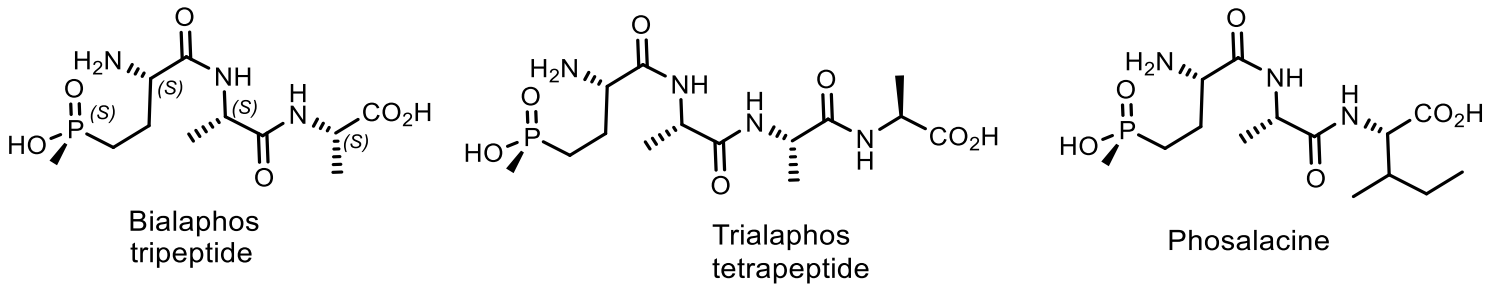{kind=link}
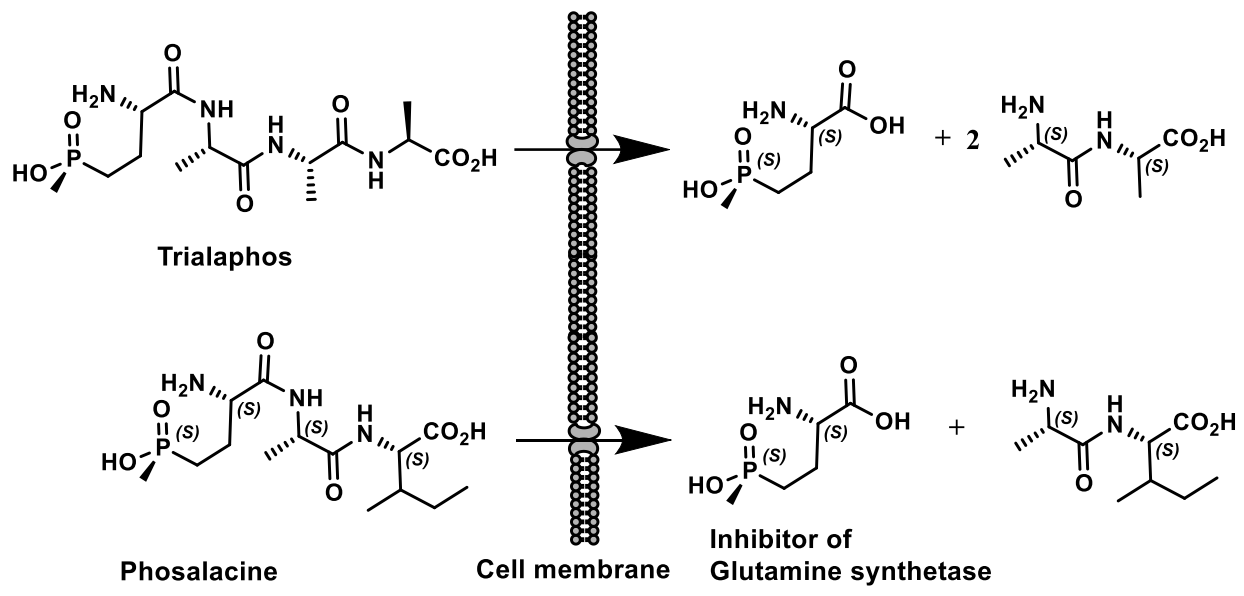{kind=link}
Table 1.
Ionization constants for orthophosphoric and citric acids
| Acid | pKa(1) | pKa(2) | pKa(3) |
|---|---|---|---|
| Phosphoric | 2.12 | 7.2 | 12.3 |
| Citric | 3.22 | 4.8 | 6.4 |
Table 2.
The Friedel–Crafts addition with D- and L-DNA.
 | |||||
| Entry | R1 | R2 | R3 | ee
[%] with D-DNA | ee
[%] with L-DNA |
| 1 | MeO | H | Me | 89 | 90 |
| 2 | H | H | Me | 76 | 76 |
| 3 | H | Me | o-BrC6H4 | 76 | 80 |
| 4 | MeO | H | 4-ClC6H4 | 72 | 74 |
Table 3.
Structures of phosmidosine, phosmidosines B and C.
 | R1 | R2 | Name |
| Me |  | Phosmidosine | |
| H |  | Phosmidosine B | |
| Me | H | Phosmidosine C |
Table 4.
The antitumor activities of phosmidosine and it stereoisomer.
| Type of tumor cells | IC50 (μM) | |
|---|---|---|
| Phosmidosine 1a | Phosmidosine 1b | |
| KB | 3.6 | 3.2 |
| KATO-III | 2.6 | 1.9 |
| MKN-28 | 9.6 | 8.5 |
| MKN-45 | 2.9 | 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kolodiazhnyi, O.I. Phosphorus Compounds of Natural Origin: Prebiotic, Stereochemistry, Application. Symmetry 2021, 13, 889. https://doi.org/10.3390/sym13050889
AMA Style
Kolodiazhnyi OI. Phosphorus Compounds of Natural Origin: Prebiotic, Stereochemistry, Application. Symmetry. 2021; 13(5):889. https://doi.org/10.3390/sym13050889
Chicago/Turabian StyleKolodiazhnyi, Oleg I. 2021. "Phosphorus Compounds of Natural Origin: Prebiotic, Stereochemistry, Application" Symmetry 13, no. 5: 889. https://doi.org/10.3390/sym13050889
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.