
Advances in the Development of Trifluoromethoxylation Reagents

 and
and {kind=link}
{kind=link}
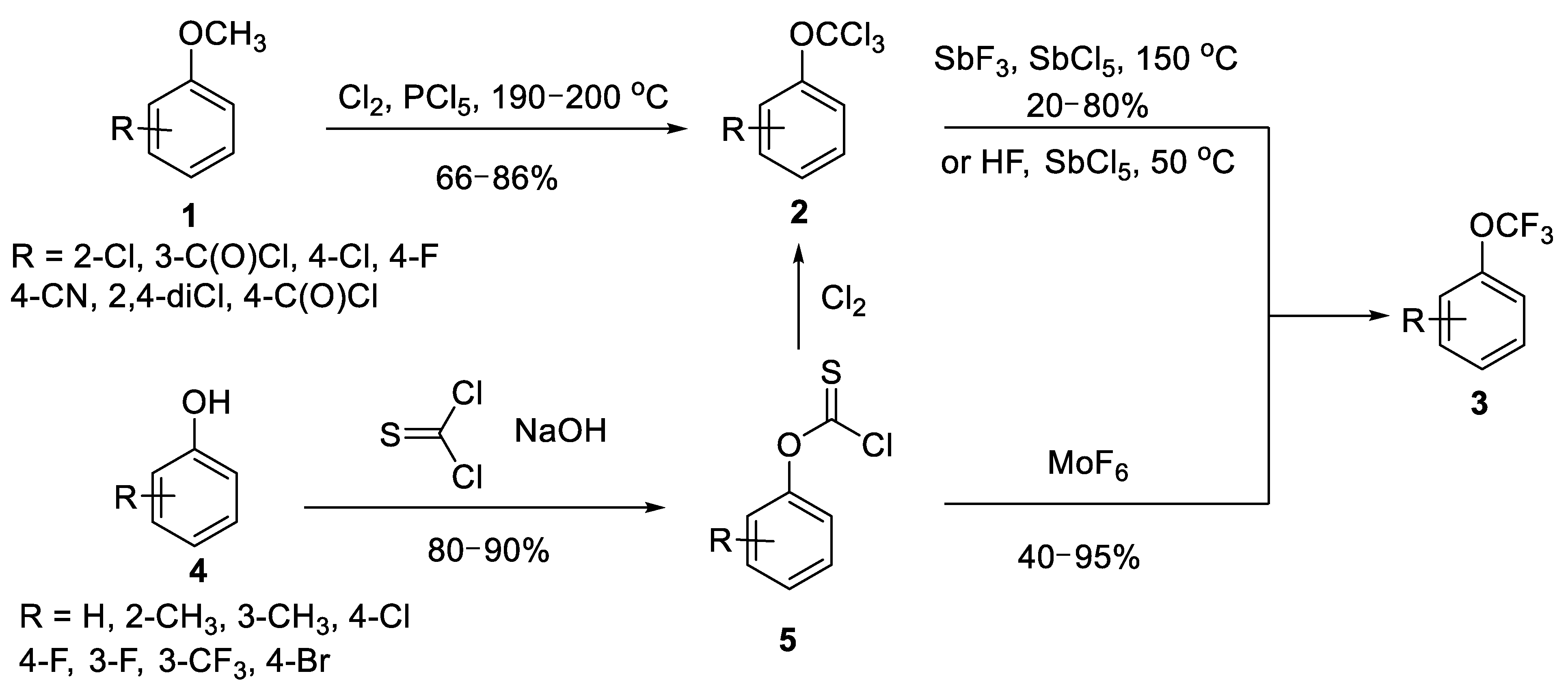{kind=link}
{kind=link}
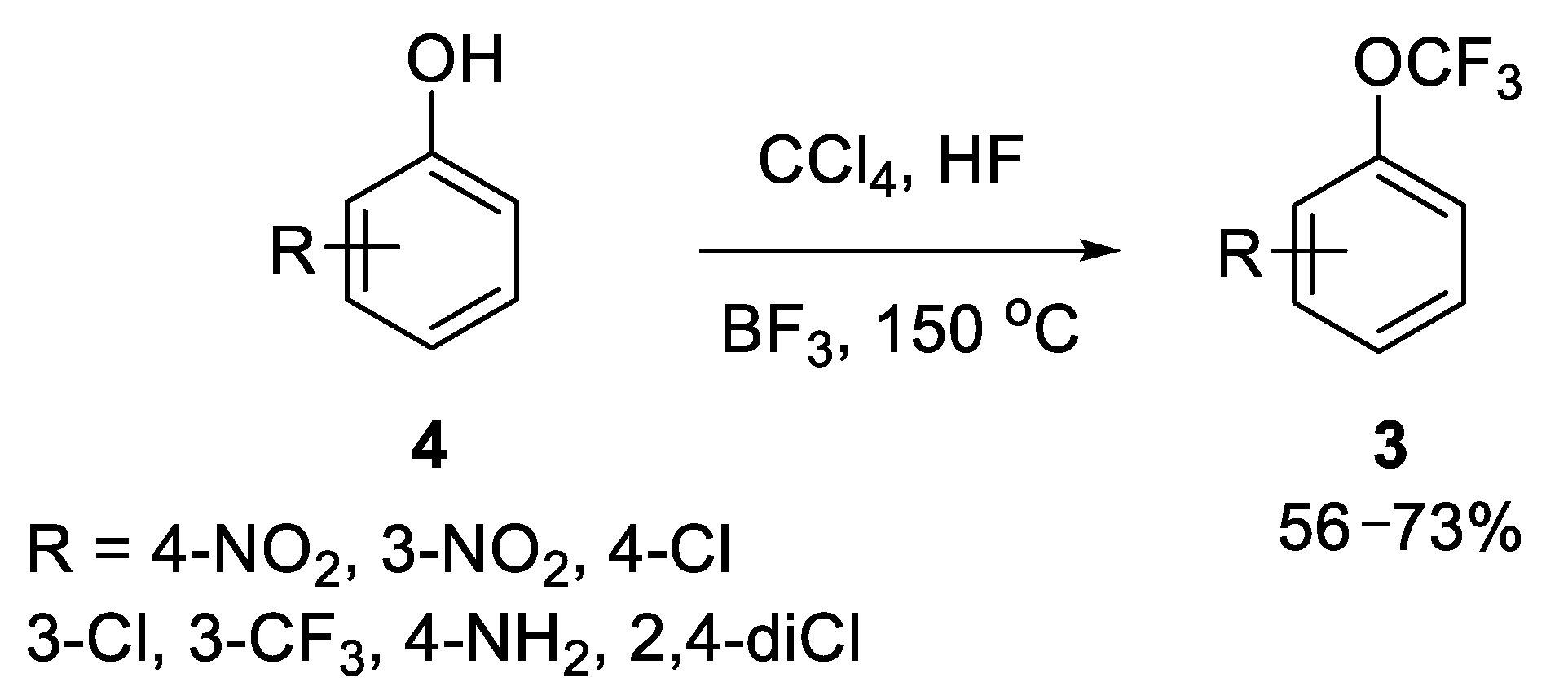{kind=link}
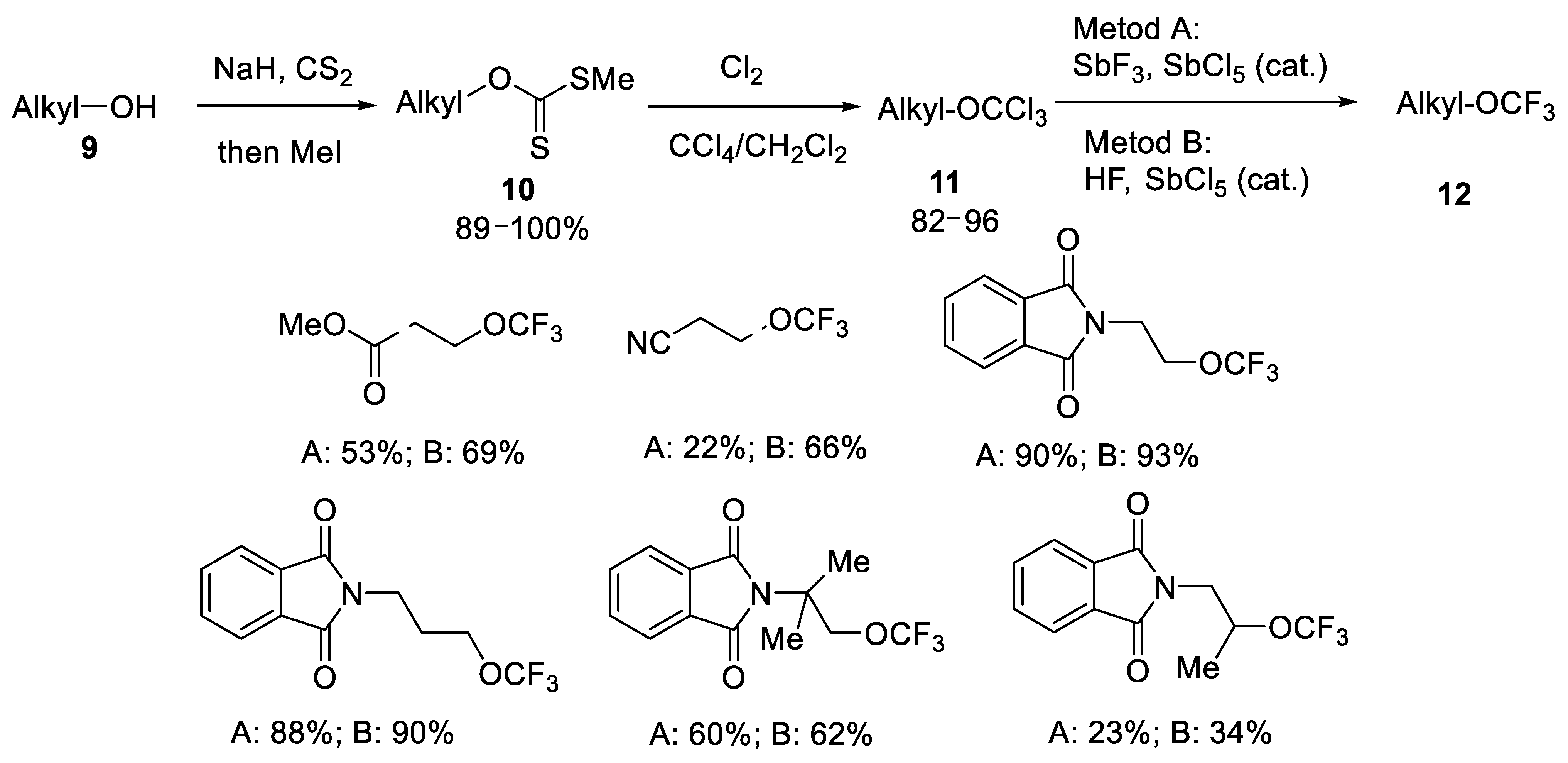{kind=link}
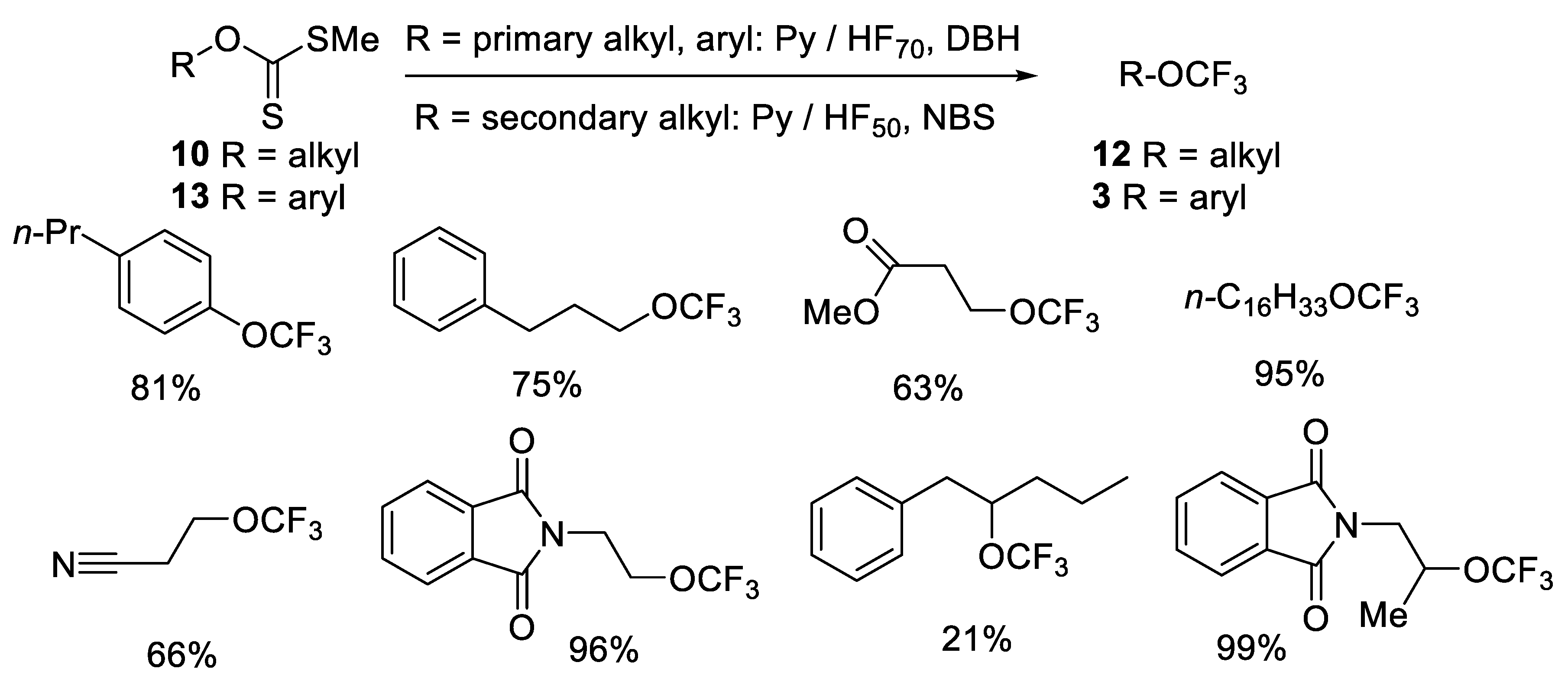{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
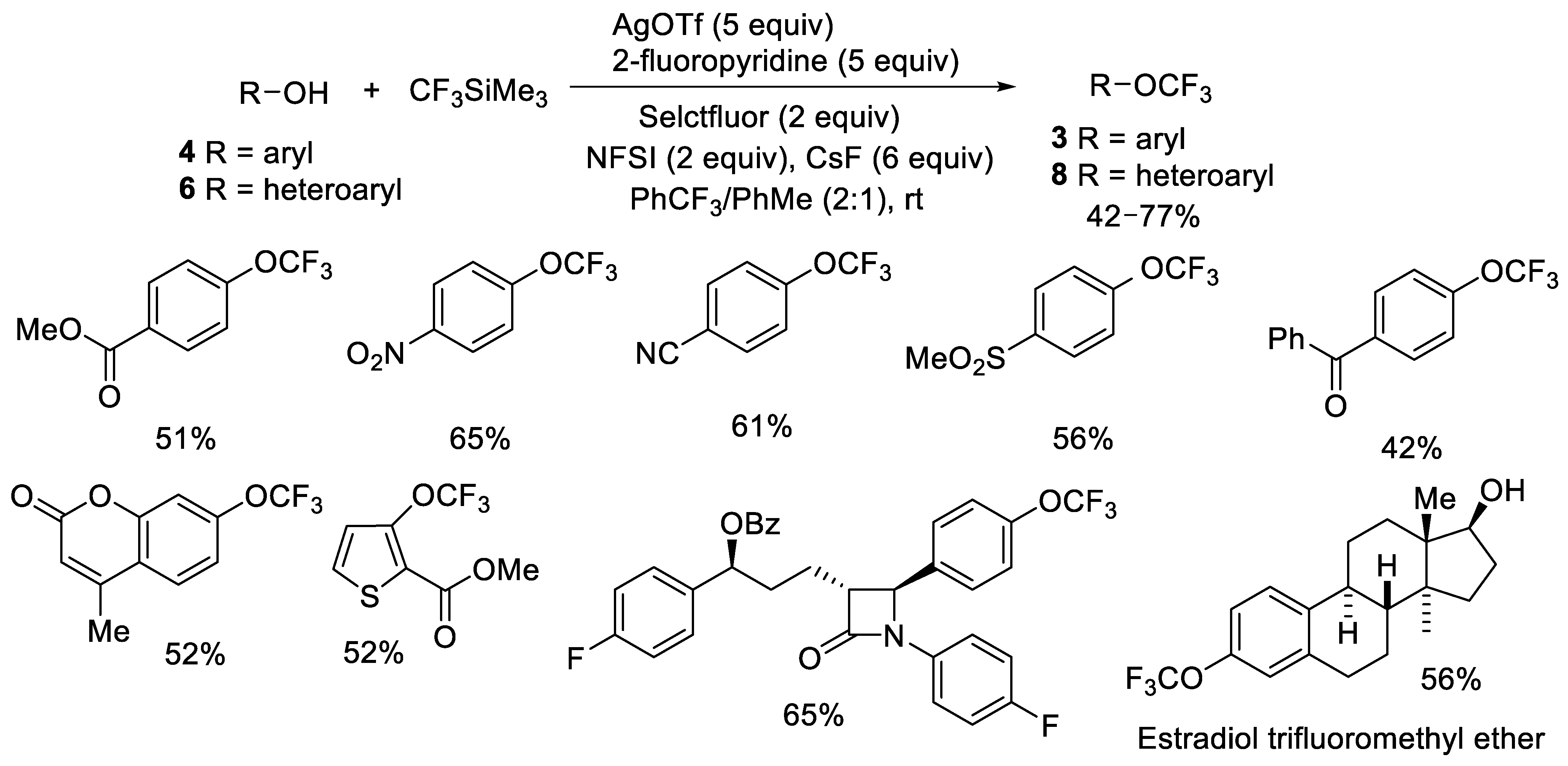{kind=link}
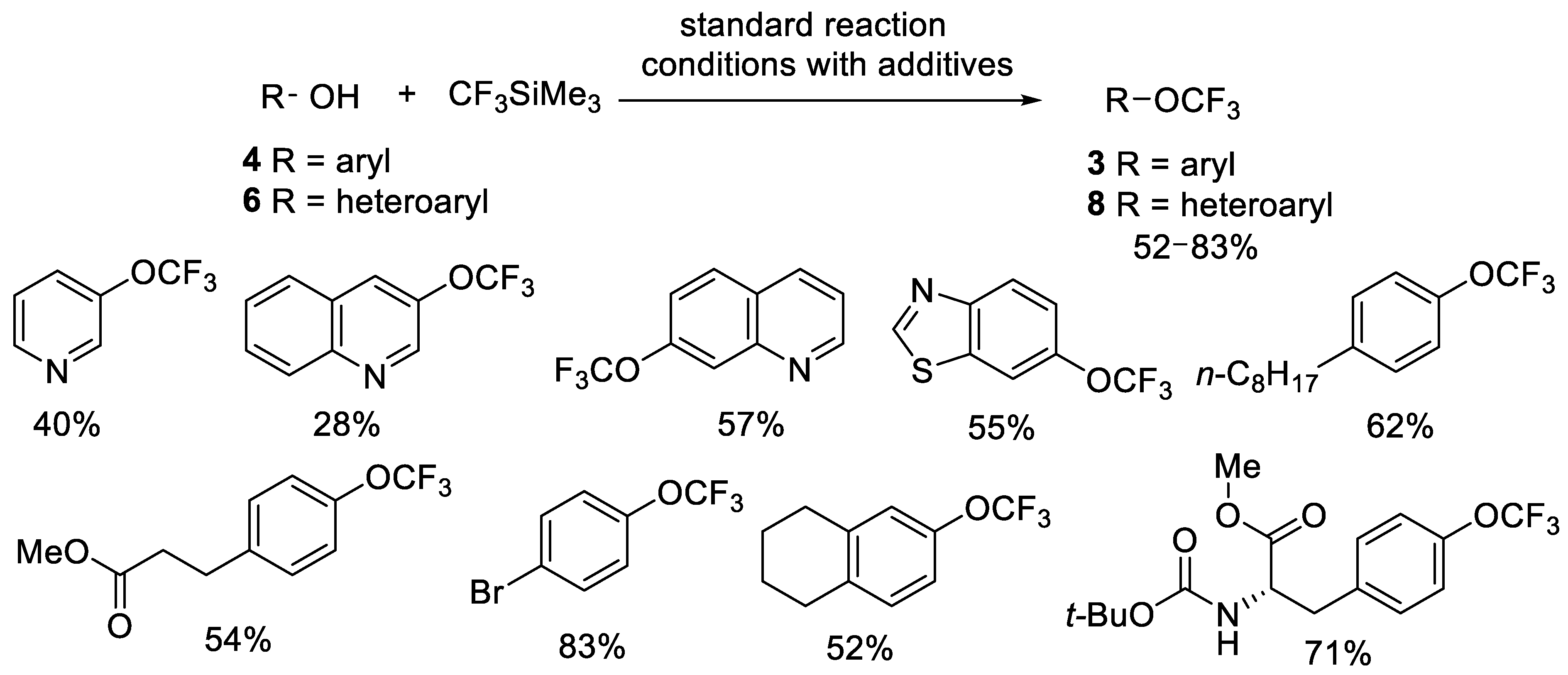{kind=link}
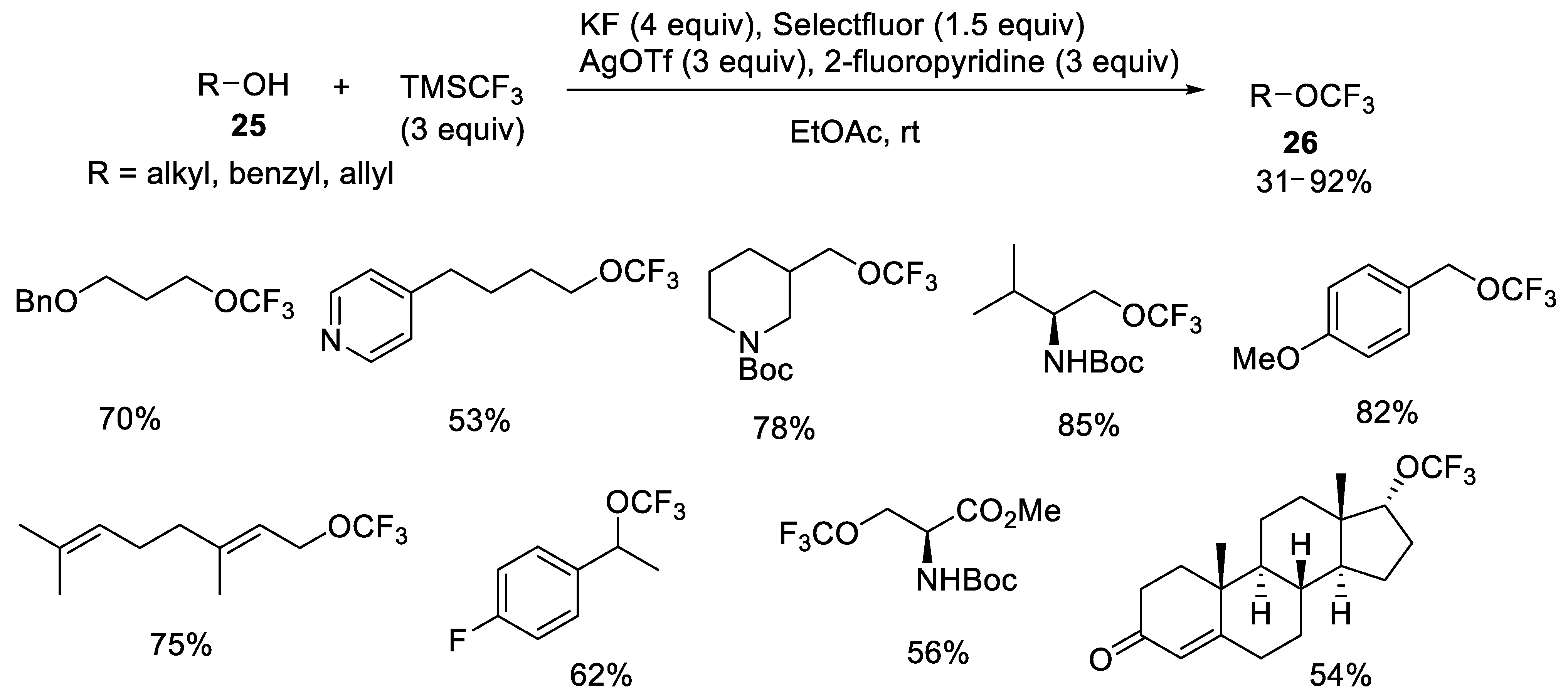{kind=link}
{kind=link}
{kind=link}
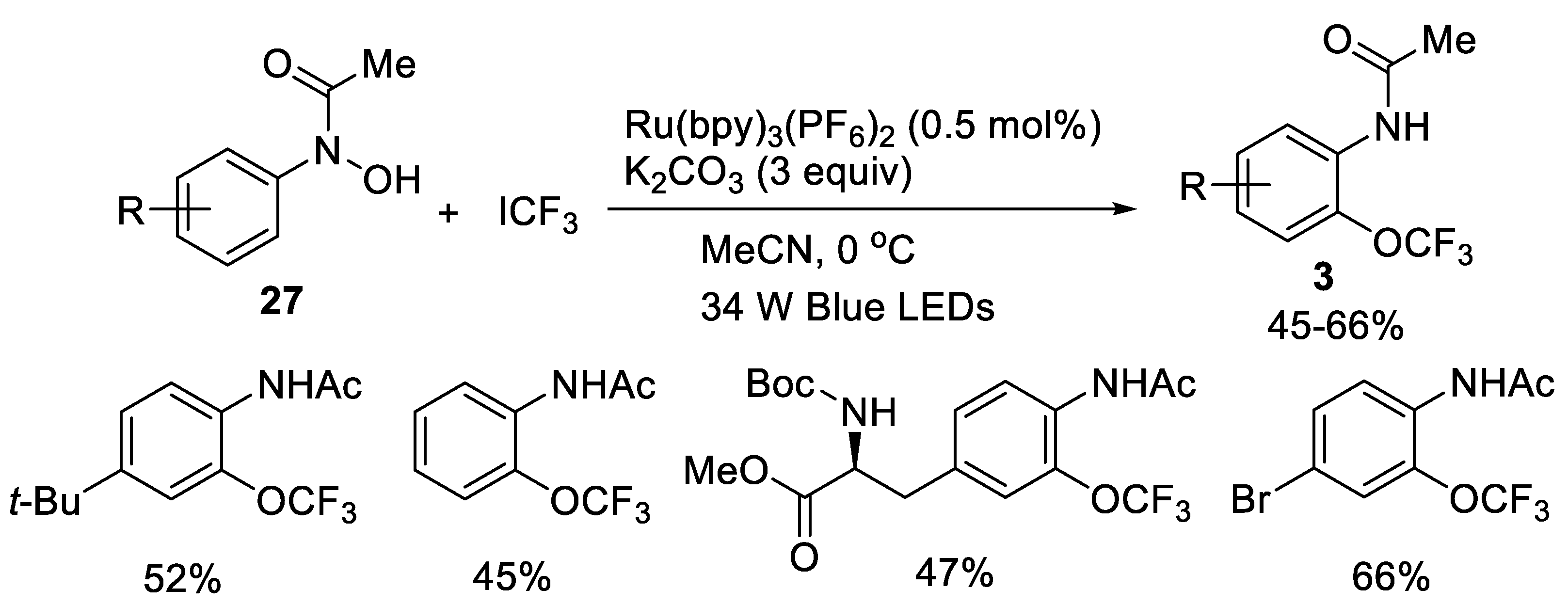{kind=link}
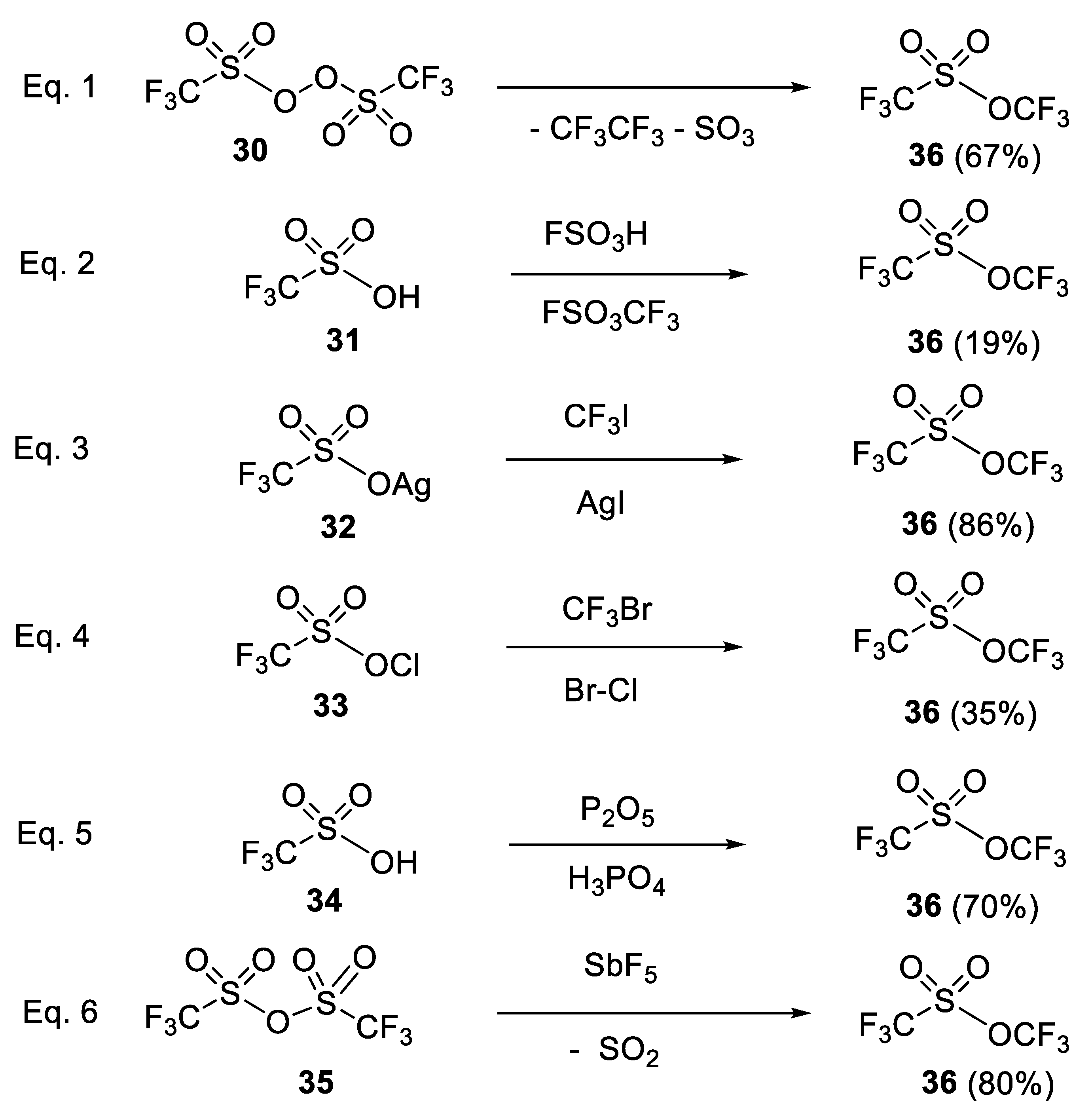{kind=link}
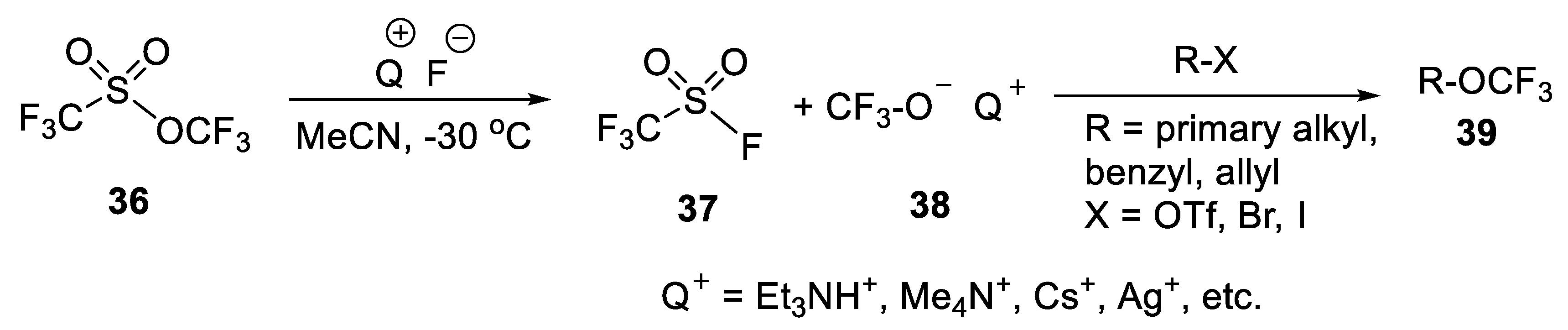{kind=link}
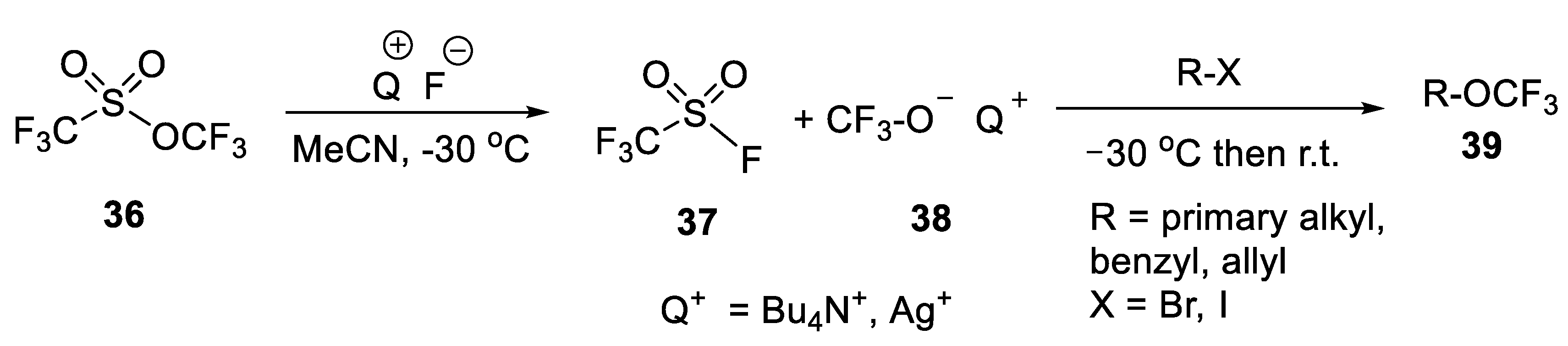{kind=link}
{kind=link}
{kind=link}
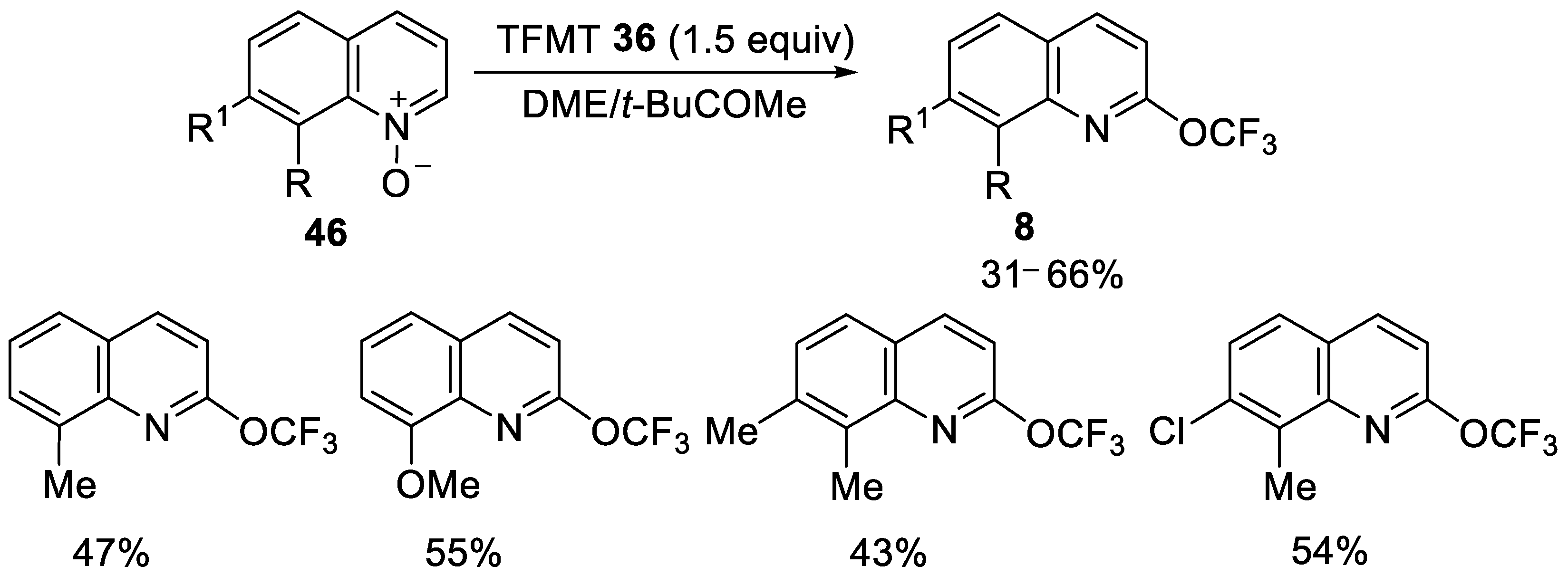{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
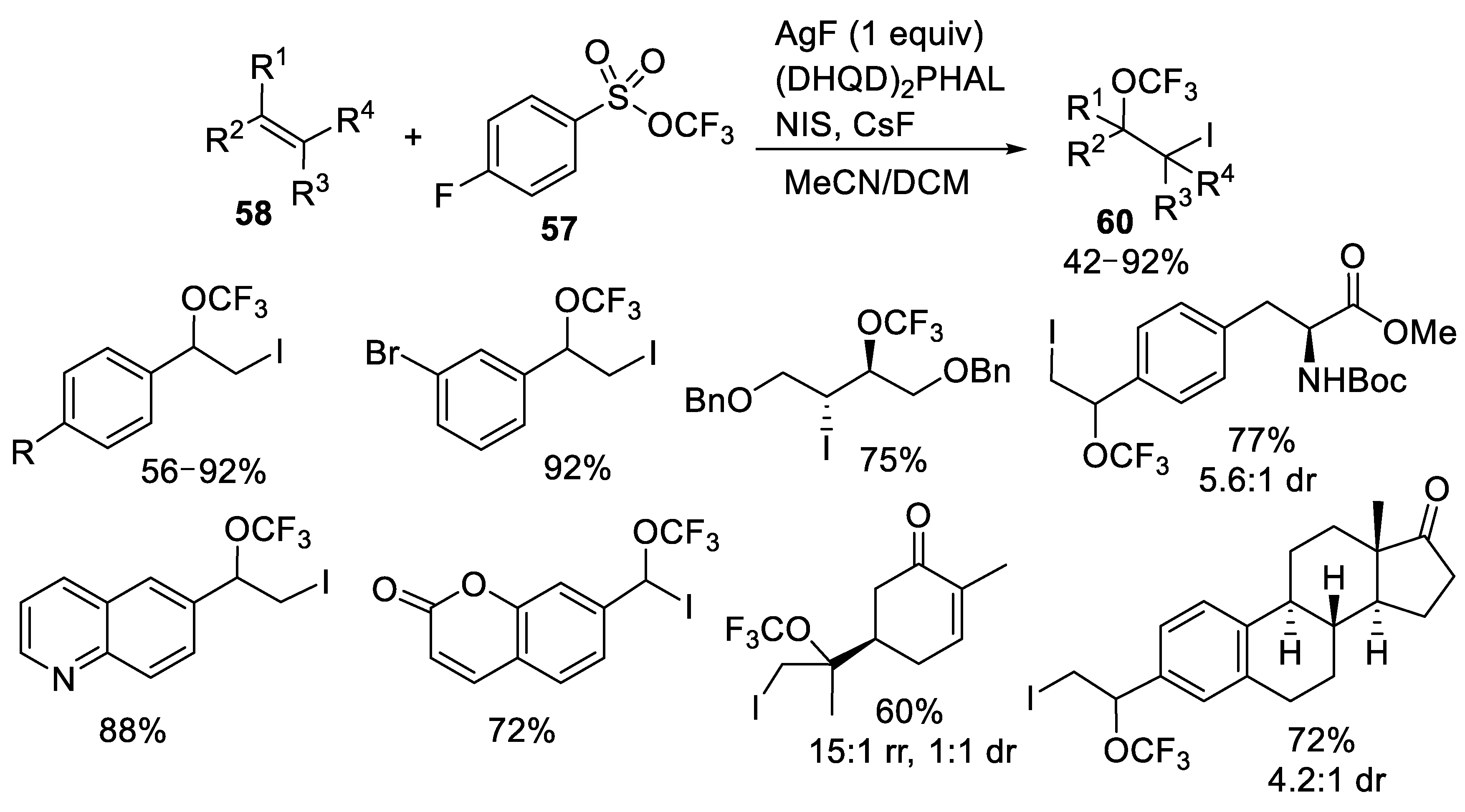{kind=link}
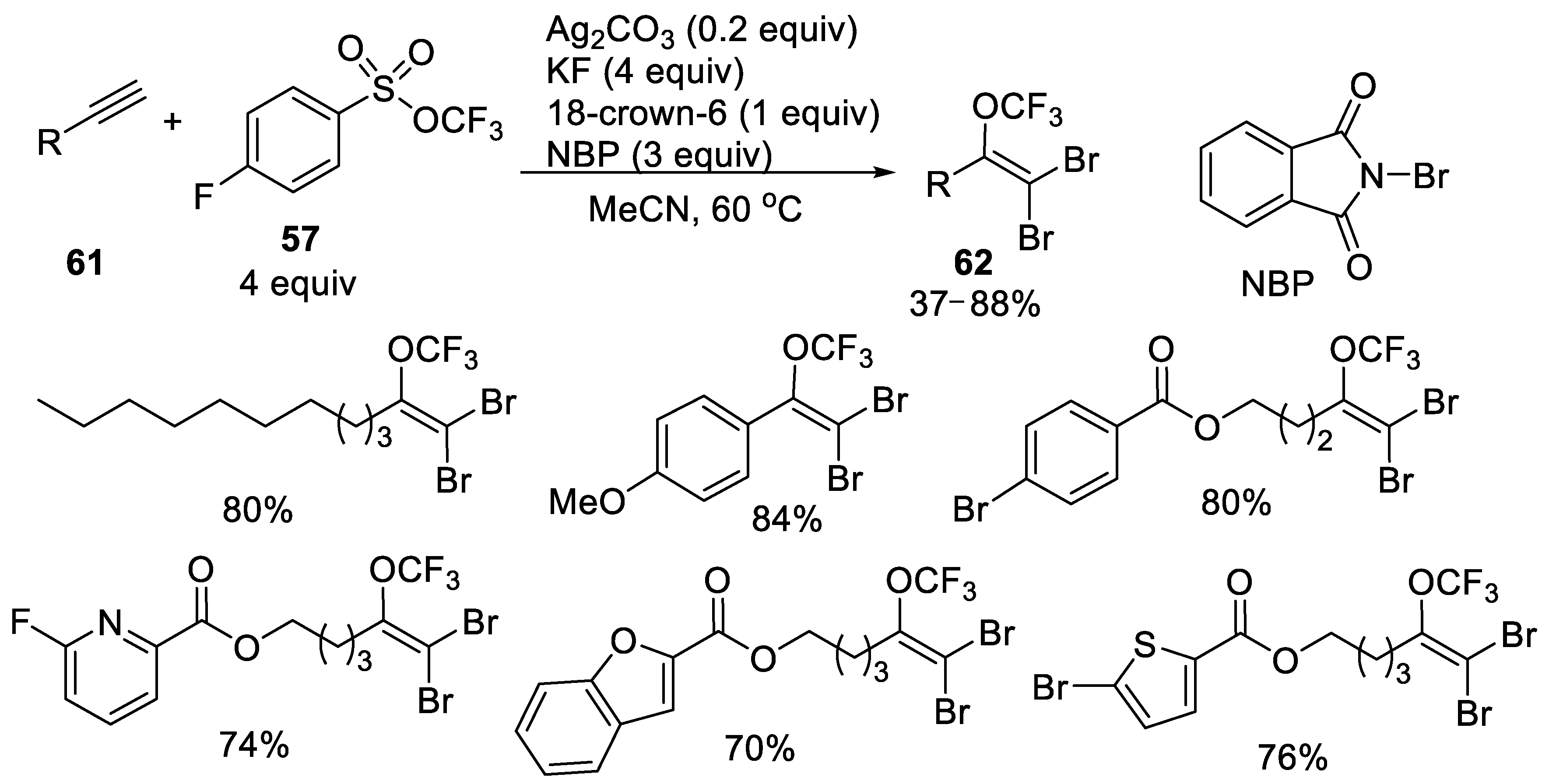{kind=link}
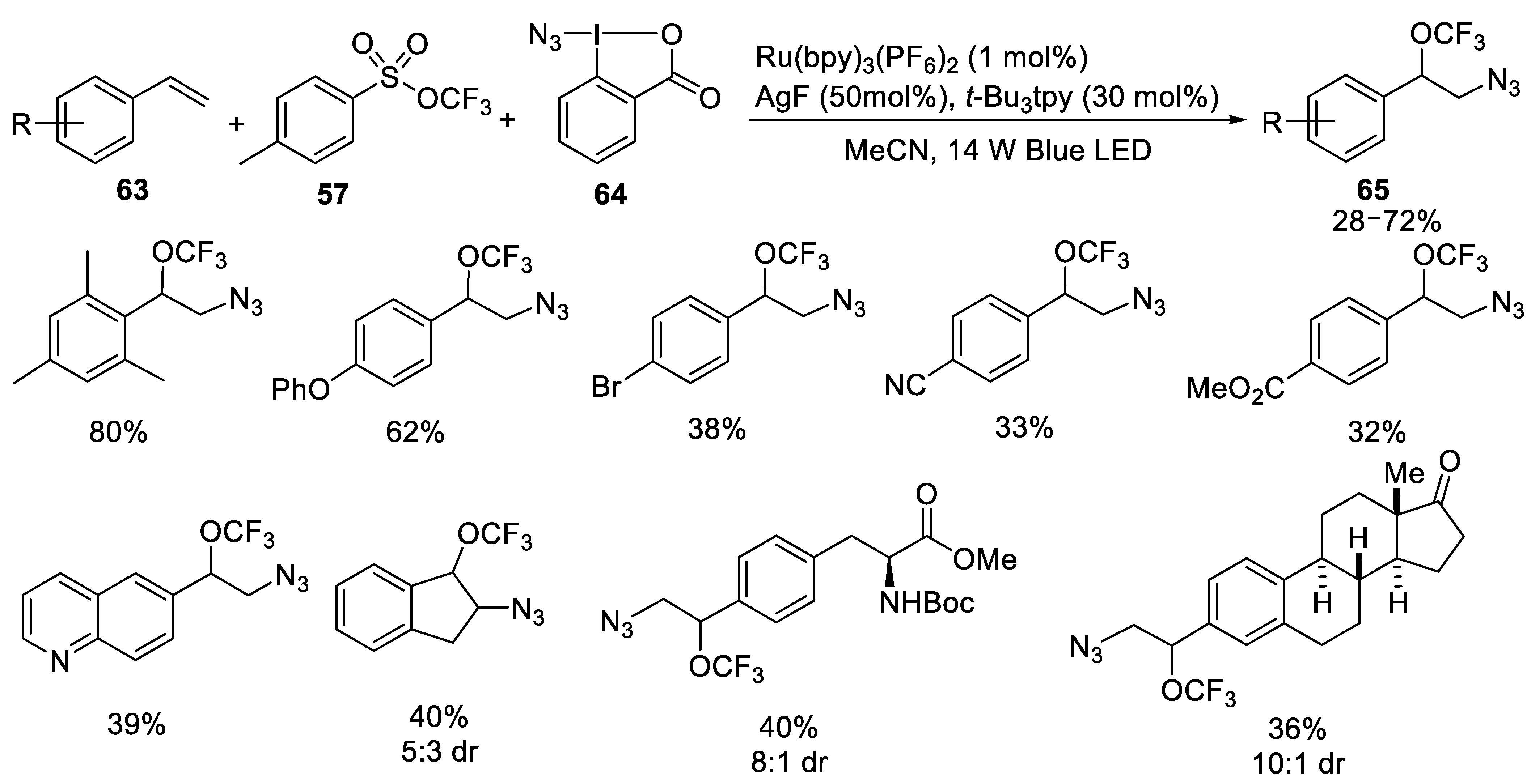{kind=link}
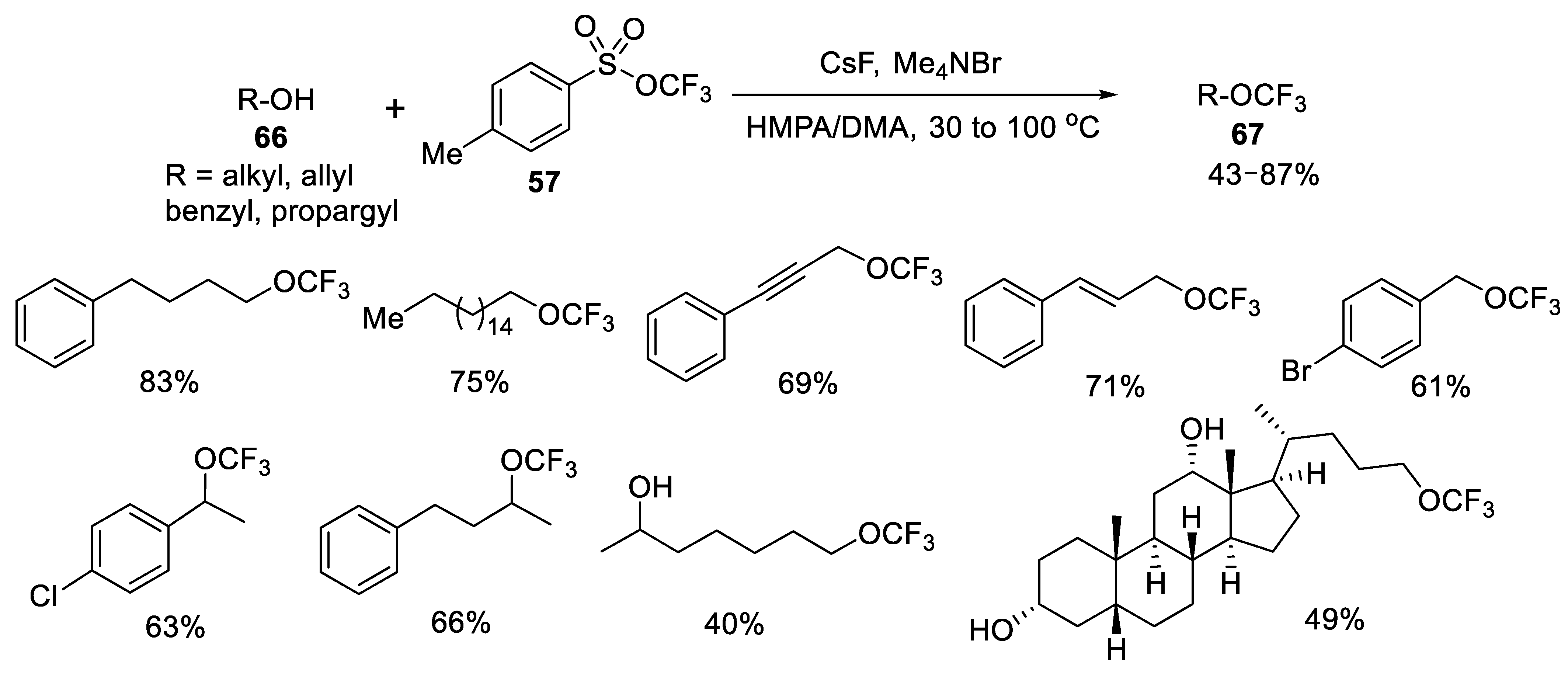{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Approaches for Preparation of Trifluoromethoxy-Containing Compounds
2.1. Nucleophilic Fluorination
2.1.1. Chlorine-Fluorine Exchange
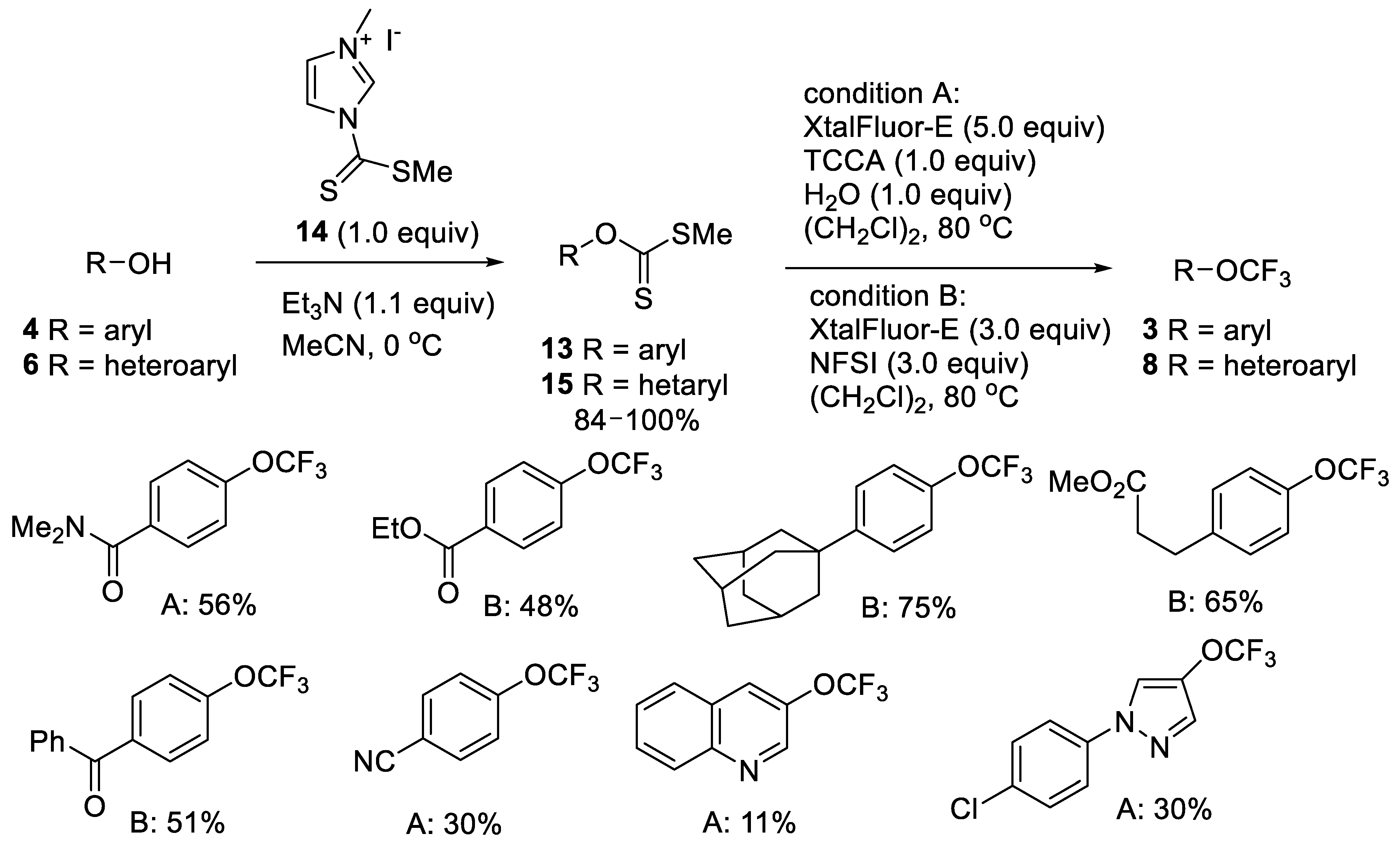
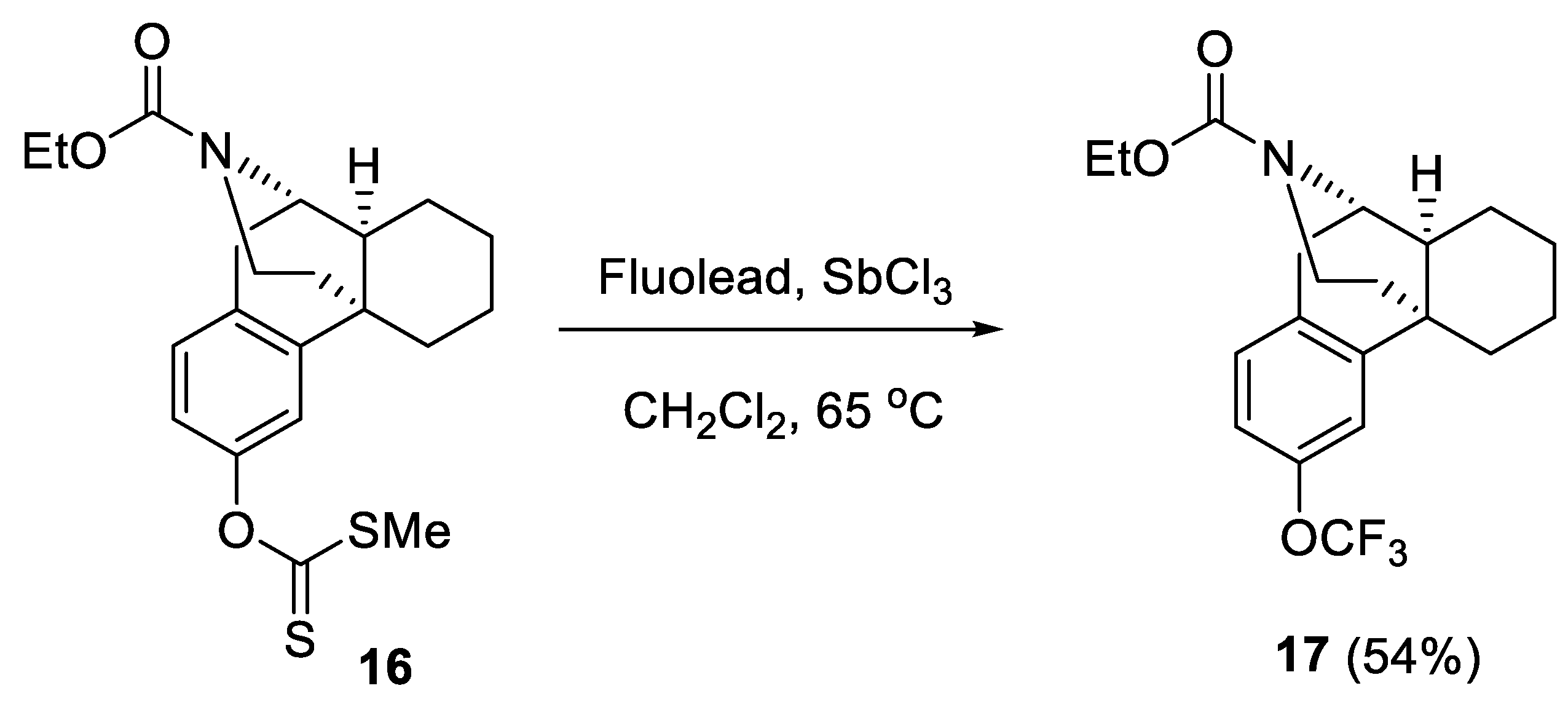
2.1.2. Oxidative Desulfurization-Fluorination
2.1.3. Deoxofluorination of Fluoroformates
2.2. Fluorodecarboxylation of Aryloxydifluoroacetic Acids
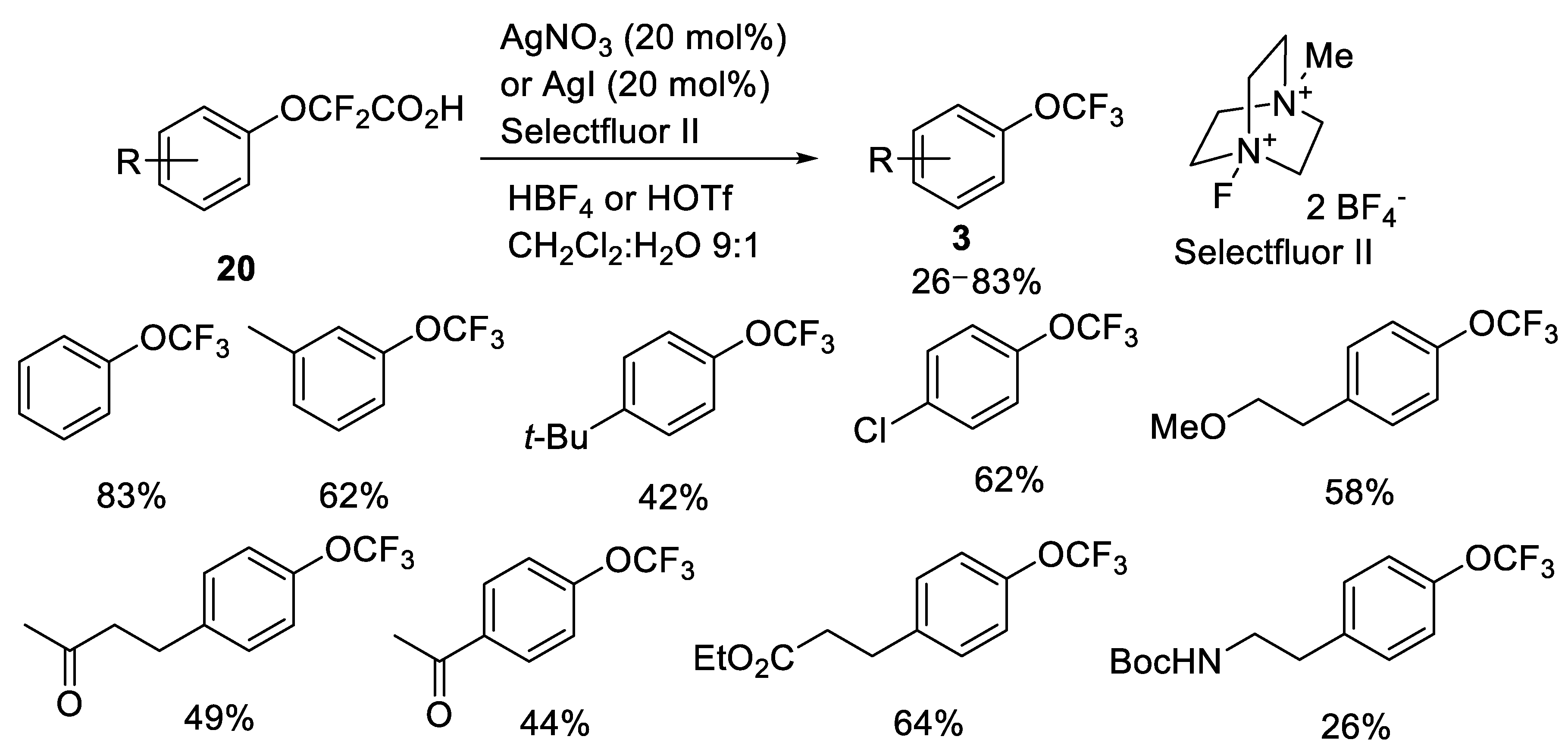
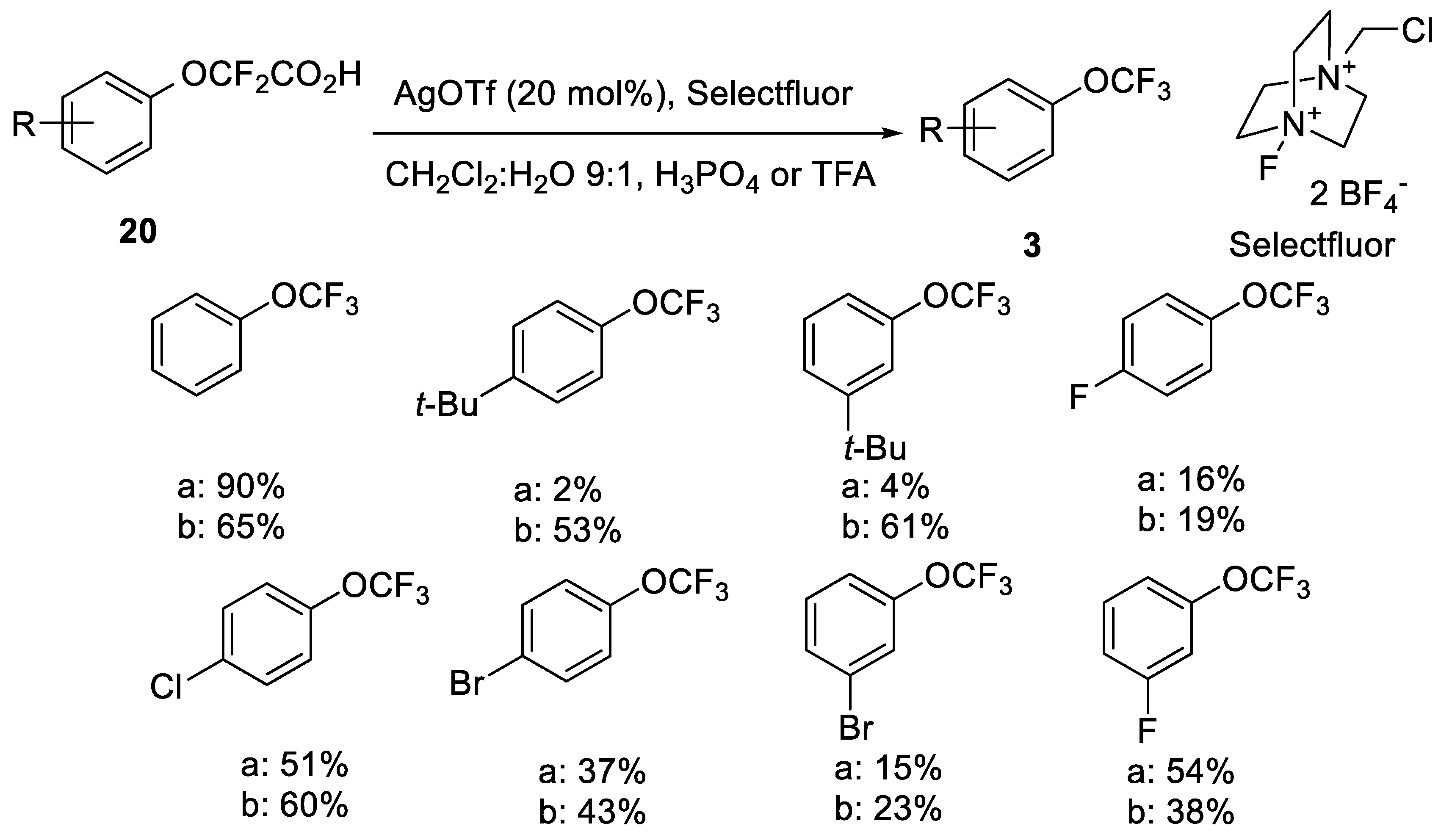
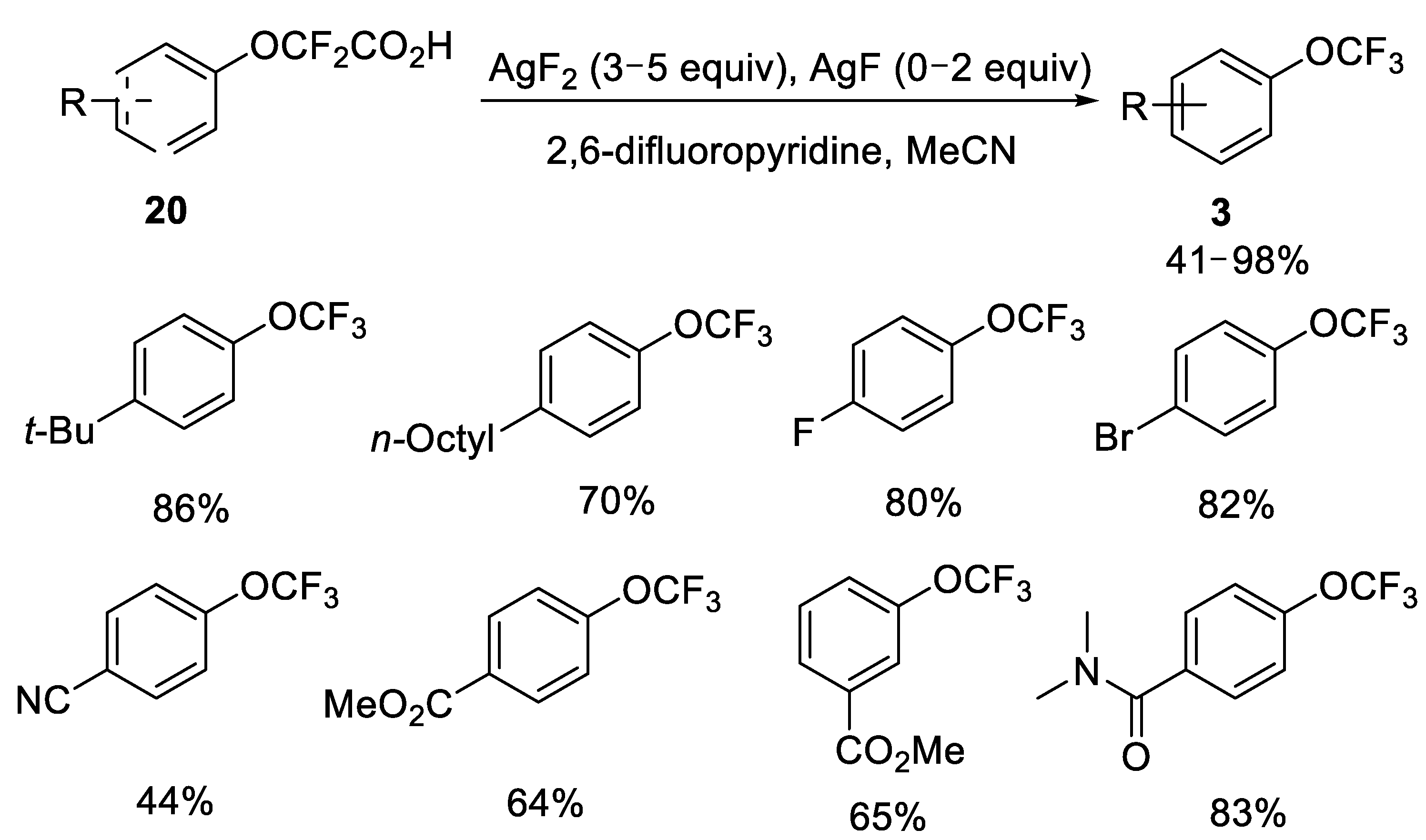
2.2.1. Silver-Catalyzed Fluorodecarboxylation
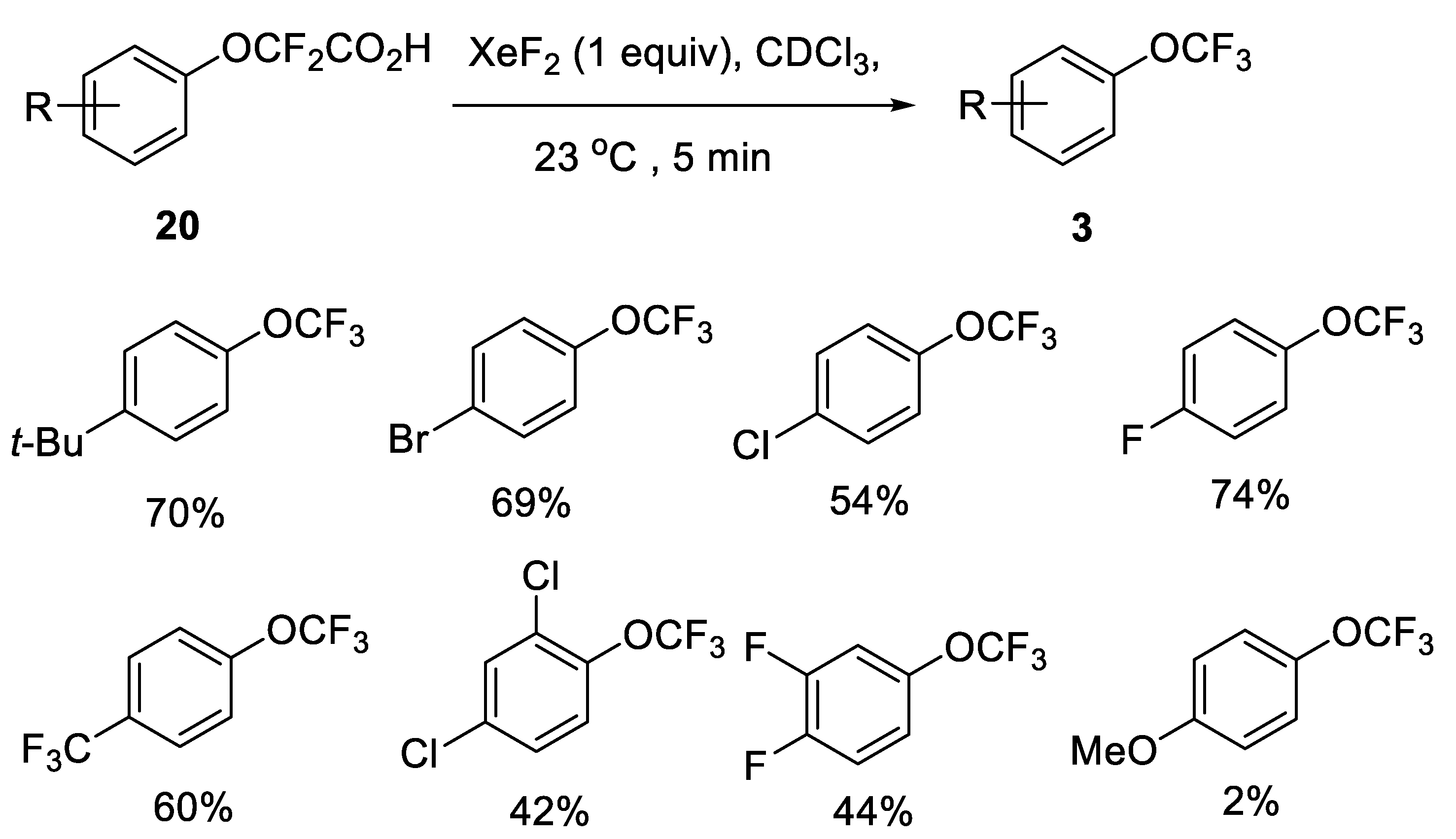
2.2.2. Fluorodecarboxylation with Silver(II) Fluoride
2.2.3. Fluorodecarboxylation with Xenon Difluoride
2.3. O-Trifluoromethylation
2.3.1. Electrophilic O-Trifluoromethylation
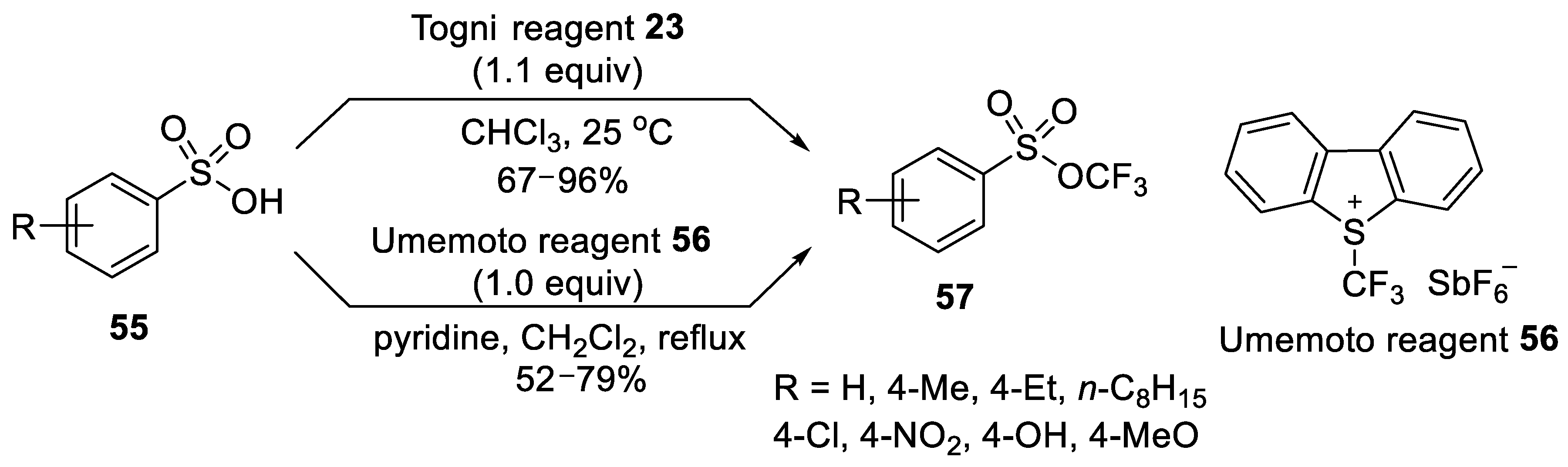
Umemoto Oxonium Reagent
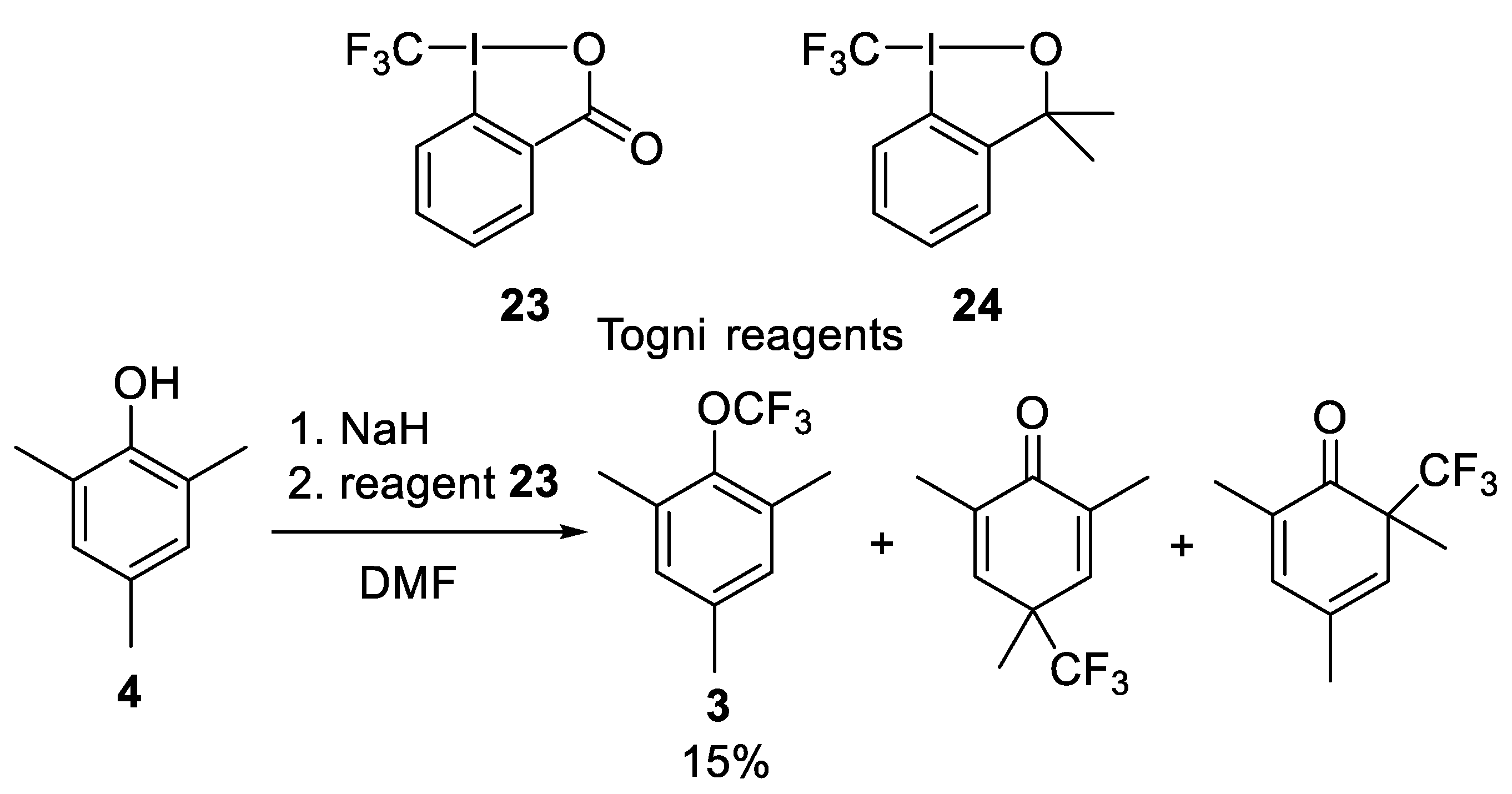
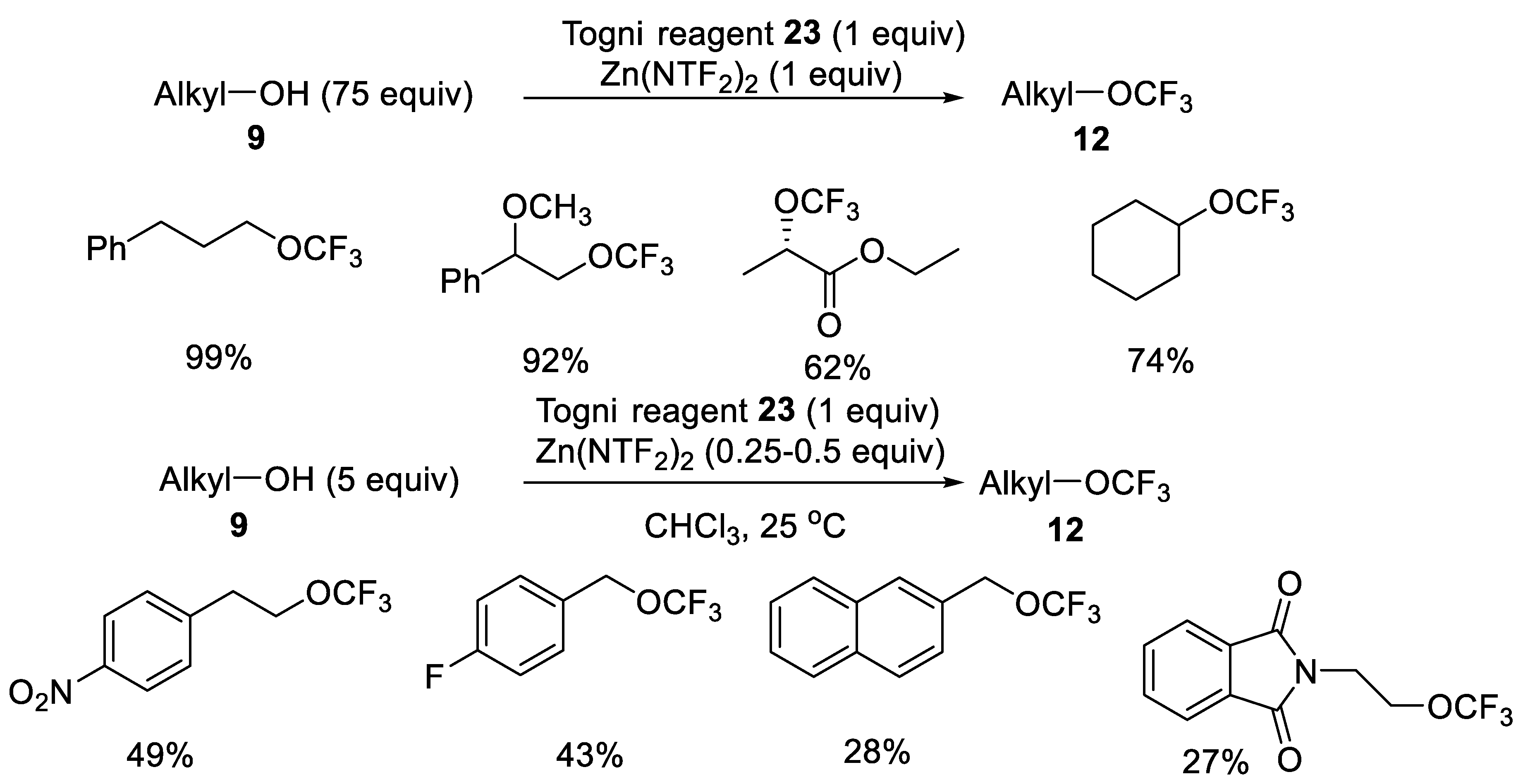
Togni Hypervalent Iodine Reagents
2.3.2. Oxidative O-Trifluoromethylation
2.3.3. Radical O-Trifluoromethylation
3. Trifluoromethoxylation Reagents
3.1. Nucleophilic Reagents
3.1.1. Trifluoromethyl Trifluoromethanesulfonate (TFMT)
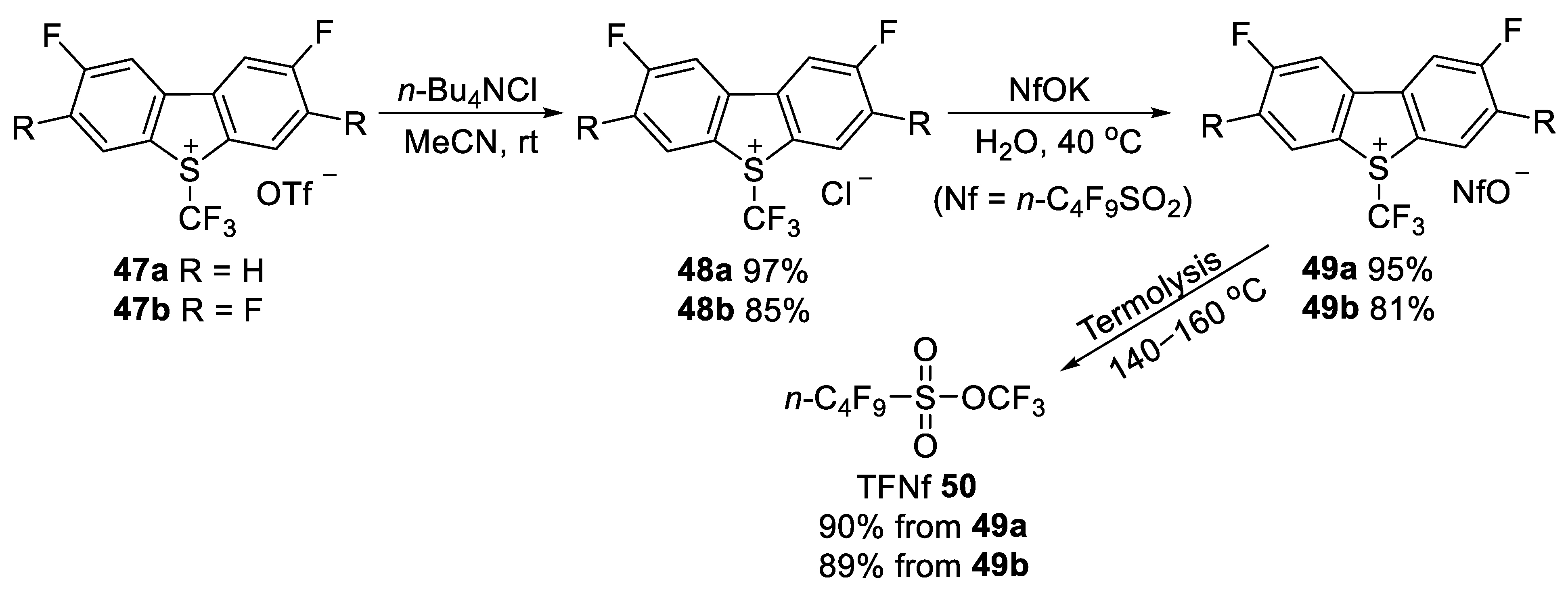
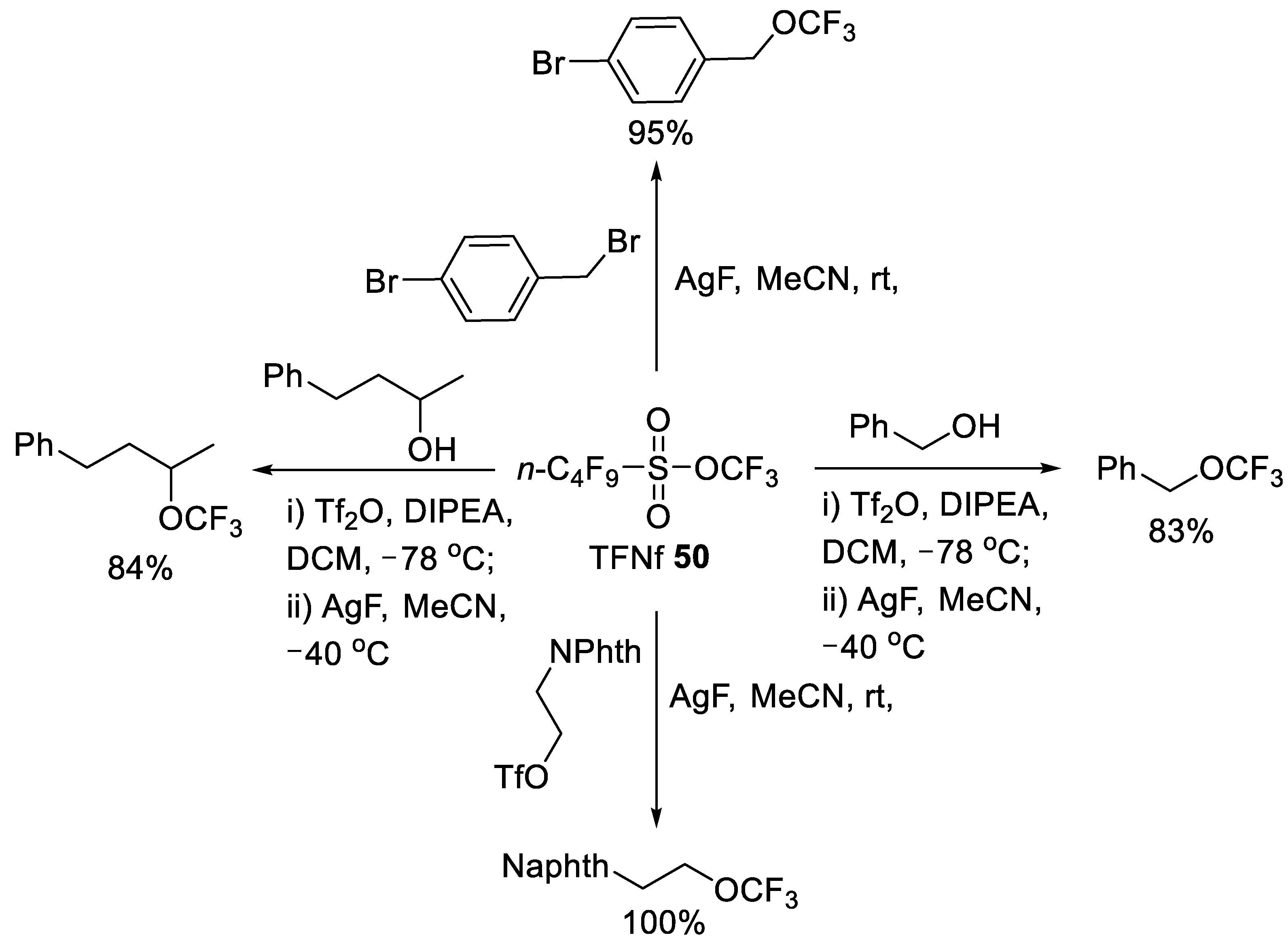
3.1.2. Trifluoromethyl Nonafluorobutanesulfonate (TFNf)
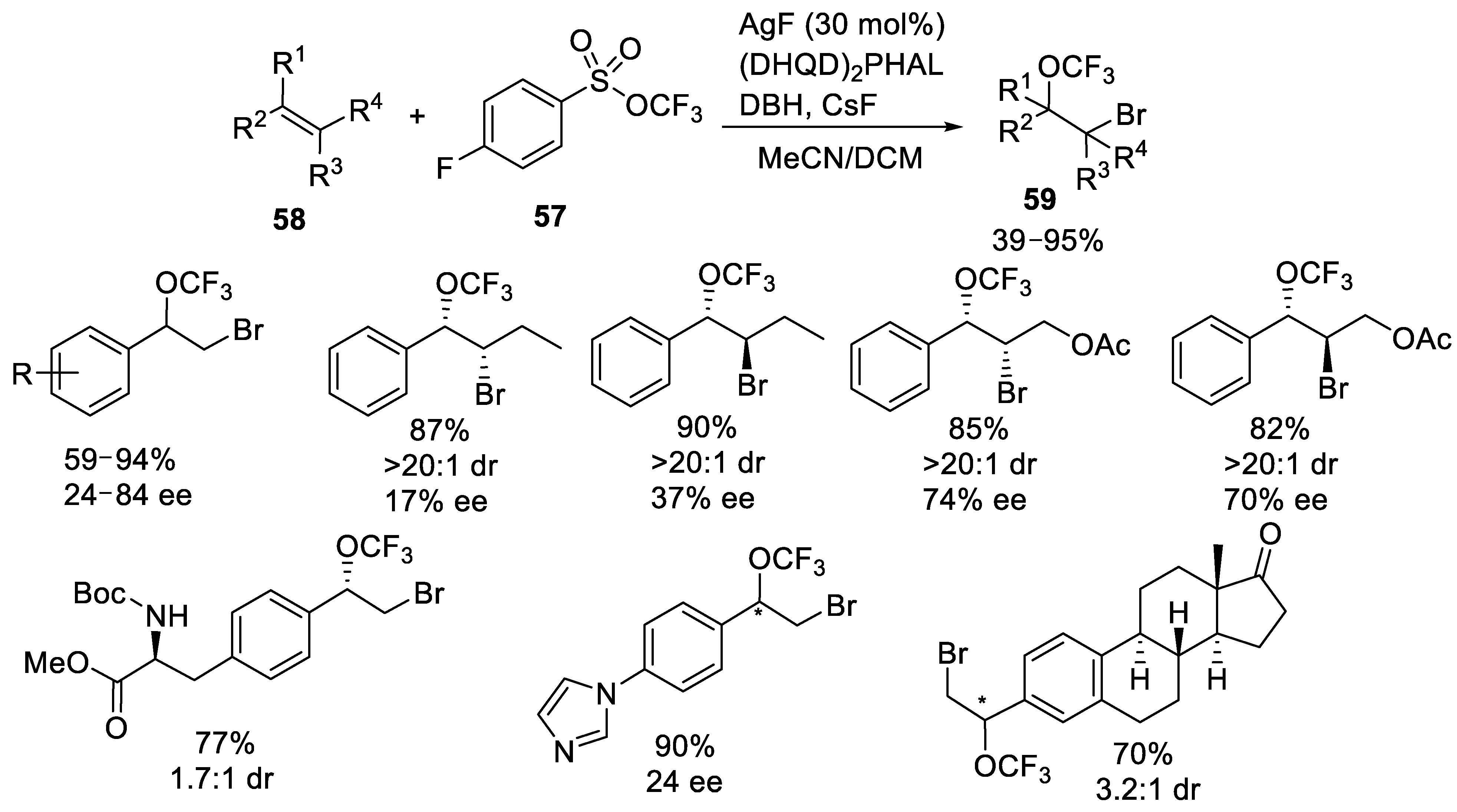
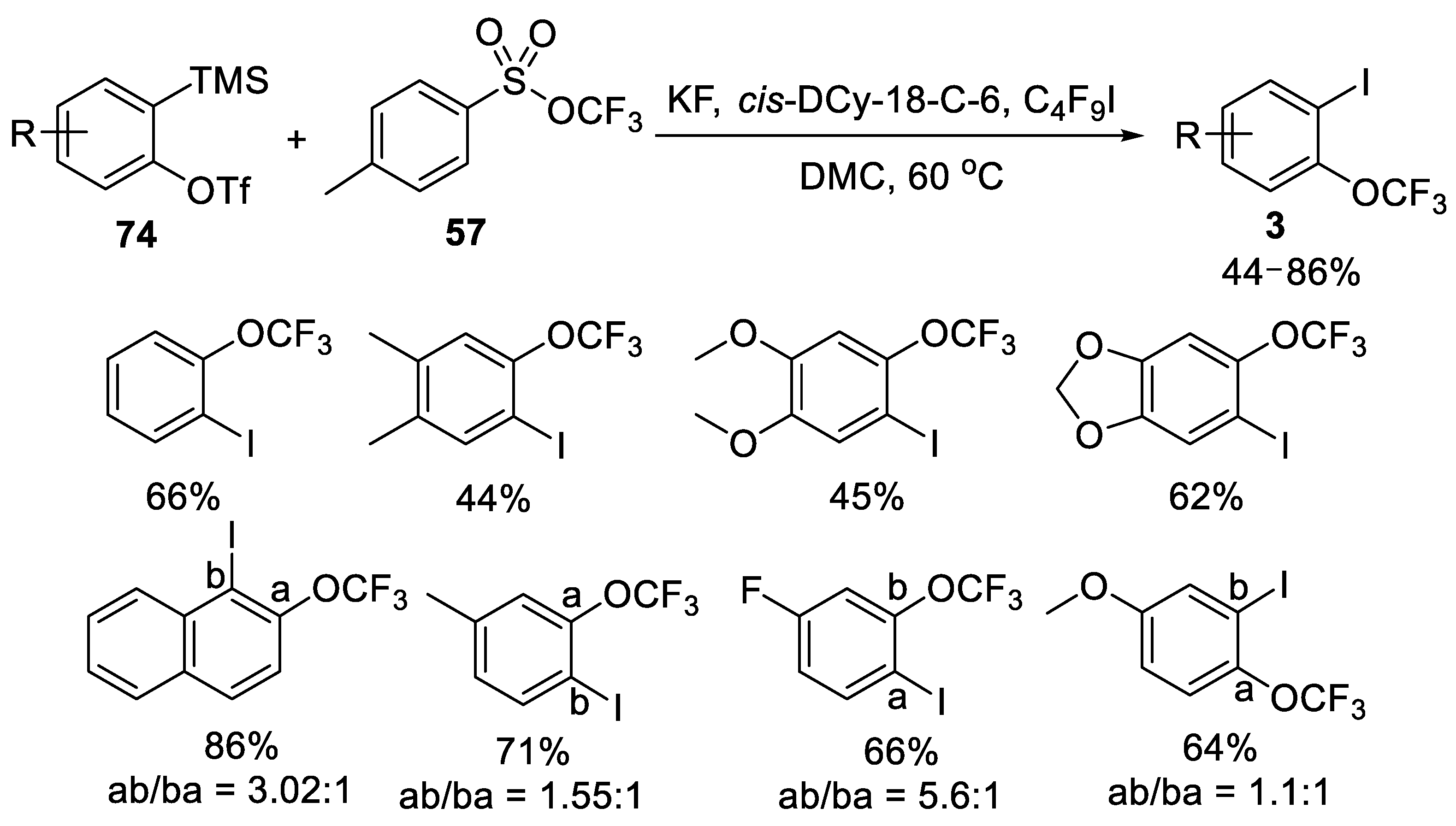
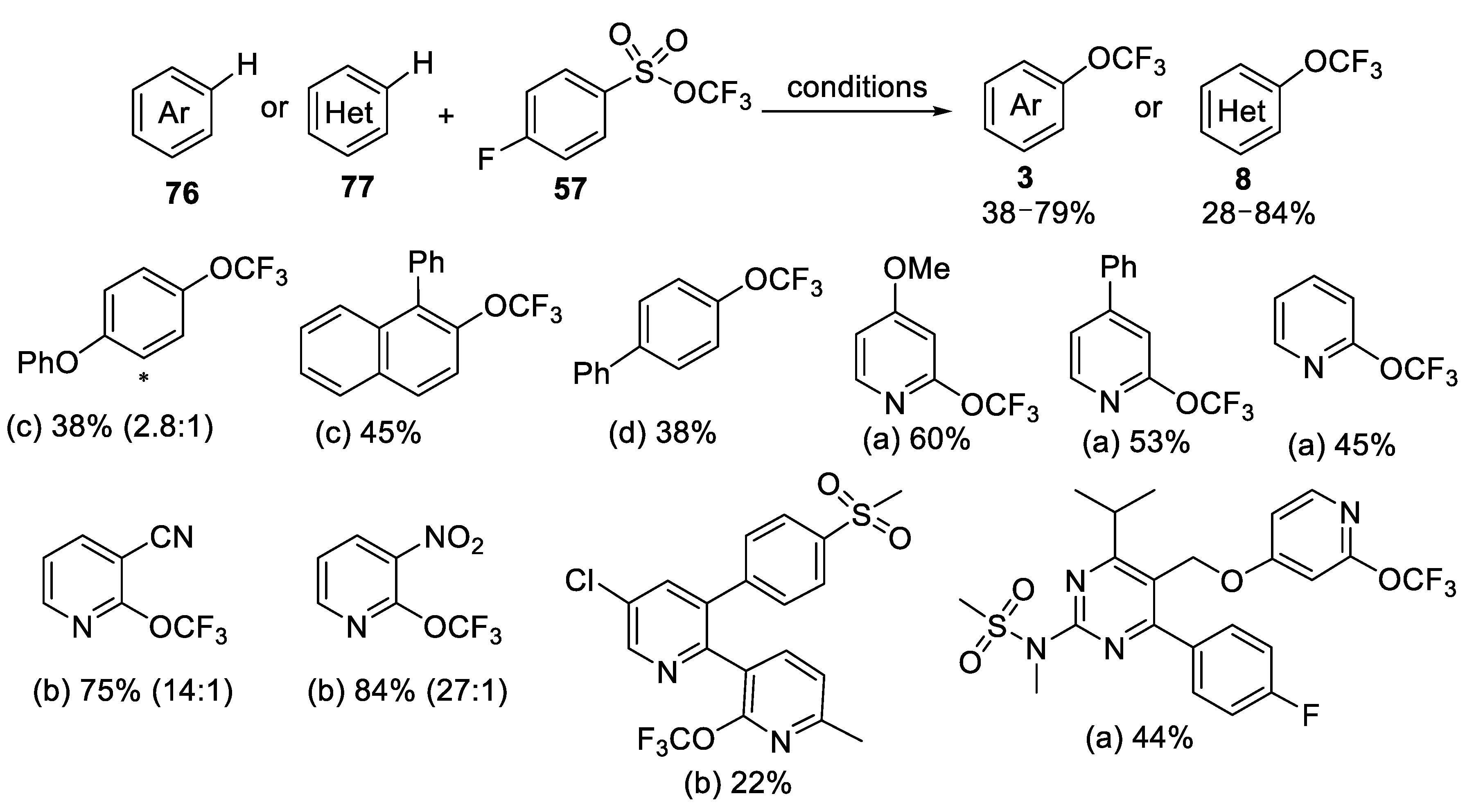
3.1.3. Trifluoromethyl Arylsulfonates (TFMS)
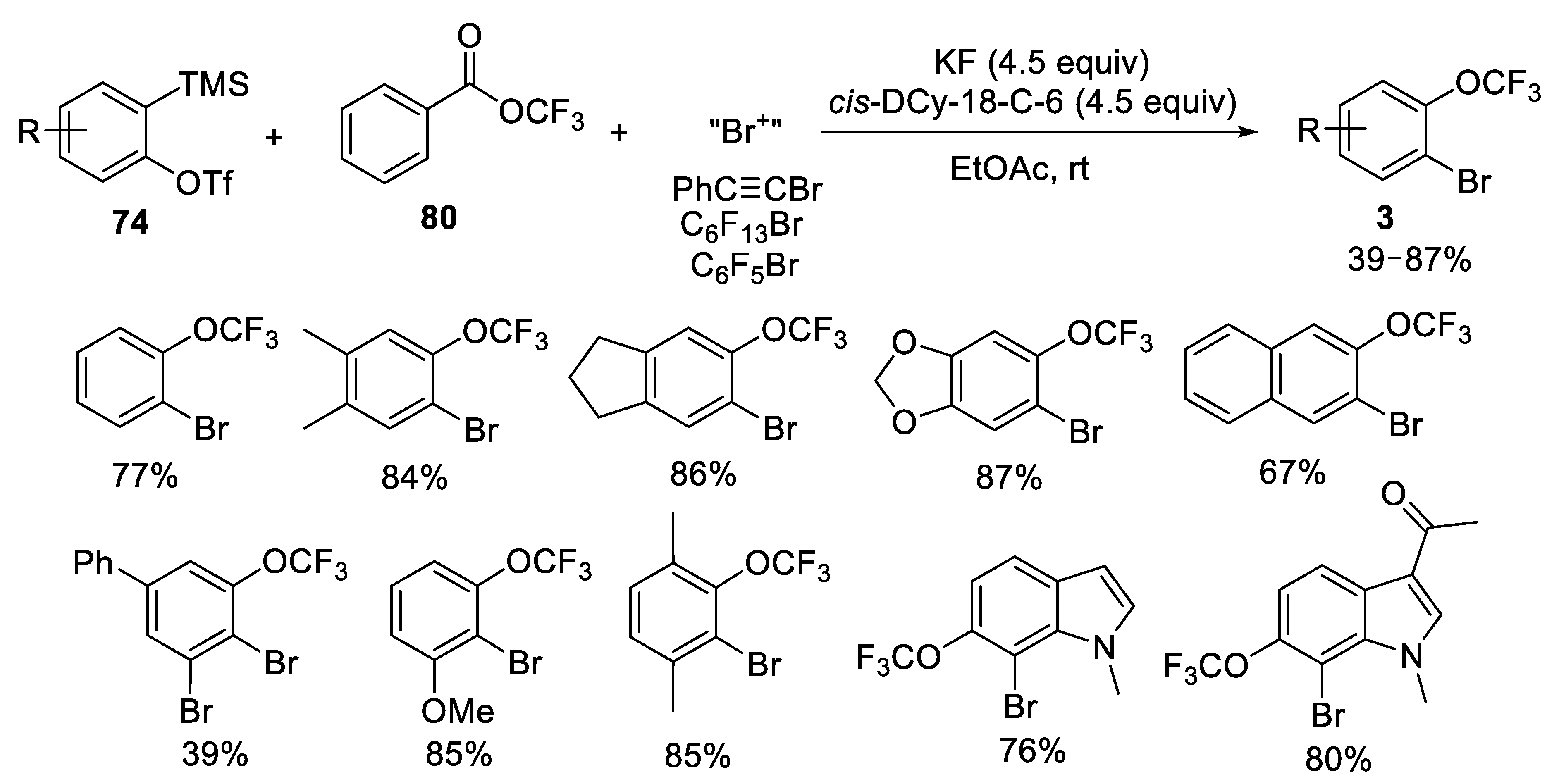
3.1.4. Trifluoromethyl Benzoate (TFBz)
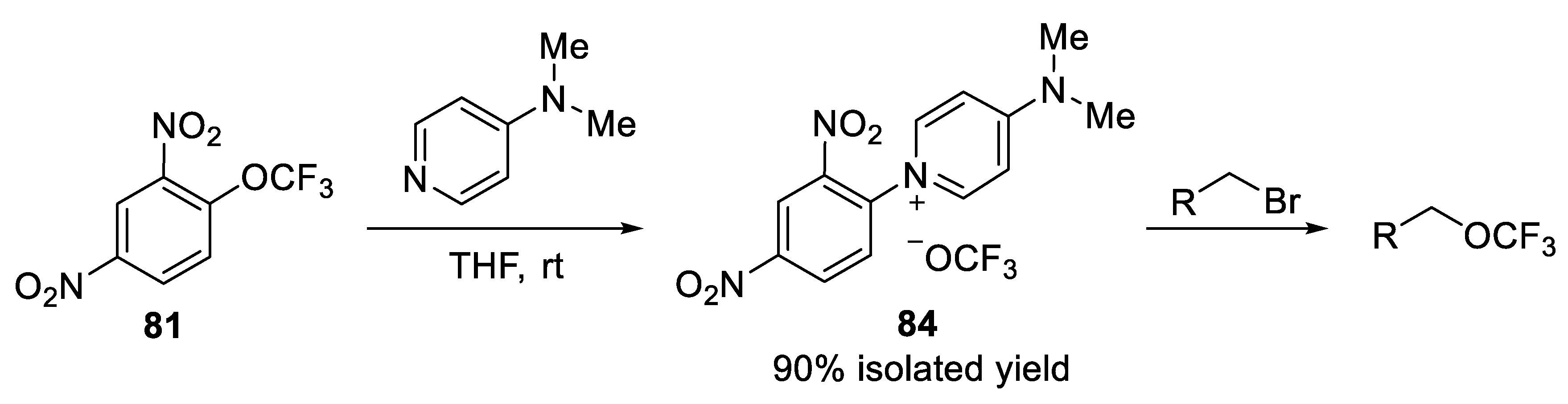
3.1.5. 2,4-Dinitro(Trifluoromethoxy)Benzene (DNTFB)
3.1.6. (E)-O-Trifluoromethylbenzaldoximes (TFBO)
3.2. Radical Reagents
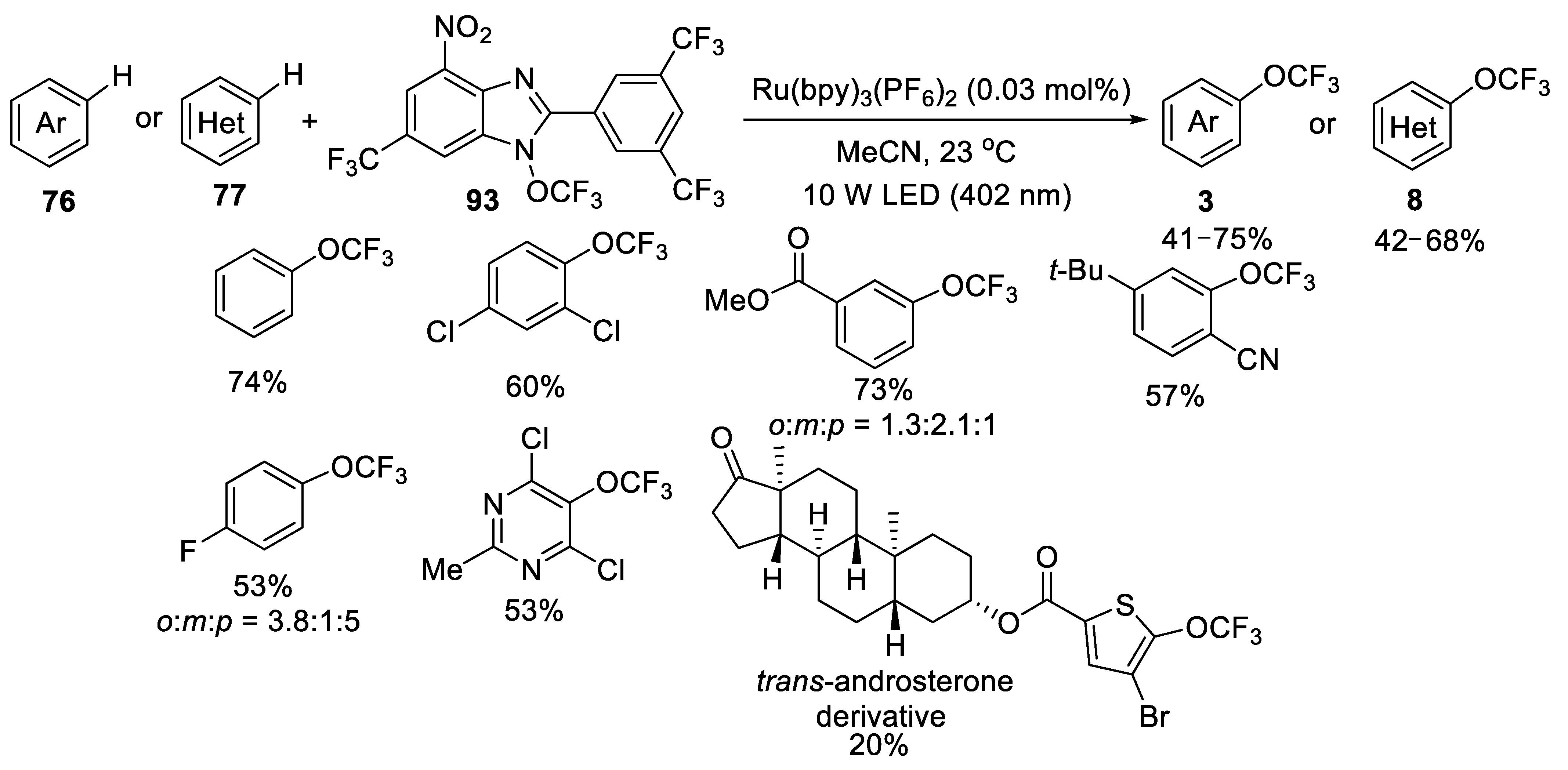
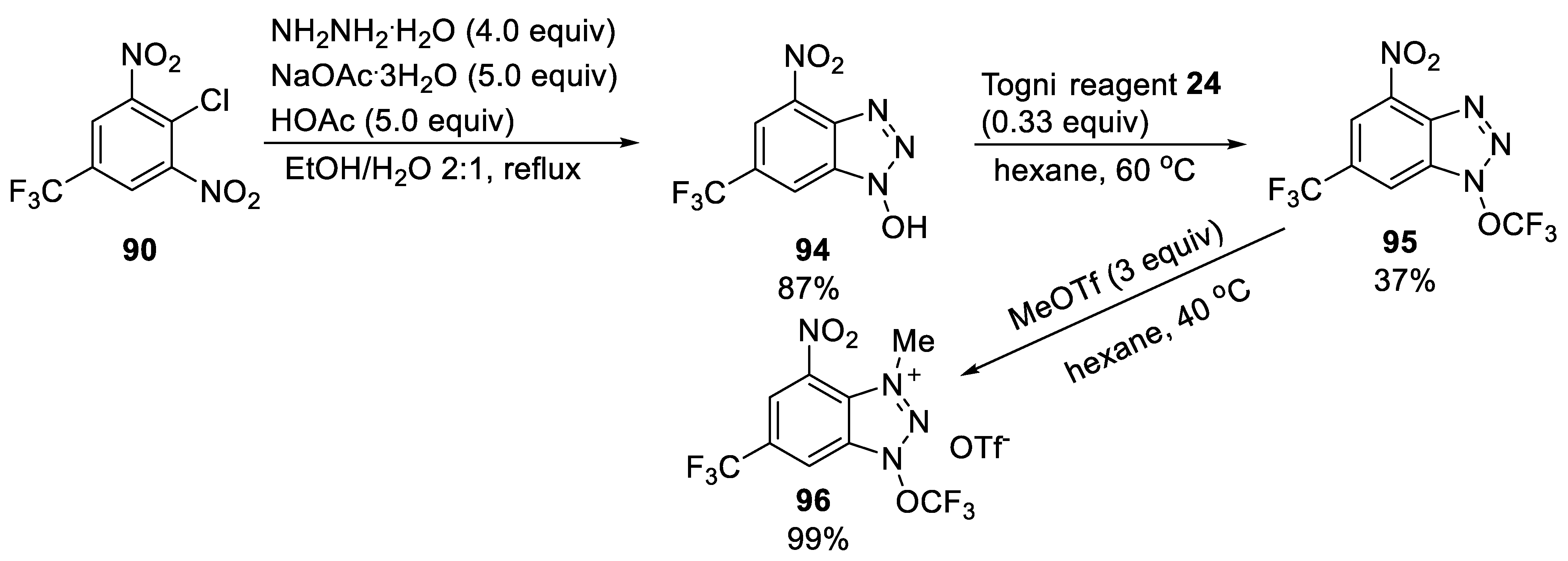
3.2.1. N-Trifluoromethoxybenzimidazole 93
3.2.2. N-Trifluoromethoxytriazolium Salt 96
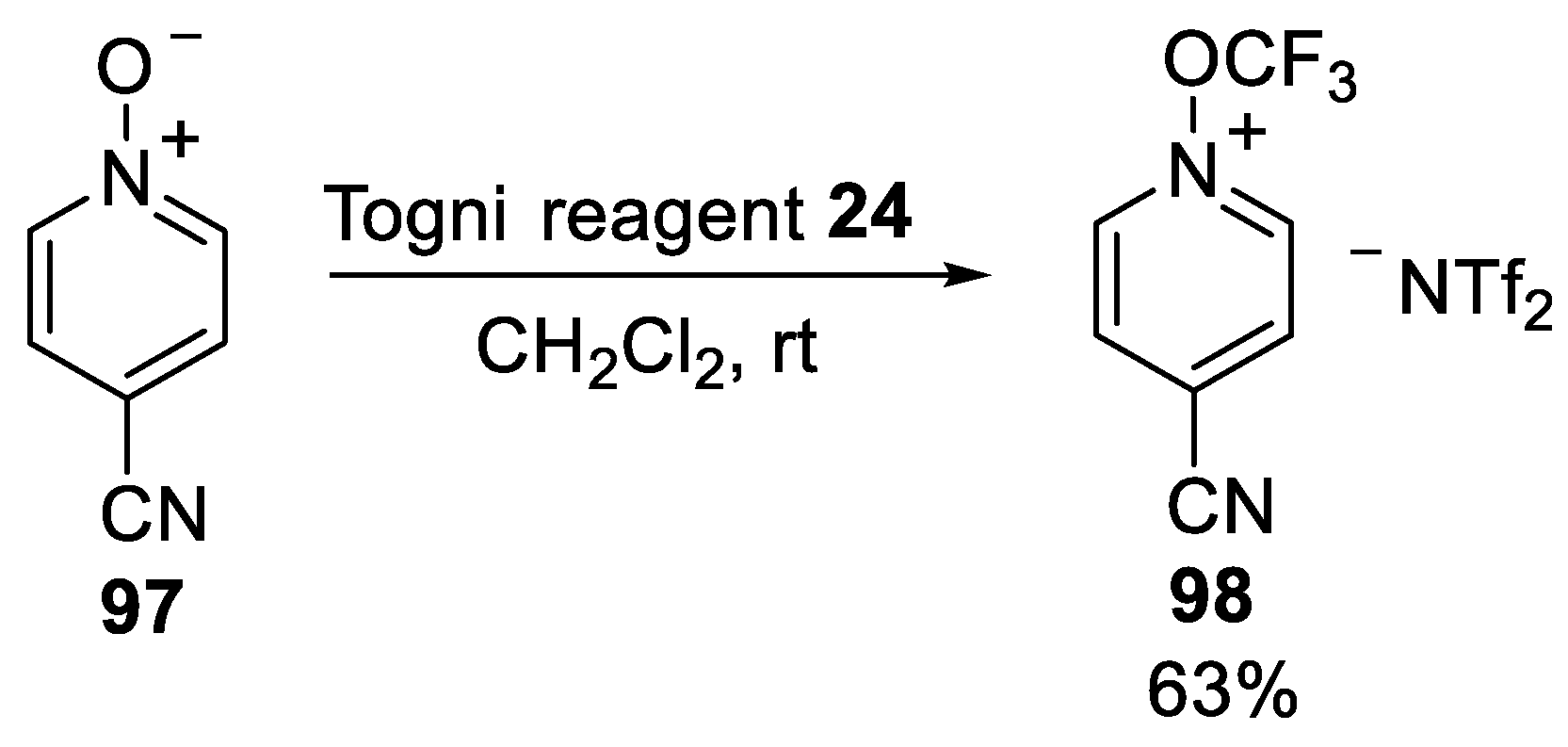
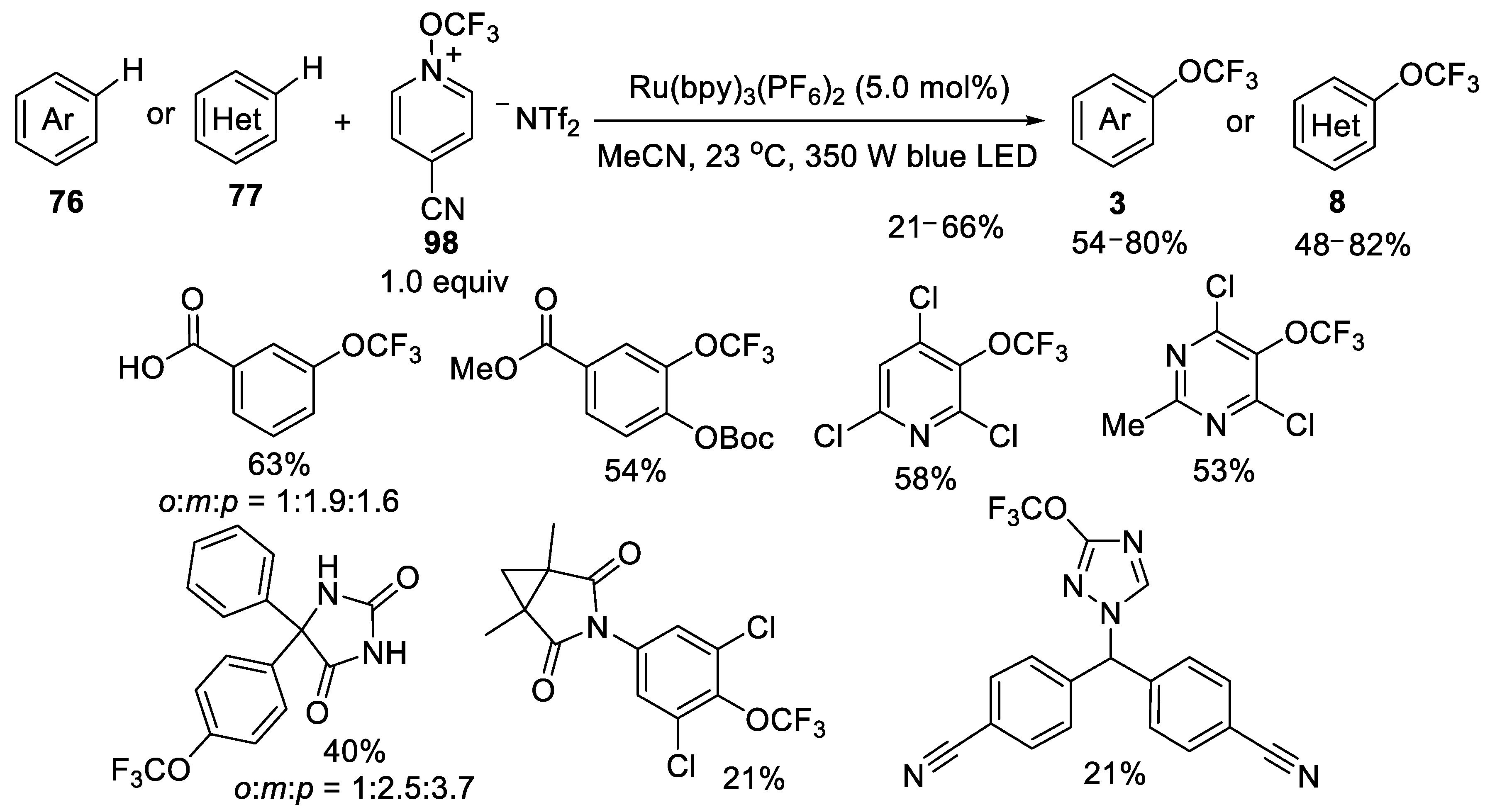
3.2.3. N-Trifluoromethoxypyridinium Salt 98
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Organic fluorine compounds: A great opportunity for enhanced materials properties. Chem. Soc. Rev. 2011, 40, 3496–3508. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; O’Hagan, D. Successful fluorine-containing herbicide agrochemicals. J. Fluor. Chem. 2014, 167, 16–29. [Google Scholar] [CrossRef]
- Cartwright, D. Recent Developments in Fluorine-Containing Agrochemicals. In Organofluorine Chemistry; Banks, R.E., Smart, B.E., Tatlow, J.C., Eds.; Springer: Singapore, 1994; pp. 237–262. [Google Scholar]
- Heodoridis, G. Fluorinecontaining agrochemicals: An overview of recent developments. In Fluorine and the Enviroment; Tressaud, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Chapter 4; pp. 121–175. [Google Scholar]
- Jeschke, P. The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection. ChemBioChem 2004, 5, 570–589. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. Current Contributions of Organofluorine Compounds to the Agrochemical Industry. Iscience 2020, 23, 101467. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluo-rine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J.L.; Izawa, K.; Liu, H.; Soloshonok, V.A. Recent advances in the trifluorometh-ylation methodology and new CF3-containing drugs. J. Fluor. Chem. 2014, 167, 37–54. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem. A Eur. J. 2019, 25, 11797–11819. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V.A.; Coelho, J.A.S.; Toste, F.D. Modern Approaches for Asymmetric Construction of Carbon–Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018, 118, 3887–3964. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Han, J.; Klika, K.D.; Izawa, K.; Sato, T.; Meanwell, N.A.; Soloshonok, V.A. Applications of fluorine-containing amino acids for drug design. Eur. J. Med. Chem. 2020, 186, 111826. [Google Scholar] [CrossRef]
- Han, J.; Kiss, L.; Mei, H.; Remete, A.M.; Ponikvar-Svet, M.; Sedgwick, D.M.; Roman, R.; Fustero, S.; Moriwaki, H.; Soloshonok, V.A. Chemical Aspects of Human and Environmental Overload with Fluorine. Chem. Rev. 2021, 121, 4678–4742. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; White, S.; Graham, D.J.; Izawa, K.; Sato, T.; Fustero, S.; Meanwell, N.A.; Soloshonok, V.A. Tailor-Made Amino Acids and Fluorinated Motifs as Prominent Traits in Modern Pharmaceuticals. Chem. A Eur. J. 2020, 26, 11349–11390. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, W.K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.; Deng, H. Enzymatic Fluorination and Biotechnological Developments of the Fluorinase. Chem. Rev. 2015, 115, 634–649. [Google Scholar] [CrossRef] [PubMed]
- Sorochinsky, A.E.; Soloshonok, V.A. Asymmetric synthesis of fluorine-containing amines, amino alcohols, α- and β-amino acids mediated by chiral sulfinyl group. J. Fluor. Chem. 2010, 131, 127–139. [Google Scholar] [CrossRef]
- Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V.A. Recent advances in asymmetric synthesis of α-(trifluoromethyl)-containing α-amino acids. Synthesis 2012, 44, 1591–1602. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Kukhar, V.P.; Röschenthaler, G.-V.; Aceña, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Recent advances in the synthesis of fluorinated aminophosphonates and aminophosphonic acids. RSC Adv. 2013, 3, 6693–6716. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Sorochinsky, A.E.; Aceña, J.L. Self-Disproportionation of Enantiomers of Chiral, Non-Racemic Fluoroorganic Compounds: Role of Fluorine as Enabling Element. Synthesis 2012, 45, 141–152. [Google Scholar] [CrossRef]
- Alonso, C.; de Marigorta, E.M.; Rubiales, G.; Palacios, F. Carbon Trifluoromethylation Reactions of Hydrocarbon Derivatives and Heteroarenes. Chem. Rev. 2015, 115, 1847–1935. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.G.; Ritter, T. Modern Carbon–Fluorine Bond Forming Reactions for Aryl Fluoride Synthesis. Chem. Rev. 2015, 115, 612–633. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, C.; Wang, M.; Liu, Q. Trifluoromethyltrimethylsilane: Nucleophilic Trifluoromethylation and Beyond. Chem. Rev. 2015, 115, 683–730. [Google Scholar] [CrossRef] [PubMed]
- Besset, T.; Poisson, T.; Pannecoucke, X. Recent Progress in Direct Introduction of Fluorinated Groups on Alkenes and Alkynes by means of C-H Bond Functionalization. Chem. A Eur. J. 2014, 20, 16830–16845. [Google Scholar] [CrossRef]
- Belhomme, M.C.; Besset, T.; Poisson, T.; Pannecoucke, X. Recent Progress toward the Introduction of Functionalized Difluo-romethylated Building Blocks onto C (sp2) and C (sp) Centers. Chem. Eur. J. 2015, 21, 12836–12865. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Jiang, X. Indirect Construction of the OCF3 Motif. In Emerging Fluorinated Motifs: Synthesis, Properties, and Applications; Cahard, D., Ma, J.A., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2020; Chapter 6; pp. 195–205. [Google Scholar]
- Tang, P.; Jiang, X. Reagents for Direct Trifluoromethoxylation. In Emerging Fluorinated Motifs: Synthesis, Properties, and Applications; Cahard, D., Ma, J.A., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2020; Chapter 7; pp. 207–224. [Google Scholar]
- Lee, W.; Lee, K.N.; Ngai, M.Y. Direct Trifluoromethoxylation of Aromatics and Heteroaromatics. In Emerging Fluorinated Motifs: Synthesis, Properties, and Applications; Cahard, D., Ma, J.A., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2020; Chapter 8; pp. 225–250. [Google Scholar]
- Chen, C.; Liu, G. Direct Trifluoromethoxylation of Aliphatic Compounds. In Emerging Fluorinated Motifs: Synthesis, Properties, and Applications; Cahard, D., Ma, J.A., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2020; Chapter 9; pp. 251–265. [Google Scholar]
- Landelle, G.; Panossian, A.; Pazenok, S.; Vors, J.-P.; Leroux, F.R. Recent advances in transition metal-catalyzed Csp2-monofluoro-, difluoro-, perfluoromethylation and trifluoromethylthiolation. Beilstein J. Org. Chem. 2013, 9, 2476–2536. [Google Scholar] [CrossRef] [Green Version]
- Landelle, G.; Panossian, A.; Leroux, F.R. Trifluoromethyl ethers and–thioethers as tools for medicinal chemistry and drug discovery. Curr. Top. Med. Chem. 2014, 14, 941–951. [Google Scholar] [CrossRef]
- Manteau, B.; Pazenok, S.; Vors, J.-P.; Leroux, F.R. New trends in the chemistry of α-fluorinated ethers, thioethers, amines and phosphines. J. Fluor. Chem. 2010, 131, 140–158. [Google Scholar] [CrossRef]
- Leroux, F.R.; Manteau, B.; Vors, J.-P.; Pazenok, S. Trifluoromethyl ethers-synthesis and properties of an unusual substituent. Beilstein J. Org. Chem. 2008, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Remete, A.M.; Dobson, L.S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V.A.; O’Hagan, D. Next generation organofluorine containing blockbuster drugs. J. Fluor. Chem. 2020, 239, 109639. [Google Scholar] [CrossRef]
- Mei, H.; Remete, A.M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V.A.; Han, J. Fluorine-containing drugs ap-proved by the FDA in 2019. Chin. Chem. Lett. 2020, 31, 2401–2413. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, A.; Dhawan, G.; Mei, H.; Zhang, W.; Izawa, K.; Soloshonok, V.A.; Han, J. Fluorine-containing pharmaceuticals approved by the FDA in 2020: Synthesis and biological activity. Chin. Chem. Lett. 2021. [Google Scholar] [CrossRef]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Unger, S.H.; Kim, K.H.; Nikaitani, D.; Lien, E.J. Aromatic substituent constants for structure-activity correlations. J. Med. Chem. 1973, 16, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Leo, A.; Jow, P.Y.C.; Silipo, C.; Hansch, C. Calculation of hydrophobic constant (log P) from.pi. and f constants. J. Med. Chem. 1975, 18, 865–868. [Google Scholar] [CrossRef]
- Lemieux, R.U. Effects of unshared pairs of electrons and their solvation on conformational equilibria. Pure Appl. Chem. 1971, 25, 527–548. [Google Scholar] [CrossRef]
- Marrec, O.; Billard, T.; Vors, J.P.; Pazenok, S.; Langlois, B.R. A deeper insight into direct trifluoromethoxylation with trifluo-romethyl triflate. J. Fluor. Chem. 2010, 131, 200–207. [Google Scholar] [CrossRef]
- Manteau, B.; Genix, P.; Brelot, L.; Vors, J.-P.; Pazenok, S.; Giornal, F.; Leuenberger, C.; Leroux, F.R. A General Approach to (Trifluoromethoxy)pyridines: First X-ray Structure Determinations and Quantum Chemistry Studies. Eur. J. Org. Chem. 2010, 2010, 6043–6066. [Google Scholar] [CrossRef]
- Sheppard, W.A. α-Fluorinated Ethers. I. Aryl Fluoroalkyl Ethers. J. Org. Chem. 1964, 29, 1–11. [Google Scholar] [CrossRef]
- Aldrich, P.E.; Sheppard, W.A. α-Fluorinated Ethers. II. Alkyl Fluoroalkyl Ethers. J. Org. Chem. 1964, 29, 11–15. [Google Scholar] [CrossRef]
- Leroux, F.; Jeschke, P.; Schlosser, M. α-Fluorinated Ethers, Thioethers, and Amines: Anomerically Biased Species. Chem. Rev. 2005, 105, 827–856. [Google Scholar] [CrossRef]
- Mathey, F.; Bensoam, J. Reaction de MoF6 avec chlorothioformiates d’aryle nouvelle synthese des aryl trifluoromethylethers ArOCF3. Tetrahedron Lett. 1973, 14, 2253–2256. [Google Scholar] [CrossRef]
- Feiring, A.E. Chemistry in hydrogen fluoride. 7. A novel synthesis of aryl trifluoromethyl ethers. J. Org. Chem. 1979, 44, 2907–2910. [Google Scholar] [CrossRef]
- Kuroboshi, M.; Suzuki, K.; Hiyama, T. Oxidative desulfurization-fluorination of xanthates. A convenient synthesis of trifluo-romethyl ethers and difluoro (methylthio) methyl ethers. Tetrahedron Lett. 1992, 33, 4173–4176. [Google Scholar] [CrossRef]
- Kanie, K.; Tanaka, Y.; Suzuki, K.; Kuroboshi, M.; Hiyama, T. A Convenient Synthesis of Trifluoromethyl Ethers by Oxidative Desulfurization-Fluorination of Dithiocarbonates. Bull. Chem. Soc. Jpn. 2000, 73, 471–484. [Google Scholar] [CrossRef]
- Kuroboshi, M.; Kanie, K.; Hiyama, T. Oxidative Desulfurization-Fluorination: A Facile Entry to a Wide Variety of Organoflu-orine Compounds Leading to Novel Liquid-Crystalline Materials. Adv. Synth. Catal. 2001, 343, 235–250. [Google Scholar] [CrossRef]
- Shimizu, M.; Hiyama, T. Modern Synthetic Methods for Fluorine-Substituted Target Molecules. Angew. Chem. Int. Ed. 2005, 44, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, T.; Adachi, K.; Ishihara, S. CF3 Oxonium Salts, O-(Trifluoromethyl)dibenzofuranium Salts: In Situ Synthesis, Properties, and Application as a Real CF3+ Species Reagent. J. Org. Chem. 2007, 72, 6905–6917. [Google Scholar] [CrossRef]
- Koller, R.; Stanek, K.; Stolz, D.; Aardoom, R.; Niedermann, K.; Togni, A. Zinc-Mediated Formation of Trifluoromethyl Ethers from Alcohols and Hypervalent Iodine Trifluoromethylation Reagents. Angew. Chem. Int. Ed. 2009, 48, 4332–4336. [Google Scholar] [CrossRef]
- Stanek, K.; Koller, R.; Togni, A. Reactivity of a 10-I-3 Hypervalent Iodine Trifluoromethylation Reagent with Phenols. J. Org. Chem. 2008, 73, 7678–7685. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Xin, J.; Tang, P. Nucleophilic trifluoromethoxylation of alkyl halides without silver. Nat. Commun. 2020, 11, 755–757. [Google Scholar] [CrossRef]
- Besset, T.; Jubault, P.; Pannecoucke, X.; Poisson, T. New entries toward the synthesis of OCF3-containing molecules. Org. Chem. Front. 2016, 3, 1004–1010. [Google Scholar] [CrossRef]
- Hardy, M.A.; Chachignon, H.; Cahard, D. Advances in Asymmetric Di-and Trifluoromethylthiolation, and Di-and Trifluo-romethoxylation Reactions. Asian J. Org. Chem. 2019, 8, 591–609. [Google Scholar] [CrossRef]
- Lin, J.-H.; Ji, Y.-L.; Xiao, J.-C. Recent Advances in C-H Trifluoromethylthiolation and Trifluoromethoxylation Reactions. Curr. Org. Chem. 2015, 19, 1541–1553. [Google Scholar] [CrossRef]
- Sahoo, B.; Hopkinson, M.N. A Radical Revolution for Trifluoromethoxylation. Angew. Chem. Int. Ed. 2018, 57, 7942–7944. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.N.; Lee, J.; Ngai, M.-Y. Recent Development of Catalytic Trifluoromethoxylation Reactions. Tetrahedron 2018, 74, 7127–7135. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, P. Recent advances in new trifluoromethoxylation reagents. Sci. China Ser. B Chem. 2019, 62, 525–532. [Google Scholar] [CrossRef]
- Jiang, X.; Tang, P. Recent Advances of Trifluoromethoxylation Reactions Using TFMS and TFBO. Chin. J. Chem. 2021, 39, 255–264. [Google Scholar] [CrossRef]
- Jiang, X.; Tang, P. Advances in Enantioselective Construction of Trifluoromethoxylated Stereogenic Carbon Centers. Chin. J. Chem. 2020, 38, 101–102. [Google Scholar] [CrossRef]
- Yagupolskii, L.M. Sintez proizvodnykh feniltriftormetilovogo efira. Dokl. Akad. Nauk. SSSR 1955, 105, 100–102. [Google Scholar]
- Yagupolskii, L.M.; Troitskaya, V.I. Synthesis of derivatives of phenyl trifluoromethyl ether. Zh. Obshch. Khim. 1961, 31, 915–924. [Google Scholar]
- Yarovenko, N.N.; Vasileva, A.S. A new method of introduction of trihalomethyl group into organic compounds. Zh. Obshch. Khim. 1958, 28, 2502–2504. [Google Scholar]
- Wang, Z. Swarts Reaction. In Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons: New York, NY, USA, 2010; pp. 2744–2747. [Google Scholar]
- Salomé, J.; Mauger, C.; Brunet, S.; Schanen, V. Synthesis conditions and activity of various Lewis acids for the fluorination of trichloromethoxy-benzene by HF in liquid phase. J. Fluor. Chem. 2004, 125, 1947–1950. [Google Scholar] [CrossRef]
- Sokolenko, T.M.; Yagupolskii, Y.L. Trifluoromethoxypyrazines: Preparation and Properties. Molecules 2020, 25, 2226. [Google Scholar] [CrossRef]
- Sokolenko, T.M.; Dronkina, M.I.; Magnier, E.; Yagupolskii, L.M.; Yagupolskii, Y.L. Evaluation of Efficient and Practical Methods for the Preparation of Functionalized Aliphatic Trifluoromethyl Ethers. Molecules 2017, 22, 804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanie, K.; Tanaka, Y.; Shimizu, M.; Kuroboshi, M.; Hiyama, T. Oxidative desulfurization–fluorination of alkanol xanthates. Control of the reaction pathway to fluorination or trifluoromethoxylation. Chem. Commun. 1997, 309–310. [Google Scholar] [CrossRef]
- Yoritate, M.; Londregan, A.T.; Lian, Y.; Hartwig, J.F. Sequential Xanthalation and O-Trifluoromethylation of Phenols: A Pro-cedure for the Synthesis of Aryl Trifluoromethyl Ethers. J. Org. Chem. 2019, 84, 15767–15776. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, P.K.; Dhar, S.; Samal, S.; Ila, H.; Junjappa, H. 1-(Methyldithiocarbonyl)imidazole: A Useful Thiocarbonyl Transfer Reagent for Synthesis of Substituted Thioureas. Tetrahedron 2000, 56, 629–637. [Google Scholar] [CrossRef]
- Mohammadkhani, L.; Heravi, M.M. XtalFluor-E: A useful and versatile reagent in organic transformations. J. Fluor. Chem. 2019, 225, 11–20. [Google Scholar] [CrossRef]
- Umemoto, T.; Singh, R.P.; Xu, Y.; Saito, N. Discovery of 4-tert-Butyl-2,6-dimethylphenylsulfur Trifluoride as a Deoxofluori-nating Agent with High Thermal Stability as Well as Unusual Resistance to Aqueous Hydrolysis, and Its Diverse Fluorination Capabilities Including Deoxofluoro-Arylsulfinylation with High Stereoselectivity. J. Am. Chem. Soc. 2010, 132, 18199–18205. [Google Scholar]
- Sorrentino, J.P.; Ambler, B.R.; Altman, R.A. Late-Stage Conversion of a Metabolically Labile Aryl Methyl Ether-Containing Natural Product to Fluoroalkyl Analogues. J. Org. Chem. 2020, 85, 5416–5427. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, I.; Rechavi, D.; Mishani, E.; Rozen, S. A novel synthesis of trifluoromethyl ethers via xanthates, utilizing BrF3. J. Fluor. Chem. 1999, 97, 75–78. [Google Scholar] [CrossRef]
- Umemoto, T.; Singh, R.P. Arylsulfur chlorotetrafluorides as useful fluorinating agents: Deoxo- and dethioxo-fluorinations. J. Fluor. Chem. 2012, 140, 17–27. [Google Scholar] [CrossRef]
- Liu, J.; Xiang, H.; Jiang, L.; Yi, W. Chemoselective desulfurization-fluorination/bromination of carbonofluoridothioates for the O-trifluoromethylation and O-bromodifluoromethylation of alcohols. Sci. China Ser. B Chem. 2021, 64, 1372–1379. [Google Scholar] [CrossRef]
- Han, J.; Butler, G.; Moriwaki, H.; Konno, H.; Soloshonok, V.A.; Kitamura, T. Kitamura Electrophilic Fluorination Using HF as a Source of Fluorine. Molecules 2020, 25, 2116. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kitamura, T.; Zhou, Y.; Butler, G.; Han, J.; Soloshonok, V.A. Electrophilic fluorination using PhIO/HF·THF reagent. J. Fluor. Chem. 2020, 240, 109670. [Google Scholar]
- Varenikov, A.; Shapiro, E.; Gandelman, M. Decarboxylative Halogenation of Organic Compounds. Chem. Rev. 2021, 121, 412–484. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Ni, C.; He, Z.; Hu, J. O-Trifluoromethylation of Phenols: Access to Aryl Trifluoromethyl Ethers by O-Carboxydifluoromethylation and Decarboxylative Fluorination. Org. Lett. 2016, 18, 3754–3757. [Google Scholar] [CrossRef] [PubMed]
- Krishanmoorthy, S.; Schnell, S.; Dang, H.; Fu, F.; Prakash, G.S. Fluorodecarboxylation: Synthesis of aryl trifluoromethyl ethers (ArOCF3) and thioethers (ArSCF3). J. Fluor. Chem. 2017, 203, 130–135. [Google Scholar] [CrossRef]
- Fier, P.S.; Hartwig, J.F. Selective C-H Fluorination of Pyridines and Diazines Inspired by a Classic Amination Reaction. Science 2013, 342, 956–960. [Google Scholar] [CrossRef]
- Zhang, Q.-W.; Brusoe, A.T.; Mascitti, V.; Hesp, K.D.; Blakemore, D.C.; Kohrt, J.T.; Hartwig, J.F. Fluorodecarboxylation for the Synthesis of Trifluoromethyl Aryl Ethers. Angew. Chem. Int. Ed. 2016, 55, 9758–9762. [Google Scholar] [CrossRef] [PubMed]
- Tius, M.A. Xenon difluoride in synthesis. Tetrahedron 1995, 51, 6605–6634. [Google Scholar] [CrossRef]
- Chatalova-Sazepin, C.; Binayeva, M.; Epifanov, M.; Zhang, W.; Foth, P.; Amador, C.; Jagdeo, M.; Boswell, B.R.; Sammis, G.M. Xenon Difluoride Mediated Fluorodecarboxylations for the Syntheses of Di- and Trifluoromethoxyarenes. Org. Lett. 2016, 18, 4570–4573. [Google Scholar] [CrossRef]
- Umemoto, T. Electrophilic Perfluoroalkylating Agents. Chem. Rev. 1996, 96, 1757–1778. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, J.; Früh, N.; Togni, A. Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents. Chem. Rev. 2015, 115, 650–682. [Google Scholar] [CrossRef]
- Brantley, J.N.; Samant, A.V.; Toste, F.D. Isolation and Reactivity of Trifluoromethyl Iodonium Salts. ACS Cent. Sci. 2016, 2, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Liang, A.; Han, S.; Liu, Z.; Wang, L.; Li, J.; Zou, D.; Wu, Y.; Wu, Y. Regioselective Synthesis of N-Heteroaromatic Trifluoromethoxy Compounds by Direct O− CF3 Bond Formation. Chem. A Eur. J. 2016, 22, 5102–5106. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, C.; Chu, L.; Chen, Z.; Xu, X.; Qing, F. Silver-Mediated Oxidative Trifluoromethylation of Phenols: Direct Synthesis of Aryl Trifluoromethyl Ethers. Angew. Chem. Int. Ed. 2015, 54, 11839–11842. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-B.; Xu, X.-H.; Qing, F.-L. Silver-Mediated Oxidative Trifluoromethylation of Alcohols to Alkyl Trifluoromethyl Ethers. Org. Lett. 2015, 17, 5048–5051. [Google Scholar] [CrossRef] [PubMed]
- Hojczyk, K.N.; Feng, P.J.; Zhan, C.B.; Ngai, M.Y. Trifluoromethoxylation of Arenes: Synthesis of ortho-Trifluoromethoxylated Aniline Derivatives by OCF3 Migration. Angew. Chem. Int. Ed. 2014, 53, 14559–14563. [Google Scholar] [CrossRef]
- Ngai, M.-Y.; Lee, K.N.; Lee, J.W. Synthesis of Trifluoromethoxylated (Hetero) Arenes via OCF3 Migration. Synlett 2016, 27, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Feng, P.; Lee, K.N.; Lee, J.W.; Zhan, C.; Ngai, M.Y. Access to a new class of synthetic building blocks via trifluoromethoxyla-tion of pyridines and pyrimidines. Chem. Sci. 2016, 7, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Spiegowski, D.N.; Ngai, M.Y. Selective C–O bond formation via a photocatalytic radical coupling strategy: Access to perfluoroalkoxylated (OR F) arenes and heteroarenes. Chem. Sci. 2017, 8, 6066–6070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Lee, K.N.; Ngai, M.Y. Synthesis of Tri-and Difluoromethoxylated Compounds by Visible-Light Photoredox Cataly-sis. Angew. Chem. Int. Ed. 2019, 58, 11171–11181. [Google Scholar] [CrossRef]
- Noftle, R.E.; Cady, G.H. Preparation and properties of bis (trifluoromethylsulfuryl) peroxide and trifluoromethyl trifluoro-methanesulfonate. Inorg. Chem. 1965, 4, 1010–1012. [Google Scholar] [CrossRef]
- Olah, G.A.; Ohyama, T. The Simple Practical Preparation of Trifluoromethyl Trifluoromethanesulfonate (Triflate) 1. Synthesis 1976, 5, 319–320. [Google Scholar] [CrossRef]
- Noftle, R.E. On the preparation of trifluoromethyl trifluoromethanesulfonate. Inorg. Nucl. Chem. Lett. 1980, 16, 195–200. [Google Scholar] [CrossRef]
- Katsuhara, Y.; DesMarteau, D.D. Synthesis of perfluoroalkyl trifluoromethanesulfonates from perfluoroalkyl halides. Substitutive electrophilic dehalogenation with chlorine (I) and bromine (I) trifluoromethanesulfonates. J. Am. Chem. Soc. 1980, 102, 2681–2686. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yoshida, T.; Kumadaki, I. Trifluoromethyl trifluoromethanesulfonate (CF3SO2OCF3). Tetrahedron Lett. 1979, 20, 3865–3866. [Google Scholar] [CrossRef]
- Engelbrecht, V.A.; Tschager, E.Z. Bor-tris-(trifluoromethansulfonat), B (OSO2CF3)3 in Trifluoromethansulfonsäure–ein neues „supersaures System”. Inorg. Allg. Chem. 1977, 433, 19–25. [Google Scholar]
- Hassani, M.O.; Germain, A.; Brunel, D.; Commeyras, A. Thermal stability of perfluoroalkanesulfonic acids and their anhy-drides. New and easy approach to RFSO2ORF esters. Tetrahedron Lett. 1981, 22, 65–68. [Google Scholar] [CrossRef]
- Taylor, S.L.; Martin, J.C. Trifluoromethyl triflate: Synthesis and reactions. J. Org. Chem. 1987, 52, 4147–4156. [Google Scholar] [CrossRef]
- Kolomeitsev, A.A.; Vorobyev, M.; Gillandt, H. Versatile application of trifluoromethyl triflate. Tetrahedron Lett. 2008, 49, 449–454. [Google Scholar] [CrossRef]
- Farnham, W.B.; Smart, B.E.; Middleton, W.J.; Calabrese, J.C.; Dixon, D.A. Crystal and molecular structure of tris (dimethylamino) sulfonium trifluoromethoxide. Evidence for negative fluorine hyperconjugation. J. Am. Chem. Soc. 1985, 107, 4565–4567. [Google Scholar] [CrossRef]
- Barbion, J.; Pazenok, S.; Vors, J.P.; Langlois, B.R.; Billard, T. Multigram laboratory scale synthesis of α-trifluoromethoxy car-bonyl compounds. Org. Process Res. Dev. 2014, 18, 1037–1040. [Google Scholar] [CrossRef]
- Sokolenko, T.M.; Davydova, Y.A.; Yagupolskii, Y. Efficient synthesis of 5′-fluoroalkoxythiazoles via α-bromo-α-fluoroalkoxyacetophenones Hantzsch type cyclization with thioureas or thioamides. J. Fluor. Chem. 2012, 136, 20–25. [Google Scholar] [CrossRef]
- Zha, G.F.; Han, J.B.; Hu, X.Q.; Qin, H.L.; Fang, W.Y.; Zhang, C.P. Silver-mediated direct trifluoromethoxylation of α-diazo esters via the−OCF3 anion. Chem. Commun. 2016, 52, 7458–7461. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Song, H.X.; Zhang, C.P. Fluoroalkylation of Diazo Compounds with Diverse Rfn Reagents. Chem. Asian J. 2020, 15, 1660–1677. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liang, T.; Harada, S.; Lee, E.; Ritter, T. Silver-Mediated Trifluoromethoxylation of Aryl Stannanes and Arylboronic Acids. J. Am. Chem. Soc. 2011, 133, 13308–13310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.-W.; Hartwig, J.F. Synthesis of heteroaromatic trifluoromethyl ethers with trifluoromethyl triflate as the source of the trifluoromethoxy group. Chem. Commun. 2018, 54, 10124–10127. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, P.; Liu, G. Palladium-Catalyzed Intramolecular Aminotrifluoromethoxylation of Alkenes. J. Am. Chem. Soc. 2015, 137, 15648–15651. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Luo, Y.; Fu, L.; Chen, P.; Lan, Y.; Liu, G. Palladium-Catalyzed Intermolecular Ditrifluoromethoxylation of Unactivated Alkenes: CF3O-Palladation Initiated by Pd (IV). J. Am. Chem. Soc. 2018, 140, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Huang, Y.; Fang, X.; Li, H.; Zhang, Z.; Hor, T.S.A.; Weng, Z. Aryl-BIAN-ligated silver (i) trifluoromethoxide complex. Dalton Trans. 2015, 44, 19682–19686. [Google Scholar] [CrossRef]
- Yang, Y.-M.; Yao, J.-F.; Yan, W.; Luo, Z.; Tang, Z.-Y. Silver-Mediated Trifluoromethoxylation of (Hetero) aryldiazonium Tetrafluoroborates. Org. Lett. 2019, 21, 8003–8007. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hou, C.; Chen, P.; Liu, G. Palladium (II)-Catalyzed Aminotrifluoromethoxylation of Alkenes: Mechanistic Insight into the Effect of N -Protecting Groups. Chin. J. Chem. 2020, 38, 346–350. [Google Scholar] [CrossRef]
- Chen, D.; Lu, L.; Shen, Q. [Ag (bpy) (PPhtBu2) (OCF3)]: A stable nucleophilic reagent for chemoselective and stereospecific trifluoromethoxylation of secondary alkyl nosylates. Org. Chem. Front. 2019, 6, 1801–1806. [Google Scholar] [CrossRef]
- Qi, X.; Chen, P.; Liu, G. Catalytic Oxidative Trifluoromethoxylation of Allylic C−H Bonds Using a Palladium Catalyst. Angew. Chem. Int. Ed. 2017, 56, 9517–9521. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Pflüger, P.M.; Chen, P.; Liu, G. Palladium (II)-Catalyzed Enantioselective Aminotrifluoromethoxylation of Unactivated Alkenes using CsOCF3 as a Trifluoromethoxide Source. Angew. Chem. Int. Ed. 2019, 58, 2392–2396. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Kumon, T.; Hammond, G.B.; Umemoto, T. Trifluoromethyl Nonaflate: A Practical Trifluoromethoxylating Reagent and its Application to the Regio- and Stereoselective Synthesis of Trifluoromethoxylated Alkenes. Angew. Chem. Int. Ed. 2021, 60, 16171–16177. [Google Scholar] [CrossRef]
- Umemoto, T.; Zhang, B.; Zhu, T.; Zhou, X.; Zhang, P.; Hu, S.; Li, Y. Powerful, Thermally Stable, One-Pot-Preparable, and Recyclable Electrophilic Trifluoromethylating Agents: 2,8-Difluoro- and 2,3,7,8-Tetrafluoro-S-(trifluoromethyl) dibenzothiophenium Salts. J. Org. Chem. 2017, 82, 7708–7719. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, T.; Zhou, X.; Li, Y. A new version of Umemoto’s reagents: A three-step one-pot preparation of 2,3,7,8-tetrafluoro-S-(trifluoromethyl) dibenzothiophenium triflate. J. Fluor. Chem. 2019, 226, 109347. [Google Scholar] [CrossRef]
- Koller, R.; Huchet, Q.; Battaglia, P.; Welch, J.M.; Togni, A. Acid-mediated formation of trifluoromethyl sulfonates from sulfonic acids and a hypervalent iodine trifluoromethylating agent. Chem. Commun. 2009, 40, 5993–5995. [Google Scholar] [CrossRef]
- Guo, S.; Cong, F.; Guo, R.; Wang, L.; Tang, P. Asymmetric silver-catalysed intermolecular bromotrifluoromethoxylation of alkenes with a new trifluoromethoxylation reagent. Nat. Chem. 2017, 9, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Tang, P. Silver-Mediated Intermolecular Iodotrifluoromethoxylation of Alkenes. J. Org. Chem. 2020, 85, 2512–2519. [Google Scholar] [CrossRef]
- Wang, F.; Guo, Y.; Zhang, Y.; Tang, P. Silver-Catalyzed Dibromotrifluoromethoxylation of Terminal Alkynes. ACS Catal. 2021, 11, 3218–3223. [Google Scholar] [CrossRef]
- Cong, F.; Wei, Y.; Tang, P. Combining photoredox and silver catalysis for azidotrifluoromethoxylation of styrenes. Chem. Commun. 2018, 54, 4473–4476. [Google Scholar] [CrossRef]
- Jiang, X.; Deng, Z.; Tang, P. Direct Dehydroxytrifluoromethoxylation of Alcohols. Angew. Chem. Int. Ed. 2018, 57, 292–295. [Google Scholar] [CrossRef]
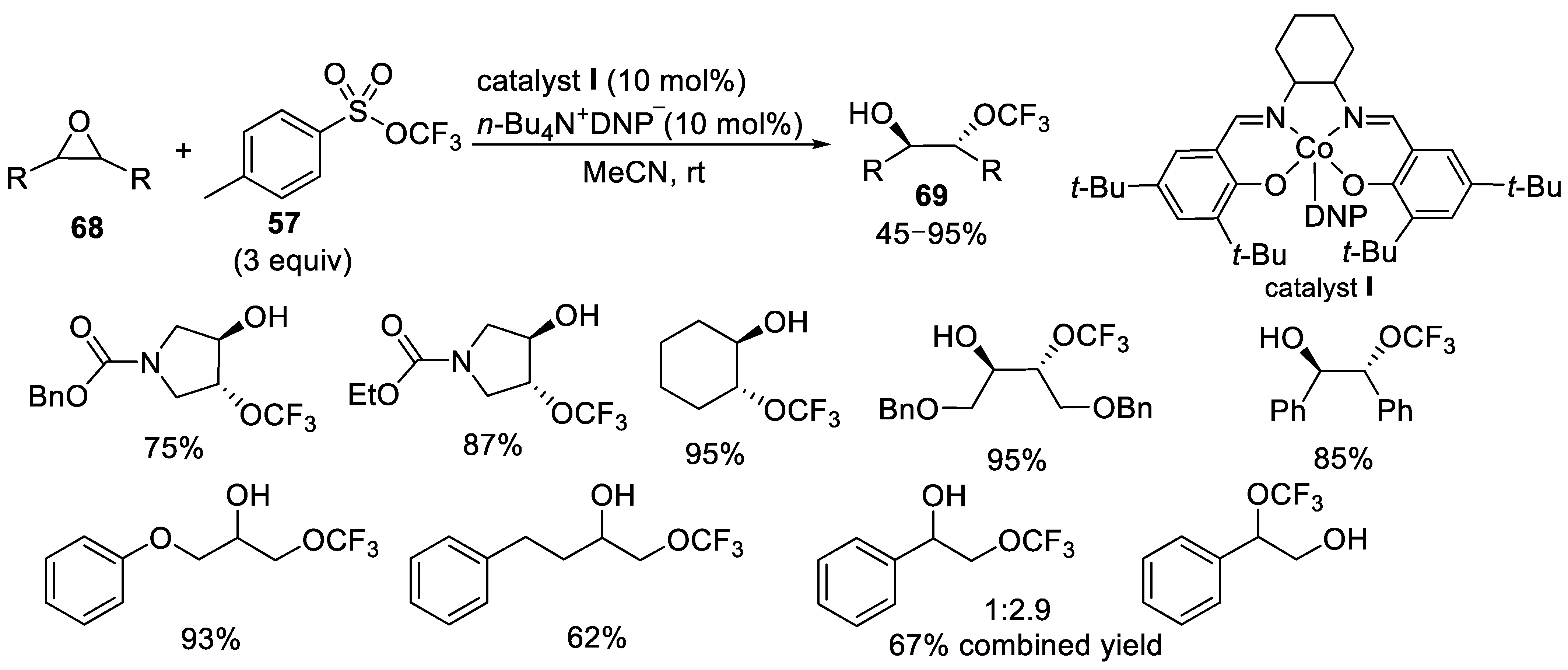
- Liu, J.; Wei, Y.; Tang, P. Cobalt-Catalyzed Trifluoromethoxylation of Epoxides. J. Am. Chem. Soc. 2018, 140, 15194–15199. [Google Scholar] [CrossRef]
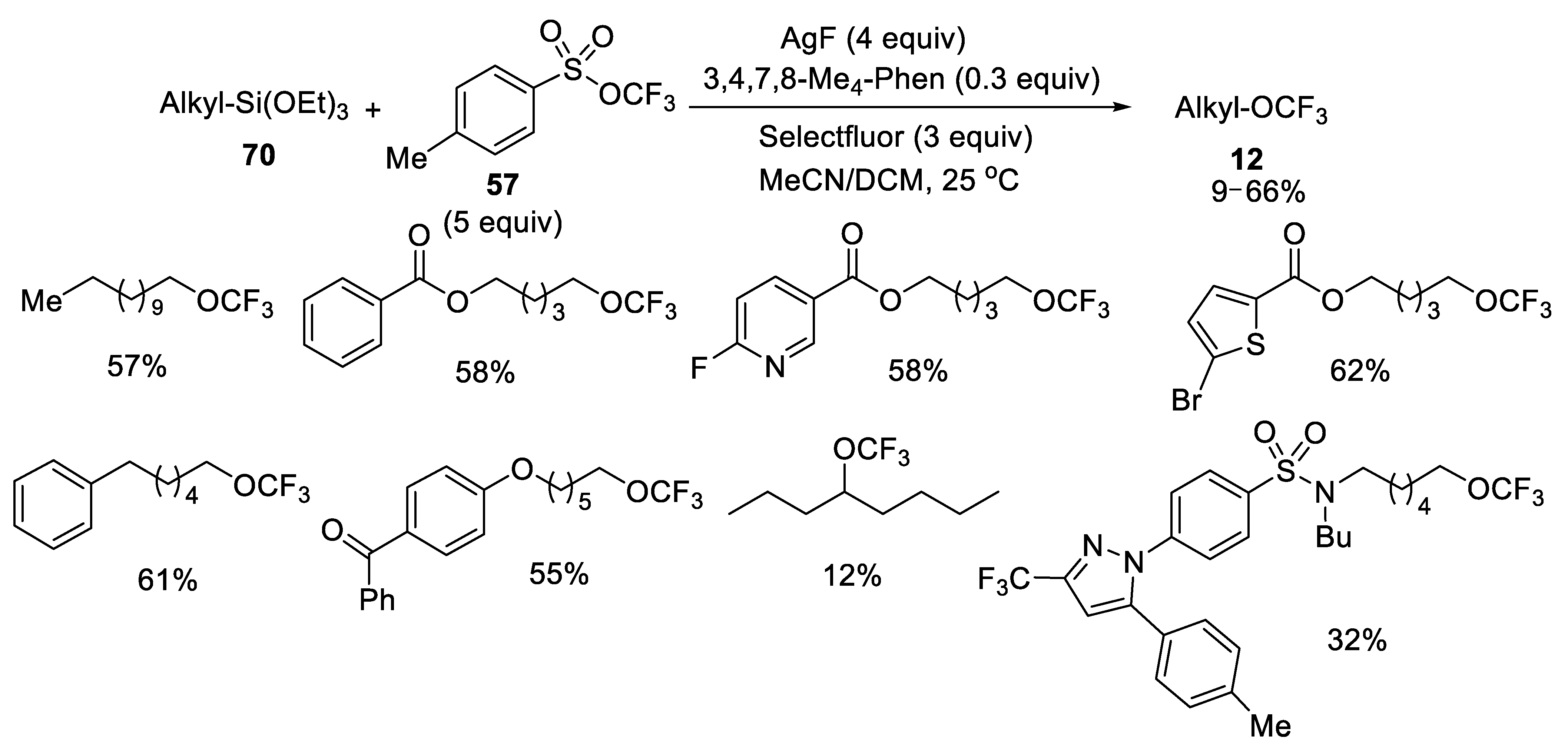
- Wang, F.; Xu, P.; Cong, F.; Tang, P. Silver-mediated oxidative functionalization of alkylsilanes. Chem. Sci. 2018, 9, 8836–8841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
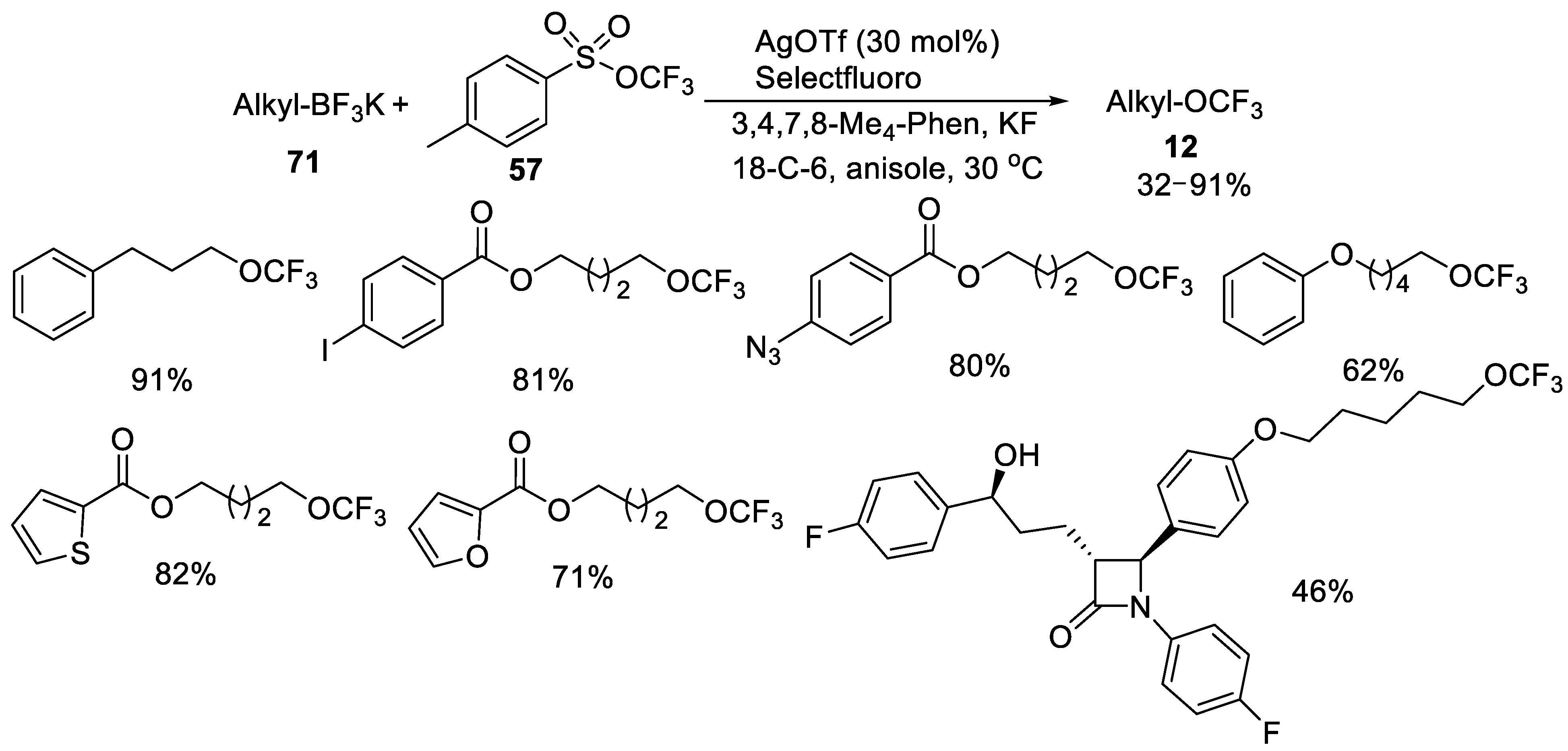
- Jiang, X.; Tang, P. Silver-Catalyzed Trifluoromethoxylation of Alkyl Trifluoroborates. Org. Lett. 2020, 22, 5135–5139. [Google Scholar] [CrossRef]
- Yang, H.; Wang, F.; Jiang, X.; Zhou, Y.; Xu, X.; Tang, P. Silver-Promoted Oxidative Benzylic C−H Trifluoromethoxylation. Angew. Chem. Int. Ed. 2018, 57, 13266–13270. [Google Scholar] [CrossRef]
- Lei, M.; Miao, H.; Wang, X.; Zhang, W.; Zhu, C.; Lu, X.; Shen, J.; Qin, Y.; Zhang, H.; Sha, S.; et al. Trifluoromethyl aryl sulfonates (TFMS): An applicable trifluoromethoxylation reagent. Tetrahedron Lett. 2019, 60, 1389–1392. [Google Scholar] [CrossRef]
- Yang, S.; Chen, M.; Tang, P. Visible-light photoredox-catalyzed and copper-promoted trifluoromethoxylation of arenediazonium tetrafluoroborates. Angew. Chem. Int. Ed. 2019, 58, 7840–7844. [Google Scholar] [CrossRef]
- Deng, Z.; Zhao, M.; Wang, F.; Tang, P. Selective C-H trifluoromethoxylation of (hetero) arenes as limiting reagent. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Zhou, M.; Ni, C.; Zeng, Y.; Hu, J. Trifluoromethyl Benzoate: A Versatile Trifluoromethoxylation Reagent. J. Am. Chem. Soc. 2018, 140, 6801–6805. [Google Scholar] [CrossRef]
- Marrec, O.; Billard, T.; Vors, J.-P.; Pazenok, S.; Langlois, B.R. A New and Direct Trifluoromethoxylation of Aliphatic Substrates with 2,4-Dinitro (trifluoromethoxy) benzene. Adv. Synth. Catal. 2010, 352, 2831–2837. [Google Scholar] [CrossRef]
- Duran-Camacho, G.; Ferguson, D.M.; Kampf, J.W.; Bland, D.C.; Sanford, M.S. Isolable Pyridinium Trifluoromethoxide Salt for Nucleophilic Trifluoromethoxylation. Org. Lett. 2021, 23, 5138–5142. [Google Scholar] [CrossRef]
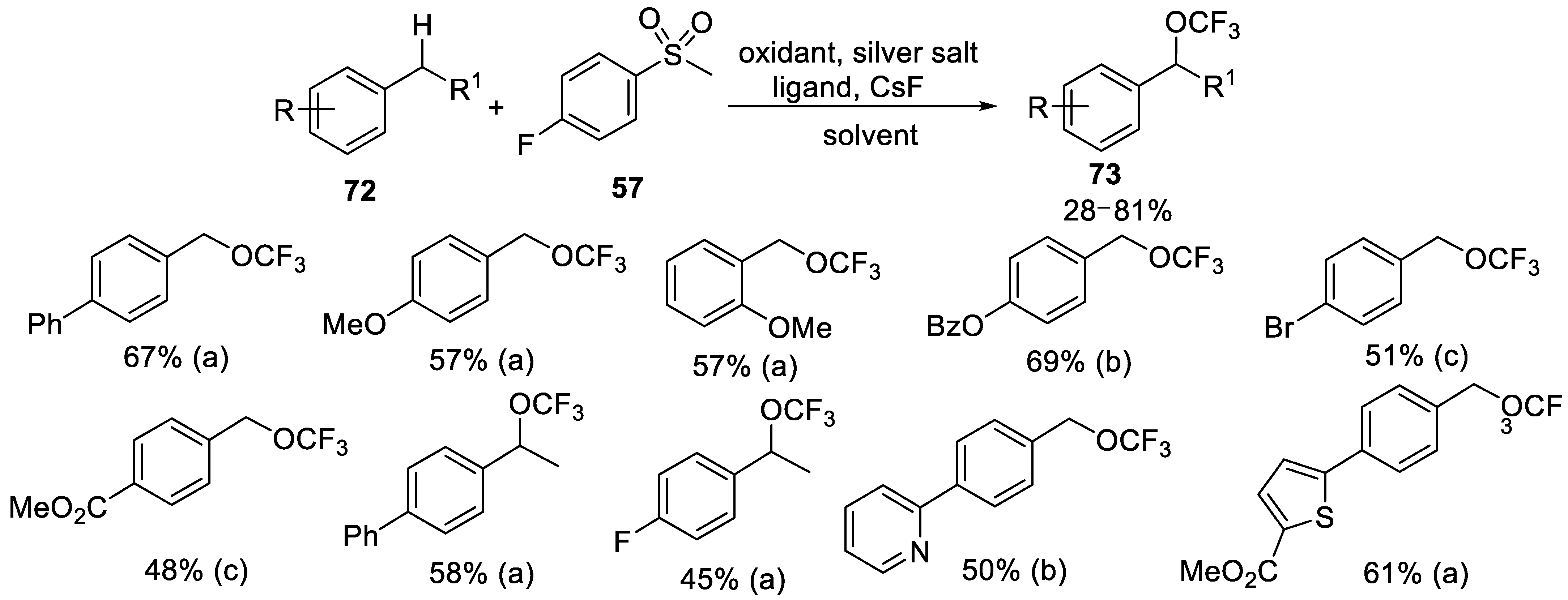
- Zheng, W.; Morales-Rivera, C.A.; Lee, J.W.; Liu, P.; Ngai, M. Catalytic C−H Trifluoromethoxylation of Arenes and Heteroarenes. Angew. Chem. Int. Ed. 2018, 57, 9645–9649. [Google Scholar] [CrossRef]
- Zheng, W.; Lee, J.W.; Morales-Rivera, C.A.; Liu, P.; Ngai, M. Redox-Active Reagents for Photocatalytic Generation of the OCF3 Radical and (Hetero) Aryl C−H Trifluoromethoxylation. Angew. Chem. Int. Ed. 2018, 57, 13795–13799. [Google Scholar] [CrossRef]
- Jelier, B.J.; Tripet, P.F.; Pietrasiak, E.; Franzoni, I.; Jeschke, G.; Togni, A. Radical Trifluoromethoxylation of Arenes Triggered by a Visible-Light-Mediated N−O Bond Redox Fragmentation. Angew. Chem. Int. Ed. 2018, 57, 13784–13789. [Google Scholar] [CrossRef]





























































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Zhang, X.; Sorochinsky, A.E.; Butler, G.; Han, J.; Soloshonok, V.A. Advances in the Development of Trifluoromethoxylation Reagents. Symmetry 2021, 13, 2380. https://doi.org/10.3390/sym13122380
Wang Q, Zhang X, Sorochinsky AE, Butler G, Han J, Soloshonok VA. Advances in the Development of Trifluoromethoxylation Reagents. Symmetry. 2021; 13(12):2380. https://doi.org/10.3390/sym13122380
Chicago/Turabian StyleWang, Qian, Xin Zhang, Alexander E. Sorochinsky, Greg Butler, Jianlin Han, and Vadim A. Soloshonok. 2021. "Advances in the Development of Trifluoromethoxylation Reagents" Symmetry 13, no. 12: 2380. https://doi.org/10.3390/sym13122380