
Cutaneous Lymphoma and Antibody-Directed Therapies


Abstract
:1. Introduction
2. Cutaneous T Cell Lymphoma
2.1. Definition and Classification
2.2. Pathophysiology of T Cells
2.3. Distinctive Molecular Alterations of CTCL
3. Cutaneous B Cell Lymphoma
3.1. Definition and Classification
3.2. Pathophysiology of B Cells
3.3. Distinctive Molecular Alterations of CBCL
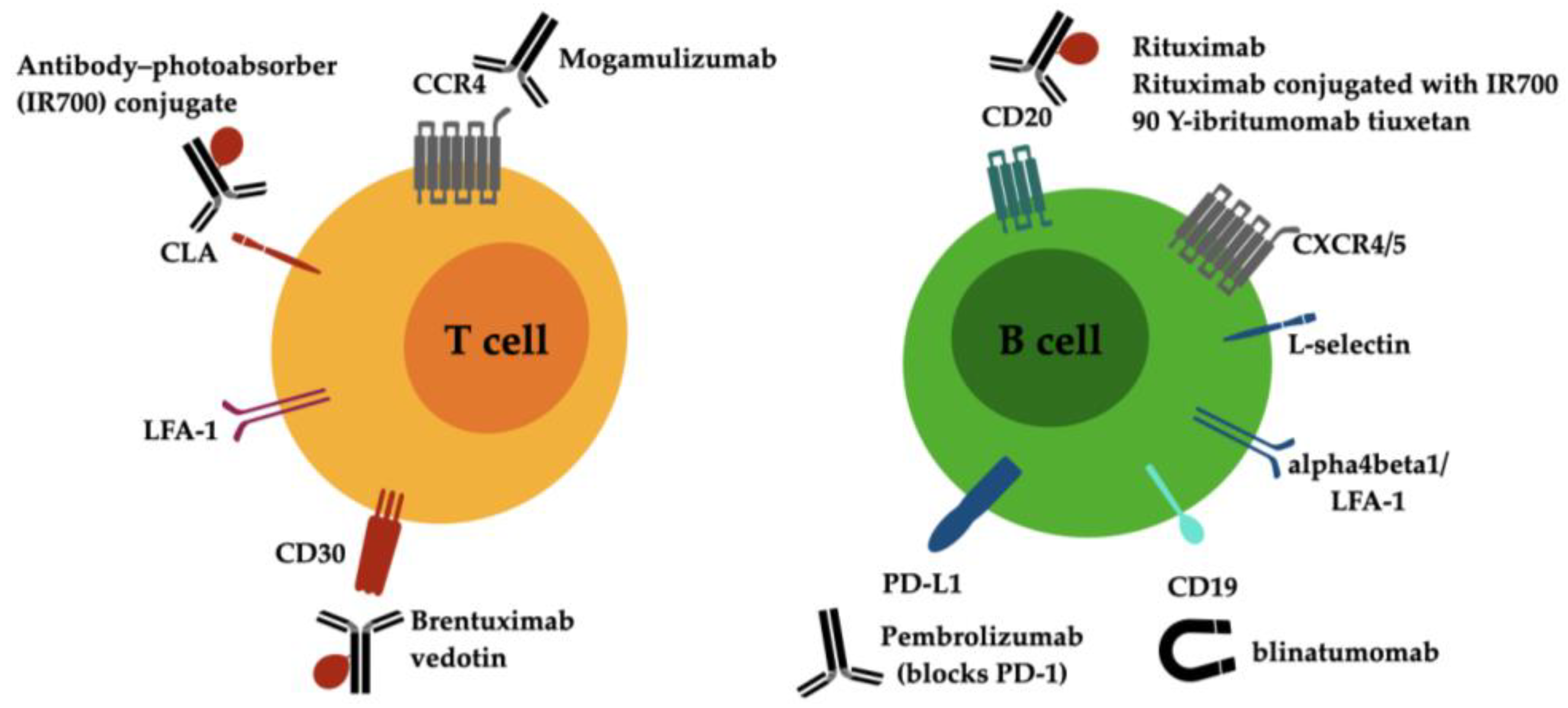
4. Antibody-Directed Therapies for the Treatment of Lymphoma
4.1. Investigative Approaches for CTCL
4.2. Investigative Approaches for CBCL
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADC, | antibody–drug conjugates |
| Akt, | serine-threonine protein kinase B |
| APC, | antibody–photoabsorber conjugate |
| BCL, | B cell lymphoma BCL2 |
| BiTE, | bi-specific T cell engager |
| B-RAF, | B-Raf proto-oncogene, serine/threonine kinase |
| BV, | brentuximab vedotin |
| B2M, | beta 2 microglobulin |
| CBCL, | cutaneous B cell lymphoma |
| CCR, | C-C chemokine receptor |
| CD, | cluster of differentiation |
| csHSP70, | cell surface heat shock protein 70 |
| CTCL, | cutaneous T cell lymphoma |
| CTLA4, | cytotoxic T-lymphocyte-associated protein 4 |
| CXCL, | chemokine (C-X-C motif) ligand |
| CXCR, | C-X-C chemokine receptor |
| DLBCL, | diffuse large B cell lymphoma |
| EBV, | Epstein–Barr virus |
| EMA, | European Medicine Agency |
| FDA, | Food and Drug Administration |
| FCL, | follicle center lymphoma |
| FISH, | fluorescence in situ hybridization |
| GNA13, | guanine nucleotide-binding protein subunit alpha-13 |
| GPCR, | G protein coupled receptor |
| HLA, | human leukocyte antigen |
| HTLV-1, | human T cell leukemia virus 1 |
| ICOS, | inducible T cell costimulator |
| IL, | interleukin |
| IR700, | IRdye 700DX® |
| JAK, | Janus kinase |
| LPD, | lymphoproliferative disorders |
| LyP, | lymphomatoid papulosis |
| mAb, | monoclonal antibody |
| MAPK, | mitogen-activated protein kinase |
| MAPK3, | mitogen-activated protein kinase 3 or extracellular signal-regulated kinase 1 (ERK1) |
| MDR1, | multidrug resistance-1 |
| MF, | mycosis fungoides and its variants—folliculotropic MF, pagetoid reticulosis, granulomatous slack skin |
| MMAE, | monomethyl auristatin E |
| mTOR, | mammalian target of rapamycin |
| MYC, | Myc proto-oncogene, bHLH transcription factor |
| NF-kB, | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK, | natural killer |
| NHL, | non-Hodgkin’s lymphoma |
| NIR-PIT, | near-infrared photoimmunotherapy |
| PCALCL, | primary cutaneous anaplastic large cell lymphoma |
| PCDLBCL, LT, | primary cutaneous diffuse large B cell lymphoma, leg type |
| PCFCL, | primary cutaneous follicle center lymphoma |
| PCMZL, | primary cutaneous marginal zone lymphoma |
| PCR, | polymerase chain reaction |
| PD-1, | programmed cell death protein 1 |
| PD-L1/PD-L2, | programmed cell death 1 ligand 1/2 |
| PI3K, | phosphatidylinositol-4,5-bisphosphate 3-kinase |
| P2RY8, | P2Y purinoceptor 8 |
| p70S6K, | ribosomal protein S6 kinase beta-1 |
| RNA, | ribonucleic acid |
| SS, | Sezary syndrome |
| STAT, | signal transducer and activator of transcription |
| S1PR2, | Sphingosine-1-phosphate receptor 2 |
| TCR, | T cell receptor |
| TLR, | Toll-like receptor |
| t(14;18)/t(8;14), | chromosomal translocation between chromosomes 14 and 18 and 8 and 14, respectively |
| UV, | ultraviolet |
| VEGFR, | vascular endothelial growth factor receptor |
| WHO-EORTC, | World Health Organization—European Organization for Research and Treatment of Cancer |
| 90Y-I, | 90 Y-ibritumomab tiuxetan |
References
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s Magic Bullet Concept: 100 Years of Progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- James, J.S.; Dubs, G. FDA Approves New Kind of Lymphoma Treatment. Food and Drug Administration. AIDS Treat News 1997, 284, 2–3. [Google Scholar]
- Saini, S.; Kumar, Y. Chapter 9—Bispecific Antibodies: A Promising Entrant in Cancer Immunotherapy. In Translational Biotechnology; Hasija, Y., Ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 233–266. ISBN 978-0-12-821972-0. [Google Scholar]
- Lundin, J.; Hagberg, H.; Repp, R.; Cavallin-Ståhl, E.; Fredén, S.; Juliusson, G.; Rosenblad, E.; Tjønnfjord, G.; Wiklund, T.; Osterborg, A. Phase 2 Study of Alemtuzumab (Anti-CD52 Monoclonal Antibody) in Patients with Advanced Mycosis Fungoides/Sezary Syndrome. Blood 2003, 101, 4267–4272. [Google Scholar] [CrossRef]
- Kim, Y.H.; Bagot, M.; Pinter-Brown, L.; Rook, A.H.; Porcu, P.; Horwitz, S.M.; Whittaker, S.; Tokura, Y.; Vermeer, M.; Zinzani, P.L.; et al. Mogamulizumab versus Vorinostat in Previously Treated Cutaneous T-Cell Lymphoma (MAVORIC): An International, Open-Label, Randomised, Controlled Phase 3 Trial. Lancet Oncol. 2018, 19, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.F.; Thurston, D.E.; Crescioli, S.; et al. Antibody Structure and Engineering Considerations for the Design and Function of Antibody Drug Conjugates (ADCs). Oncoimmunology 2018, 7, e1395127. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Schrama, D.; Houben, R.; Guyétant, S.; Desgranges, A.; Martin, C.; Berthon, P.; Viaud-Massuard, M.-C.; Touzé, A.; Kervarrec, T.; et al. Antibody-Drug Conjugates as an Emerging Therapy in Oncodermatology. Cancers 2022, 14, 778. [Google Scholar] [CrossRef]
- Birrer, M.J.; Moore, K.N.; Betella, I.; Bates, R.C. Antibody-Drug Conjugate-Based Therapeutics: State of the Science. J. Natl. Cancer Inst. 2019, 111, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Chia, C.S.B. A Patent Review on FDA-Approved Antibody-Drug Conjugates, Their Linkers and Drug Payloads. ChemMedChem 2022, 17, e202200032. [Google Scholar] [CrossRef] [PubMed]
- Staudacher, A.H.; Brown, M.P. Antibody Drug Conjugates and Bystander Killing: Is Antigen-Dependent Internalisation Required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.P.; Sharma, S.; Shah, D.K. Quantitative Characterization of in Vitro Bystander Effect of Antibody-Drug Conjugates. J. Pharm. Pharm. 2016, 43, 567–582. [Google Scholar] [CrossRef] [Green Version]
- Taheri-Ledari, R.; Zhang, W.; Radmanesh, M.; Cathcart, N.; Maleki, A.; Kitaev, V. Plasmonic Photothermal Release of Docetaxel by Gold Nanoparticles Incorporated onto Halloysite Nanotubes with Conjugated 2D8-E3 Antibodies for Selective Cancer Therapy. J. Nanobiotechnol. 2021, 19, 239. [Google Scholar] [CrossRef]
- Parvaz, S.; Taheri-Ledari, R.; Esmaeili, M.S.; Rabbani, M.; Maleki, A. A Brief Survey on the Advanced Brain Drug Administration by Nanoscale Carriers: With a Particular Focus on AChE Reactivators. Life Sci. 2020, 240, 117099. [Google Scholar] [CrossRef]
- Zhang, W.; Taheri-Ledari, R.; Ganjali, F.; Afruzi, F.H.; Hajizadeh, Z.; Saeidirad, M.; Qazi, F.S.; Kashtiaray, A.; Sehat, S.S.; Hamblin, M.R.; et al. Nanoscale Bioconjugates: A Review of the Structural Attributes of Drug-Loaded Nanocarrier Conjugates for Selective Cancer Therapy. Heliyon 2022, 8, e09577. [Google Scholar] [CrossRef]
- Taheri-Ledari, R.; Zolfaghari, E.; Zarei-Shokat, S.; Kashtiaray, A.; Maleki, A. A Magnetic Antibody-Conjugated Nano-System for Selective Delivery of Ca(OH)2 and Taxotere in Ovarian Cancer Cells. Commun. Biol. 2022, 5, 995. [Google Scholar] [CrossRef]
- Wilcox, R.A. Cutaneous T-Cell Lymphoma: 2017 Update on Diagnosis, Risk-Stratification, and Management. Am. J. Hematol. 2017, 92, 1085–1102. [Google Scholar] [CrossRef] [Green Version]
- Duvic, M.; Hymes, K.; Heald, P.; Breneman, D.; Martin, A.G.; Myskowski, P.; Crowley, C.; Yocum, R.C. Bexarotene Worldwide Study Group Bexarotene Is Effective and Safe for Treatment of Refractory Advanced-Stage Cutaneous T-Cell Lymphoma: Multinational Phase II-III Trial Results. J. Clin. Oncol. 2001, 19, 2456–2471. [Google Scholar] [CrossRef]
- Heider, U.; Rademacher, J.; Lamottke, B.; Mieth, M.; Moebs, M.; von Metzler, I.; Assaf, C.; Sezer, O. Synergistic Interaction of the Histone Deacetylase Inhibitor SAHA with the Proteasome Inhibitor Bortezomib in Cutaneous T Cell Lymphoma. Eur. J. Haematol. 2009, 82, 440–449. [Google Scholar] [CrossRef]
- Dummer, R.; Beyer, M.; Hymes, K.; Epping, M.T.; Bernards, R.; Steinhoff, M.; Sterry, W.; Kerl, H.; Heath, K.; Ahern, J.D.; et al. Vorinostat Combined with Bexarotene for Treatment of Cutaneous T-Cell Lymphoma: In Vitro and Phase I Clinical Evidence Supporting Augmentation of Retinoic Acid Receptor/Retinoid X Receptor Activation by Histone Deacetylase Inhibition. Leuk. Lymphoma 2012, 53, 1501–1508. [Google Scholar] [CrossRef]
- Edelson, R.; Berger, C.; Gasparro, F.; Jegasothy, B.; Heald, P.; Wintroub, B.; Vonderheid, E.; Knobler, R.; Wolff, K.; Plewig, G. Treatment of Cutaneous T-Cell Lymphoma by Extracorporeal Photochemotherapy. Preliminary Results. N. Engl. J. Med. 1987, 316, 297–303. [Google Scholar] [CrossRef]
- Hughes, C.F.M.; Khot, A.; McCormack, C.; Lade, S.; Westerman, D.A.; Twigger, R.; Buelens, O.; Newland, K.; Tam, C.; Dickinson, M.; et al. Lack of Durable Disease Control with Chemotherapy for Mycosis Fungoides and Sézary Syndrome: A Comparative Study of Systemic Therapy. Blood 2015, 125, 71–81. [Google Scholar] [CrossRef]
- Hanel, W.; Briski, R.; Ross, C.W.; Anderson, T.F.; Kaminski, M.S.; Hristov, A.C.; Wilcox, R.A. A Retrospective Comparative Outcome Analysis Following Systemic Therapy in Mycosis Fungoides and Sezary Syndrome. Am. J. Hematol. 2016, 91, E491–E495. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.A.; Kim, Y.H.; Lavori, P.W.; Hoppe, R.T.; Stockerl-Goldstein, K.E. A Meta-Analysis of Patients Receiving Allogeneic or Autologous Hematopoietic Stem Cell Transplant in Mycosis Fungoides and Sézary Syndrome. Biol. Blood Marrow Transplant. 2009, 15, 982–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, R.F.; Schmitz, N.; Servitje, O.; Sureda, A. Haematopoietic Stem Cell Transplantation for Patients with Primary Cutaneous T-Cell Lymphoma. Bone Marrow Transplant. 2008, 41, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Devata, S.; Wilcox, R.A. Cutaneous T-Cell Lymphoma: A Review with a Focus on Targeted Agents. Am. J. Clin. Dermatol. 2016, 17, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, R.A. A Three-Signal Model of T-Cell Lymphoma Pathogenesis. Am. J. Hematol. 2016, 91, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamijo, H.; Miyagaki, T. Mycosis Fungoides and Sézary Syndrome: Updates and Review of Current Therapy. Curr. Treat. Options Oncol. 2021, 22, 10. [Google Scholar] [CrossRef] [PubMed]
- Willemze, R.; Jaffe, E.S.; Burg, G.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Diaz-Perez, J.L.; Duncan, L.M.; et al. WHO-EORTC Classification for Cutaneous Lymphomas. Blood 2005, 105, 3768–3785. [Google Scholar] [CrossRef] [Green Version]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 Update of the WHO-EORTC Classification for Primary Cutaneous Lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef]
- Pulitzer, M. Cutaneous T-Cell Lymphoma. Clin. Lab. Med. 2017, 37, 527–546. [Google Scholar] [CrossRef]
- Ferenczi, K.; Makkar, H.S. Cutaneous Lymphoma: Kids Are Not Just Little People. Clin. Dermatol. 2016, 34, 749–759. [Google Scholar] [CrossRef]
- Pals, S.T.; de Gorter, D.J.J.; Spaargaren, M. Lymphoma Dissemination: The Other Face of Lymphocyte Homing. Blood 2007, 110, 3102–3111. [Google Scholar] [CrossRef] [Green Version]
- Woodland, D.L.; Kohlmeier, J.E. Migration, Maintenance and Recall of Memory T Cells in Peripheral Tissues. Nat. Rev. Immunol. 2009, 9, 153–161. [Google Scholar] [CrossRef]
- Kieffer, J.D.; Fuhlbrigge, R.C.; Armerding, D.; Robert, C.; Ferenczi, K.; Camphausen, R.T.; Kupper, T.S. Neutrophils, Monocytes, and Dendritic Cells Express the Same Specialized Form of PSGL-1 as Do Skin-Homing Memory T Cells: Cutaneous Lymphocyte Antigen. Biochem. Biophys. Res. Commun. 2001, 285, 577–587. [Google Scholar] [CrossRef]
- Chang, S.-E.; Kim, M.-J.; Lee, W.-S.; Kang, Y.-K.; Moon, K.-C.; Koh, J.-K.; Choi, J.-H. Natural Killer Cells in Human Peripheral Blood and Primary Cutaneous Natural Killer Cell Lymphomas May Express Cutaneous Lymphocyte Antigen. Acta Derm. Venereol. 2003, 83, 162–166. [Google Scholar] [CrossRef]
- Kantele, A.; Savilahti, E.; Tiimonen, H.; Iikkanen, K.; Autio, S.; Kantele, J.M. Cutaneous Lymphocyte Antigen Expression on Human Effector B Cells Depends on the Site and on the Nature of Antigen Encounter. Eur. J. Immunol. 2003, 33, 3275–3283. [Google Scholar] [CrossRef]
- Magro, C.M.; Dyrsen, M.E. Cutaneous Lymphocyte Antigen Expression in Benign and Neoplastic Cutaneous B- and T-Cell Lymphoid Infiltrates. J. Cutan. Pathol. 2008, 35, 1040–1049. [Google Scholar] [CrossRef]
- Campbell, J.J.; Clark, R.A.; Watanabe, R.; Kupper, T.S. Sezary Syndrome and Mycosis Fungoides Arise from Distinct T-Cell Subsets: A Biologic Rationale for Their Distinct Clinical Behaviors. Blood 2010, 116, 767–771. [Google Scholar] [CrossRef]
- Beyer, M.; Möbs, M.; Humme, D.; Sterry, W. Pathogenesis of Mycosis Fungoides. J. Dtsch. Dermatol. Ges. 2011, 9, 594–598. [Google Scholar] [CrossRef]
- Sokolowska-Wojdylo, M.; Wenzel, J.; Gaffal, E.; Lenz, J.; Speuser, P.; Erdmann, S.; Abuzahra, F.; Bowman, E.; Roszkiewicz, J.; Bieber, T.; et al. Circulating Clonal CLA(+) and CD4(+) T Cells in Sezary Syndrome Express the Skin-Homing Chemokine Receptors CCR4 and CCR10 as Well as the Lymph Node-Homing Chemokine Receptor CCR7. Br. J. Dermatol. 2005, 152, 258–264. [Google Scholar] [CrossRef]
- Kallinich, T.; Muche, J.M.; Qin, S.; Sterry, W.; Audring, H.; Kroczek, R.A. Chemokine Receptor Expression on Neoplastic and Reactive T Cells in the Skin at Different Stages of Mycosis Fungoides. J. Investig. Dermatol. 2003, 121, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Karenko, L.; Hahtola, S.; Päivinen, S.; Karhu, R.; Syrjä, S.; Kähkönen, M.; Nedoszytko, B.; Kytölä, S.; Zhou, Y.; Blazevic, V.; et al. Primary Cutaneous T-Cell Lymphomas Show a Deletion or Translocation Affecting NAV3, the Human UNC-53 Homologue. Cancer Res. 2005, 65, 8101–8110. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Yang, J.; Wenzel, A.T.; Ramachandran, A.; Lee, W.J.; Daniels, J.C.; Kim, J.; Martinez-Escala, E.; Amankulor, N.; Pro, B.; et al. Genomic Analysis of 220 CTCLs Identifies a Novel Recurrent Gain-of-Function Alteration in RLTPR (p.Q575E). Blood 2017, 130, 1430–1440. [Google Scholar] [CrossRef]
- de la Garza Bravo, M.M.; Patel, K.P.; Loghavi, S.; Curry, J.L.; Torres Cabala, C.A.; Cason, R.C.; Gangar, P.; Prieto, V.G.; Medeiros, L.J.; Duvic, M.; et al. Shared Clonality in Distinctive Lesions of Lymphomatoid Papulosis and Mycosis Fungoides Occurring in the Same Patients Suggests a Common Origin. Hum. Pathol. 2015, 46, 558–569. [Google Scholar] [CrossRef]
- Damsky, W.E.; Choi, J. Genetics of Cutaneous T Cell Lymphoma: From Bench to Bedside. Curr. Treat. Options Oncol. 2016, 17, 33. [Google Scholar] [CrossRef]
- da Silva Almeida, A.C.; Abate, F.; Khiabanian, H.; Martinez-Escala, E.; Guitart, J.; Tensen, C.P.; Vermeer, M.H.; Rabadan, R.; Ferrando, A.; Palomero, T. The Mutational Landscape of Cutaneous T Cell Lymphoma and Sézary Syndrome. Nat. Genet. 2015, 47, 1465–1470. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Goh, G.; Walradt, T.; Hong, B.S.; Bunick, C.G.; Chen, K.; Bjornson, R.D.; Maman, Y.; Wang, T.; Tordoff, J.; et al. Genomic Landscape of Cutaneous T Cell Lymphoma. Nat. Genet. 2015, 47, 1011–1019. [Google Scholar] [CrossRef]
- McGirt, L.Y.; Jia, P.; Baerenwald, D.A.; Duszynski, R.J.; Dahlman, K.B.; Zic, J.A.; Zwerner, J.P.; Hucks, D.; Dave, U.; Zhao, Z.; et al. Whole-Genome Sequencing Reveals Oncogenic Mutations in Mycosis Fungoides. Blood 2015, 126, 508–519. [Google Scholar] [CrossRef] [Green Version]
- Rendón-Serna, N.; Correa-Londoño, L.A.; Velásquez-Lopera, M.M.; Bermudez-Muñoz, M. Cell Signaling in Cutaneous T-Cell Lymphoma Microenvironment: Promising Targets for Molecular-Specific Treatment. Int. J. Dermatol. 2021, 60, 1462–1480. [Google Scholar] [CrossRef]
- Bos, J.D.; de Boer, O.J.; Tibosch, E.; Das, P.K.; Pals, S.T. Skin-Homing T Lymphocytes: Detection of Cutaneous Lymphocyte-Associated Antigen (CLA) by HECA-452 in Normal Human Skin. Arch. Dermatol. Res. 1993, 285, 179–183. [Google Scholar] [CrossRef]
- Pals, S.T.; Horst, E.; Scheper, R.J.; Meijer, C.J. Mechanisms of Human Lymphocyte Migration and Their Role in the Pathogenesis of Disease. Immunol. Rev. 1989, 108, 111–133. [Google Scholar] [CrossRef]
- Aloisi, F.; Pujol-Borrell, R. Lymphoid Neogenesis in Chronic Inflammatory Diseases. Nat. Rev. Immunol. 2006, 6, 205–217. [Google Scholar] [CrossRef]
- Mori, M.; Manuelli, C.; Pimpinelli, N.; Bianchi, B.; Orlando, C.; Mavilia, C.; Cappugi, P.; Maggi, E.; Giannotti, B.; Santucci, M. BCA-1, A B-Cell Chemoattractant Signal, Is Constantly Expressed in Cutaneous Lymphoproliferative B-Cell Disorders. Eur. J. Cancer 2003, 39, 1625–1631. [Google Scholar] [CrossRef]
- Gunn, M.D.; Ngo, V.N.; Ansel, K.M.; Ekland, E.H.; Cyster, J.G.; Williams, L.T. A B-Cell-Homing Chemokine Made in Lymphoid Follicles Activates Burkitt’s Lymphoma Receptor-1. Nature 1998, 391, 799–803. [Google Scholar] [CrossRef]
- Förster, R.; Mattis, A.E.; Kremmer, E.; Wolf, E.; Brem, G.; Lipp, M. A Putative Chemokine Receptor, BLR1, Directs B Cell Migration to Defined Lymphoid Organs and Specific Anatomic Compartments of the Spleen. Cell 1996, 87, 1037–1047. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.D.C.; Ansel, K.M.; Low, C.; Lesley, R.; Tamamura, H.; Fujii, N.; Cyster, J.G. Germinal Center Dark and Light Zone Organization Is Mediated by CXCR4 and CXCR5. Nat. Immunol. 2004, 5, 943–952. [Google Scholar] [CrossRef]
- Hoefnagel, J.J.; Vermeer, M.H.; Jansen, P.M.; Fleuren, G.J.; Meijer, C.J.L.M.; Willemze, R. Bcl-2, Bcl-6 and CD10 Expression in Cutaneous B-Cell Lymphoma: Further Support for a Follicle Centre Cell Origin and Differential Diagnostic Significance. Br. J. Dermatol. 2003, 149, 1183–1191. [Google Scholar] [CrossRef]
- Lawnicki, L.C.; Weisenburger, D.D.; Aoun, P.; Chan, W.C.; Wickert, R.S.; Greiner, T.C. The t(14;18) and Bcl-2 Expression Are Present in a Subset of Primary Cutaneous Follicular Lymphoma: Association with Lower Grade. Am. J. Clin. Pathol. 2002, 118, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Servitje, O.; Climent, F.; Colomo, L.; Ruiz, N.; García-Herrera, A.; Gallardo, F.; Mercadal, S.; Pomares, H.; Muniesa, C.; Martin-Callizo, C.; et al. Primary Cutaneous vs Secondary Cutaneous Follicular Lymphomas: A Comparative Study Focused on BCL2, CD10, and t(14;18) Expression. J. Cutan. Pathol. 2019, 46, 182–189. [Google Scholar] [CrossRef]
- Abdul-Wahab, A.; Tang, S.-Y.; Robson, A.; Morris, S.; Agar, N.; Wain, E.M.; Child, F.; Scarisbrick, J.; Neat, M.; Whittaker, S. Chromosomal Anomalies in Primary Cutaneous Follicle Center Cell Lymphoma Do Not Portend a Poor Prognosis. J. Am. Acad. Dermatol. 2014, 70, 1010–1020. [Google Scholar] [CrossRef]
- Phan, R.T.; Dalla-Favera, R. The BCL6 Proto-Oncogene Suppresses P53 Expression in Germinal-Centre B Cells. Nature 2004, 432, 635–639. [Google Scholar] [CrossRef]
- Basso, K.; Dalla-Favera, R. Germinal Centres and B Cell Lymphomagenesis. Nat. Rev. Immunol. 2015, 15, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Capello, D.; Vitolo, U.; Pasqualucci, L.; Quattrone, S.; Migliaretti, G.; Fassone, L.; Ariatti, C.; Vivenza, D.; Gloghini, A.; Pastore, C.; et al. Distribution and Pattern of BCL-6 Mutations throughout the Spectrum of B-Cell Neoplasia. Blood 2000, 95, 651–659. [Google Scholar] [PubMed]
- Ye, B.H.; Chaganti, S.; Chang, C.C.; Niu, H.; Corradini, P.; Chaganti, R.S.; Dalla-Favera, R. Chromosomal Translocations Cause Deregulated BCL6 Expression by Promoter Substitution in B Cell Lymphoma. EMBO J. 1995, 14, 6209–6217. [Google Scholar] [CrossRef]
- Ci, W.; Polo, J.M.; Cerchietti, L.; Shaknovich, R.; Wang, L.; Yang, S.N.; Ye, K.; Farinha, P.; Horsman, D.E.; Gascoyne, R.D.; et al. The BCL6 Transcriptional Program Features Repression of Multiple Oncogenes in Primary B Cells and Is Deregulated in DLBCL. Blood 2009, 113, 5536–5548. [Google Scholar] [CrossRef] [Green Version]
- Cattoretti, G.; Mandelbaum, J.; Lee, N.; Chaves, A.H.; Mahler, A.M.; Chadburn, A.; Dalla-Favera, R.; Pasqualucci, L.; MacLennan, A.J. Targeted Disruption of the S1P2 Sphingosine 1-Phosphate Receptor Gene Leads to Diffuse Large B-Cell Lymphoma Formation. Cancer Res. 2009, 69, 8686–8692. [Google Scholar] [CrossRef] [Green Version]
- Muppidi, J.R.; Schmitz, R.; Green, J.A.; Xiao, W.; Larsen, A.B.; Braun, S.E.; An, J.; Xu, Y.; Rosenwald, A.; Ott, G.; et al. Loss of Signalling via Gα13 in Germinal Centre B-Cell-Derived Lymphoma. Nature 2014, 516, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Challa-Malladi, M.; Lieu, Y.K.; Califano, O.; Holmes, A.B.; Bhagat, G.; Murty, V.V.; Dominguez-Sola, D.; Pasqualucci, L.; Dalla-Favera, R. Combined Genetic Inactivation of Β2-Microglobulin and CD58 Reveals Frequent Escape from Immune Recognition in Diffuse Large B Cell Lymphoma. Cancer Cell 2011, 20, 728–740. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.A.; Louissaint, A.; Wenzel, A.; Yang, J.; Martinez-Escala, M.E.; Moy, A.P.; Morgan, E.A.; Paxton, C.N.; Hong, B.; Andersen, E.F.; et al. Genomic Analyses Identify Recurrent Alterations in Immune Evasion Genes in Diffuse Large B-Cell Lymphoma, Leg Type. J. Investig. Dermatol. 2018, 138, 2365–2376. [Google Scholar] [CrossRef] [Green Version]
- Ginaldi, L.; De Martinis, M.; Matutes, E.; Farahat, N.; Morilla, R.; Dyer, M.J.; Catovsky, D. Levels of Expression of CD52 in Normal and Leukemic B and T Cells: Correlation with in Vivo Therapeutic Responses to Campath-1H. Leuk. Res. 1998, 22, 185–191. [Google Scholar] [CrossRef]
- Watanabe, R.; Teague, J.E.; Fisher, D.C.; Kupper, T.S.; Clark, R.A. Alemtuzumab Therapy for Leukemic Cutaneous T-Cell Lymphoma: Diffuse Erythema as a Positive Predictor of Complete Remission. JAMA Dermatol. 2014, 150, 776–779. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Almazan, T.H.; Hong, E.K.; Khodadoust, M.S.; Arai, S.; Weng, W.-K.; Kim, Y.H. Potential Association of Anti-CCR4 Antibody Mogamulizumab and Graft-vs-Host Disease in Patients With Mycosis Fungoides and Sézary Syndrome. JAMA Dermatol. 2018, 154, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, R.A. Mogamulizumab: 2 Birds, 1 Stone. Blood 2015, 125, 1847–1848. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.J. Brentuximab Vedotin: A Review in CD30-Positive Hodgkin Lymphoma. Drugs 2017, 77, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.M.R.; Mehta, A. Diagnostic, Prognostic and Therapeutic Role of CD30 in Lymphoma. Expert Rev. Hematol. 2017, 10, 29–37. [Google Scholar] [CrossRef]
- Kampa, F.; Mitteldorf, C. A Review of CD30 Expression in Cutaneous Neoplasms. J. Cutan. Pathol. 2021, 48, 495–510. [Google Scholar] [CrossRef]
- Kim, Y.H.; Tavallaee, M.; Sundram, U.; Salva, K.A.; Wood, G.S.; Li, S.; Rozati, S.; Nagpal, S.; Krathen, M.; Reddy, S.; et al. Phase II Investigator-Initiated Study of Brentuximab Vedotin in Mycosis Fungoides and Sézary Syndrome With Variable CD30 Expression Level: A Multi-Institution Collaborative Project. J. Clin. Oncol. 2015, 33, 3750–3758. [Google Scholar] [CrossRef]
- Duvic, M.; Tetzlaff, M.T.; Gangar, P.; Clos, A.L.; Sui, D.; Talpur, R. Results of a Phase II Trial of Brentuximab Vedotin for CD30+ Cutaneous T-Cell Lymphoma and Lymphomatoid Papulosis. J. Clin. Oncol. 2015, 33, 3759–3765. [Google Scholar] [CrossRef] [Green Version]
- Prince, H.M.; Kim, Y.H.; Horwitz, S.M.; Dummer, R.; Scarisbrick, J.; Quaglino, P.; Zinzani, P.L.; Wolter, P.; Sanches, J.A.; Ortiz-Romero, P.L.; et al. Brentuximab Vedotin or Physician’s Choice in CD30-Positive Cutaneous T-Cell Lymphoma (ALCANZA): An International, Open-Label, Randomised, Phase 3, Multicentre Trial. Lancet 2017, 390, 555–566. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Scarisbrick, J.J.; Dummer, R.; Whittaker, S.; Duvic, M.; Kim, Y.H.; Quaglino, P.; Zinzani, P.L.; Bechter, O.; Eradat, H.; et al. Randomized Phase 3 ALCANZA Study of Brentuximab Vedotin vs Physician’s Choice in Cutaneous T-Cell Lymphoma: Final Data. Blood Adv. 2021, 5, 5098–5106. [Google Scholar] [CrossRef]
- Van Der Weyden, C.; Dickinson, M.; Whisstock, J.; Prince, H.M. Brentuximab Vedotin in T-Cell Lymphoma. Expert Rev. Hematol. 2019, 12, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Velasco, R.; Domingo-Domenech, E.; Sureda, A. Brentuximab-Induced Peripheral Neurotoxicity: A Multidisciplinary Approach to Manage an Emerging Challenge in Hodgkin Lymphoma Therapy. Cancers 2021, 13, 6125. [Google Scholar] [CrossRef]
- Chen, R.; Hou, J.; Newman, E.; Kim, Y.; Donohue, C.; Liu, X.; Thomas, S.H.; Forman, S.J.; Kane, S.E. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Mol. Cancer Ther. 2015, 14, 1376–1384. [Google Scholar] [CrossRef] [Green Version]
- Liu-Kreyche, P.; Shen, H.; Marino, A.M.; Iyer, R.A.; Humphreys, W.G.; Lai, Y. Lysosomal P-Gp-MDR1 Confers Drug Resistance of Brentuximab Vedotin and Its Cytotoxic Payload Monomethyl Auristatin E in Tumor Cells. Front. Pharmacol. 2019, 10, 749. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Herrera, A.F.; Hou, J.; Chen, L.; Wu, J.; Guo, Y.; Synold, T.W.; Ngo, V.N.; Puverel, S.; Mei, M.; et al. Inhibition of MDR1 Overcomes Resistance to Brentuximab Vedotin in Hodgkin Lymphoma. Clin. Cancer Res. 2020, 26, 1034–1044. [Google Scholar] [CrossRef]
- Hasanali, Z.S.; Saroya, B.S.; Stuart, A.; Shimko, S.; Evans, J.; Vinod Shah, M.; Sharma, K.; Leshchenko, V.V.; Parekh, S.; Loughran, T.P.; et al. Epigenetic Therapy Overcomes Treatment Resistance in T Cell Prolymphocytic Leukemia. Sci. Transl. Med. 2015, 7, 293ra102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barta, S.K.; Feldman, T.A.; DeSimone, J.A.; Kim, E.; Devajaran, K.; Yang, Y.; Fung, H.C.; Mahvish, T.; Nakhoda, S.; Khan, N. A Phase I Trial Assessing the Feasibility of Romidepsin Combined with Brentuximab Vedotin for Patients Requiring Systemic Therapy for Cutaneous T-Cell Lymphoma. Blood 2022, 140, 6543–6544. [Google Scholar] [CrossRef]
- Amatore, F.; Ortonne, N.; Lopez, M.; Orlanducci, F.; Castellano, R.; Ingen-Housz-Oro, S.; De Croos, A.; Salvado, C.; Gorvel, L.; Goubard, A.; et al. ICOS Is Widely Expressed in Cutaneous T-Cell Lymphoma, and Its Targeting Promotes Potent Killing of Malignant Cells. Blood Adv. 2020, 4, 5203–5214. [Google Scholar] [CrossRef]
- Choudhary, R.K.; Jones, R.J.; Kuiatse, I.; Wang, H.; Vega, F.; Bouska, A.C.; Lone, W.G.; Iqbal, J.; Orlowski, R.Z. A Novel Antibody Drug Conjugate (ADC) Targeting Cell Surface Heat Shock Protein 70 (CsHSP70) Is Active Against Pre-Clinical Models of Peripheral T-Cell Lymphoma (PTCL). Blood 2021, 138, 870. [Google Scholar] [CrossRef]
- Grossbard, M.L.; Press, O.W.; Appelbaum, F.R.; Bernstein, I.D.; Nadler, L.M. Monoclonal Antibody-Based Therapies of Leukemia and Lymphoma. Blood 1992, 80, 863–878. [Google Scholar] [CrossRef] [Green Version]
- Tadiotto Cicogna, G.; Ferranti, M.; Lazzarotto, A.; Alaibac, M. Biological Approaches to Aggressive Cutaneous B-Cell Lymphomas. Front. Oncol. 2019, 9, 1238. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Kufer, P.; Bargou, R. BiTE: Teaching Antibodies to Engage T-Cells for Cancer Therapy. Curr. Opin. Mol. Ther. 2009, 11, 22–30. [Google Scholar]
- Yu, J.; Wang, W.; Huang, H. Efficacy and Safety of Bispecific T-Cell Engager (BiTE) Antibody Blinatumomab for the Treatment of Relapsed/Refractory Acute Lymphoblastic Leukemia and Non-Hodgkin’s Lymphoma: A Systemic Review and Meta-Analysis. Hematology 2019, 24, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Raimondo, C.; Abdulla, F.R.; Zain, J.; Querfeld, C.; Rosen, S.T. Rituximab, Lenalidomide and Pembrolizumab in Refractory Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type. Br. J. Haematol. 2019, 187, e79–e82. [Google Scholar] [CrossRef]
- Gupta, E.; Accurso, J.; Sluzevich, J.; Menke, D.M.; Tun, H.W. Excellent Outcome of Immunomodulation or Bruton’s Tyrosine Kinase Inhibition in Highly Refractory Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type. Rare Tumors 2015, 7, 6067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancebo, S.E.; Smith, J.R.; Intlekofer, A.M.; Zelenetz, A.D.; Myskowski, P.L. Treatment Response of Cutaneous Mantle Cell Lymphoma to Ibrutinib and Radiotherapy. Clin. Lymphoma Myeloma Leuk. 2015, 15, e113–e115. [Google Scholar] [CrossRef]
- Mondello, P.; Cuzzocrea, S.; Navarra, M.; Mian, M. 90 Y-Ibritumomab Tiuxetan: A Nearly Forgotten Opportunityr. Oncotarget 2016, 7, 7597–7609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morschhauser, F.; Illidge, T.; Huglo, D.; Martinelli, G.; Paganelli, G.; Zinzani, P.L.; Rule, S.; Liberati, A.M.; Milpied, N.; Hess, G.; et al. Efficacy and Safety of Yttrium-90 Ibritumomab Tiuxetan in Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma Not Appropriate for Autologous Stem-Cell Transplantation. Blood 2007, 110, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, M.S.; Fig, L.M.; Zasadny, K.R.; Koral, K.F.; DelRosario, R.B.; Francis, I.R.; Hanson, C.A.; Normolle, D.P.; Mudgett, E.; Liu, C.P. Imaging, Dosimetry, and Radioimmunotherapy with Iodine 131-Labeled Anti-CD37 Antibody in B-Cell Lymphoma. J. Clin. Oncol. 1992, 10, 1696–1711. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Rossi, G.; Franceschetti, S.; Botto, B.; Di Rocco, A.; Cabras, M.G.; Petti, M.C.; Stefoni, V.; Broccoli, A.; Fanti, S.; et al. Phase II Trial of Short-Course R-CHOP Followed by 90Y-Ibritumomab Tiuxetan in Previously Untreated High-Risk Elderly Diffuse Large B-Cell Lymphoma Patients. Clin. Cancer Res. 2010, 16, 3998–4004. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Griffiths, G.L.; Choyke, P.L. Near-Infrared Photoimmunotherapy: Photoactivatable Antibody-Drug Conjugates (ADCs). Bioconjug. Chem. 2020, 31, 28–36. [Google Scholar] [CrossRef]
- Kato, T.; Wakiyama, H.; Furusawa, A.; Choyke, P.L.; Kobayashi, H. Near Infrared Photoimmunotherapy; A Review of Targets for Cancer Therapy. Cancers 2021, 13, 2535. [Google Scholar] [CrossRef] [PubMed]
- Silic-Benussi, M.; Saponeri, A.; Michelotto, A.; Russo, I.; Colombo, A.; Pelizzo, M.G.; Ciminale, V.; Alaibac, M. Near Infrared Photoimmunotherapy Targeting the Cutaneous Lymphocyte Antigen for Mycosis Fungoides. Expert Opin. Biol. Ther. 2021, 21, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Nagaya, T.; Nakamura, Y.; Sato, K.; Harada, T.; Choyke, P.L.; Kobayashi, H. Near Infrared Photoimmunotherapy of B-Cell Lymphoma. Mol. Oncol. 2016, 10, 1404–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Clinical Behavior | CTCL | CBCL |
|---|---|---|
| Indolent | MF PCALCL LyP Subcutaneous panniculitis-like T cell lymphoma Rare subtypes * | PCMZL PCFCL EBV+ mucocutaneous ulcer |
| Intermediate | PCALCL | PCDLBCL, LT PCDLBCL, other Primary cutaneous intravascular large B cell lymphoma |
| Aggressive | SS Rare subtypes ** | PCDLBCL, LT |
| Pathway | Drug | Target | Evidence |
|---|---|---|---|
| MAPK | Sorafenib | Multikinase (B-RAF, VEGFR) | Pilot study |
| PI3K/Akt/mTOR | Duvelisib | PI3K-delta/gamma | Phase I |
| Everolimus | mTOR | Pilot study | |
| JAK/STAT | Ruxolitinib | JAK1/2 | In vitro |
| Tofacitinib | JAK1/2/3 | In vitro | |
| T cell | Nivolumab | PD-1 | Phase I |
| Pembrolizumab | PD-1 | Phase II | |
| Ipilimumab | CTLA4 | Case report | |
| Mogamulizumab | CCR4 | Approved | |
| Alemtuzumab | CD52 | (withdrawn) | |
| Resimmune | CD3 | Phase II |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sernicola, A.; Ciolfi, C.; Miceli, P.; Alaibac, M. Cutaneous Lymphoma and Antibody-Directed Therapies. Antibodies 2023, 12, 21. https://doi.org/10.3390/antib12010021
Sernicola A, Ciolfi C, Miceli P, Alaibac M. Cutaneous Lymphoma and Antibody-Directed Therapies. Antibodies. 2023; 12(1):21. https://doi.org/10.3390/antib12010021
Chicago/Turabian StyleSernicola, Alvise, Christian Ciolfi, Paola Miceli, and Mauro Alaibac. 2023. "Cutaneous Lymphoma and Antibody-Directed Therapies" Antibodies 12, no. 1: 21. https://doi.org/10.3390/antib12010021