
Aerosols in Northern Morocco-2: Chemical Characterization and PMF Source Apportionment of Ambient PM2.5
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
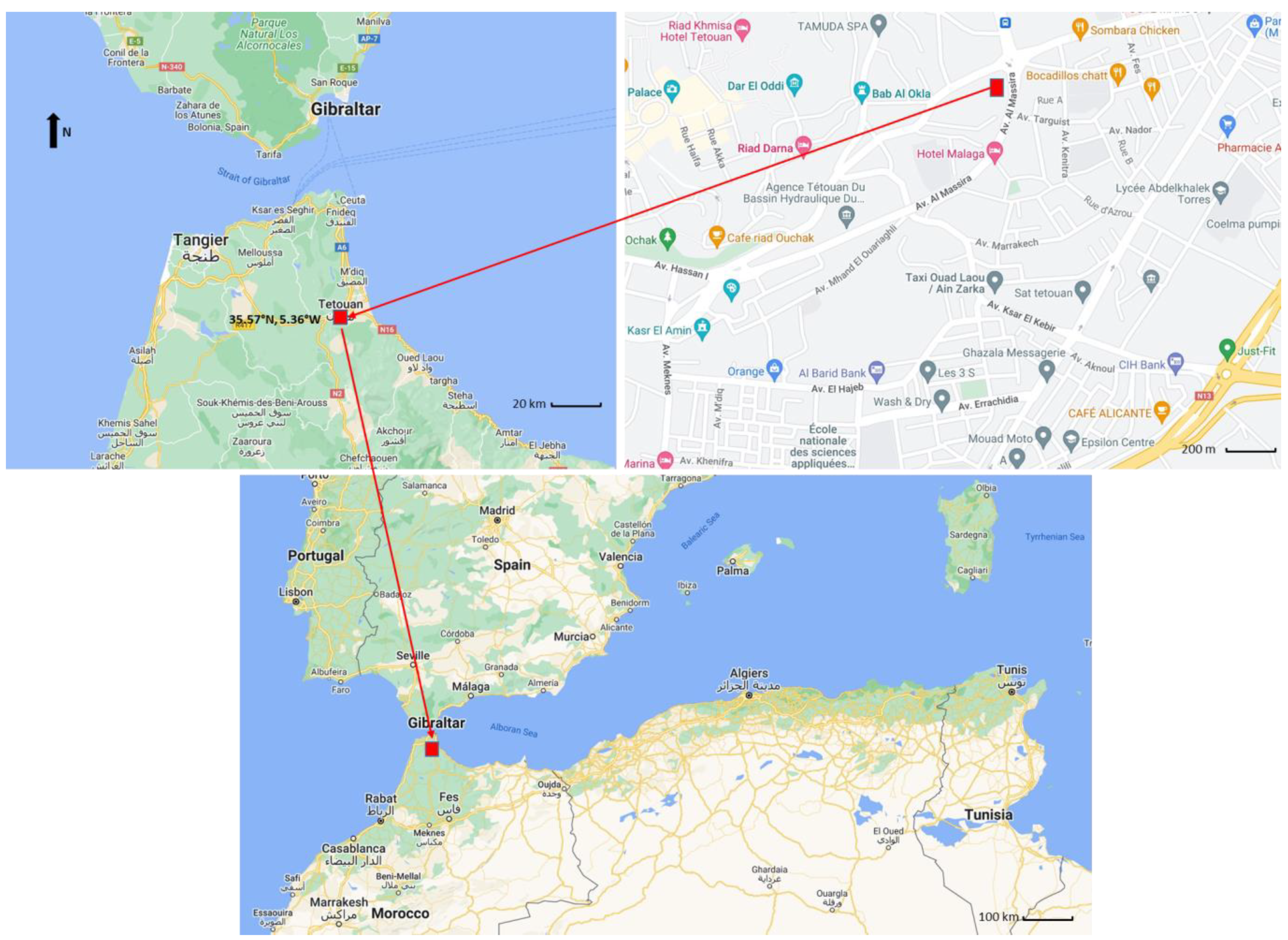
2.1. Sampling and Aerosol Chemical Analyses
2.2. Aerosol Chemical Closure Methodology
- Sea salt component estimation:
- Inorganic ions:
- Carbonaceous fractions, BC and OC:
- POM calculation:
- Dust calculation:
2.3. Positive Matrix Factorization (PMF) Model
2.4. Settings and Diagnostics for a PMF Optimum Solution
3. Results
3.1. Aerosol Chemical Mass Closure
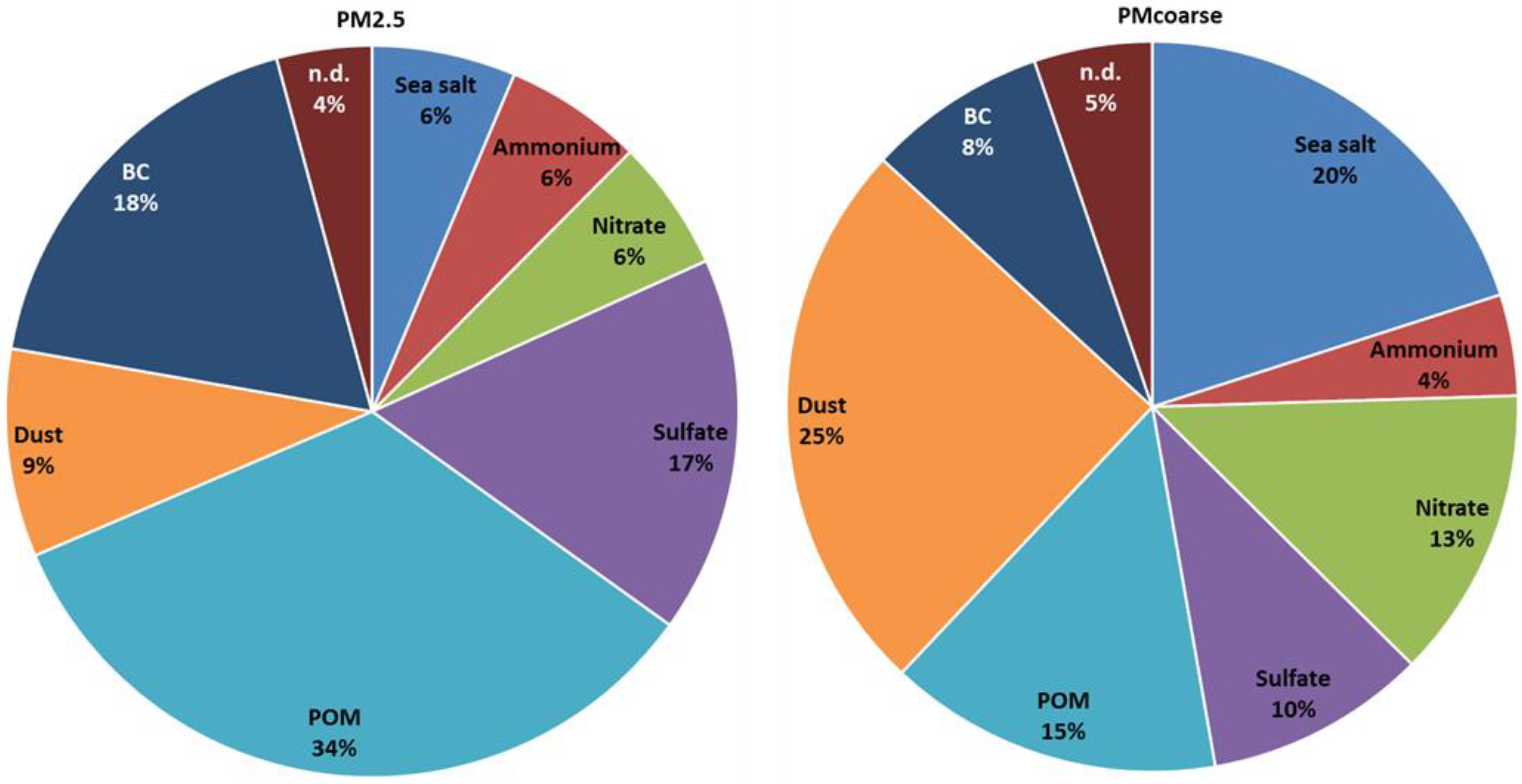
3.1.1. Overview of PM2.5 and Its Chemical Components
3.1.2. Mass Closure and Differences among PM Size Fractions
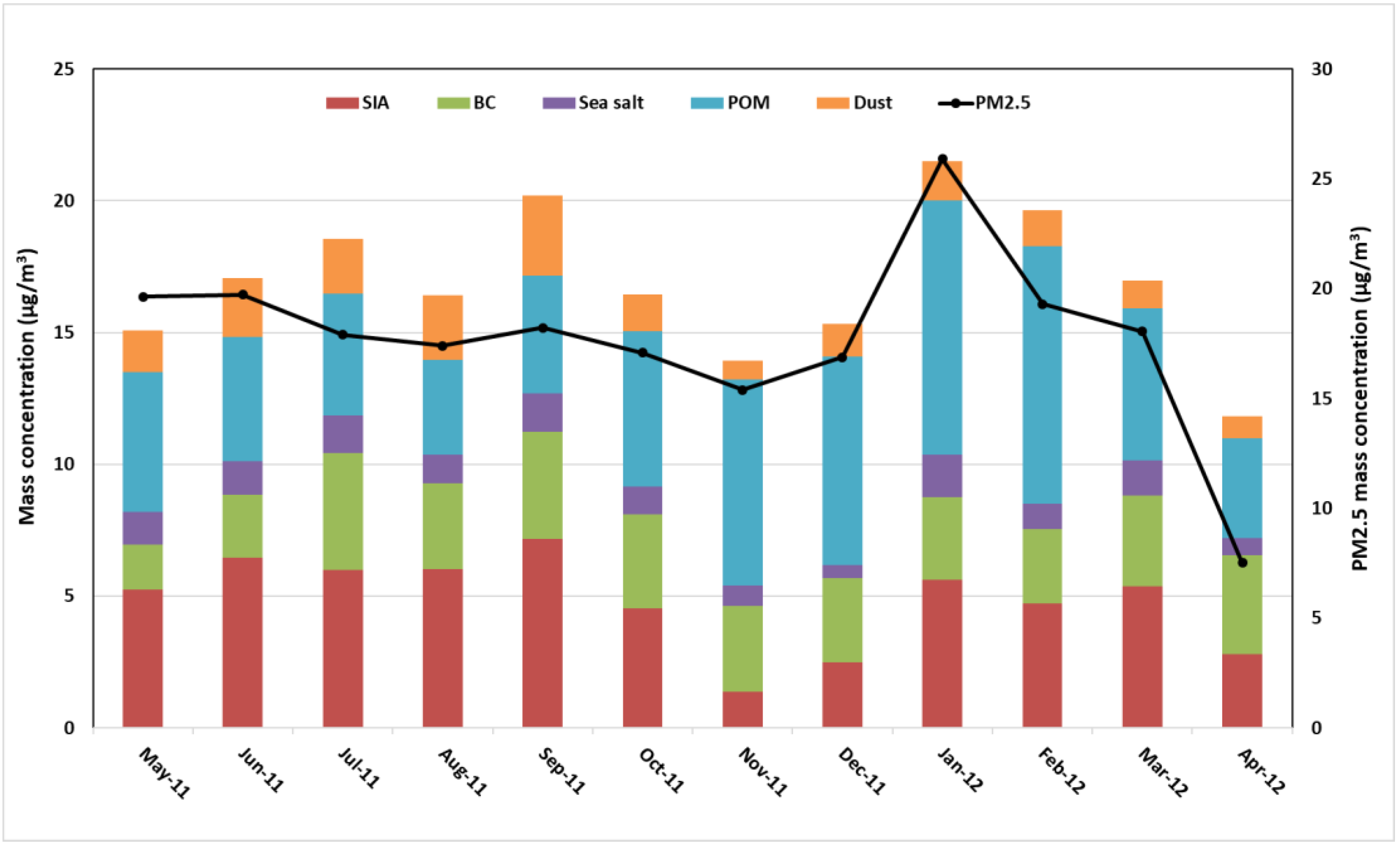
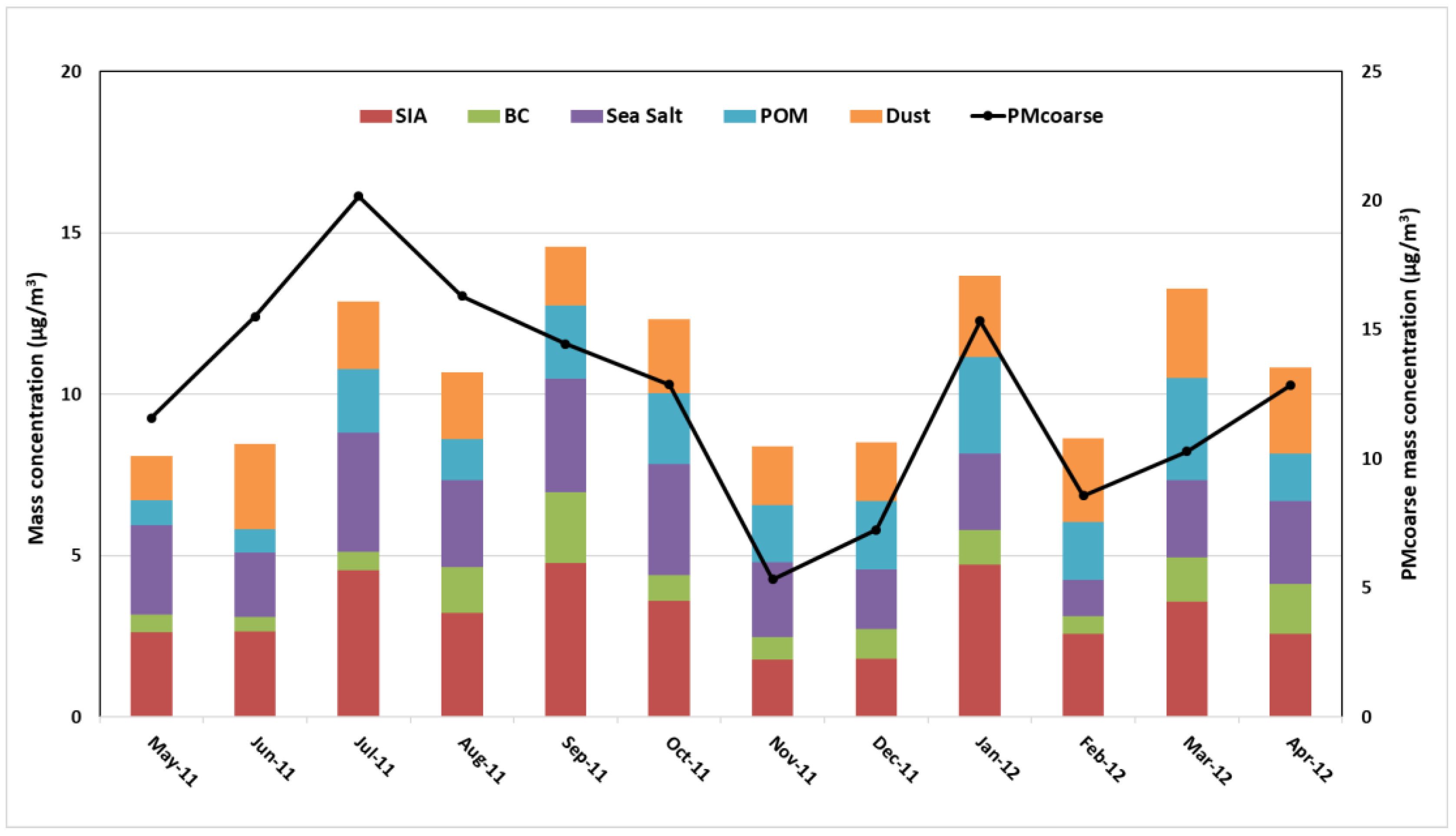
3.1.3. Seasonal Variability of the Reconstructed PM2.5 and PMcoarse
3.1.4. Non-Determined Mass
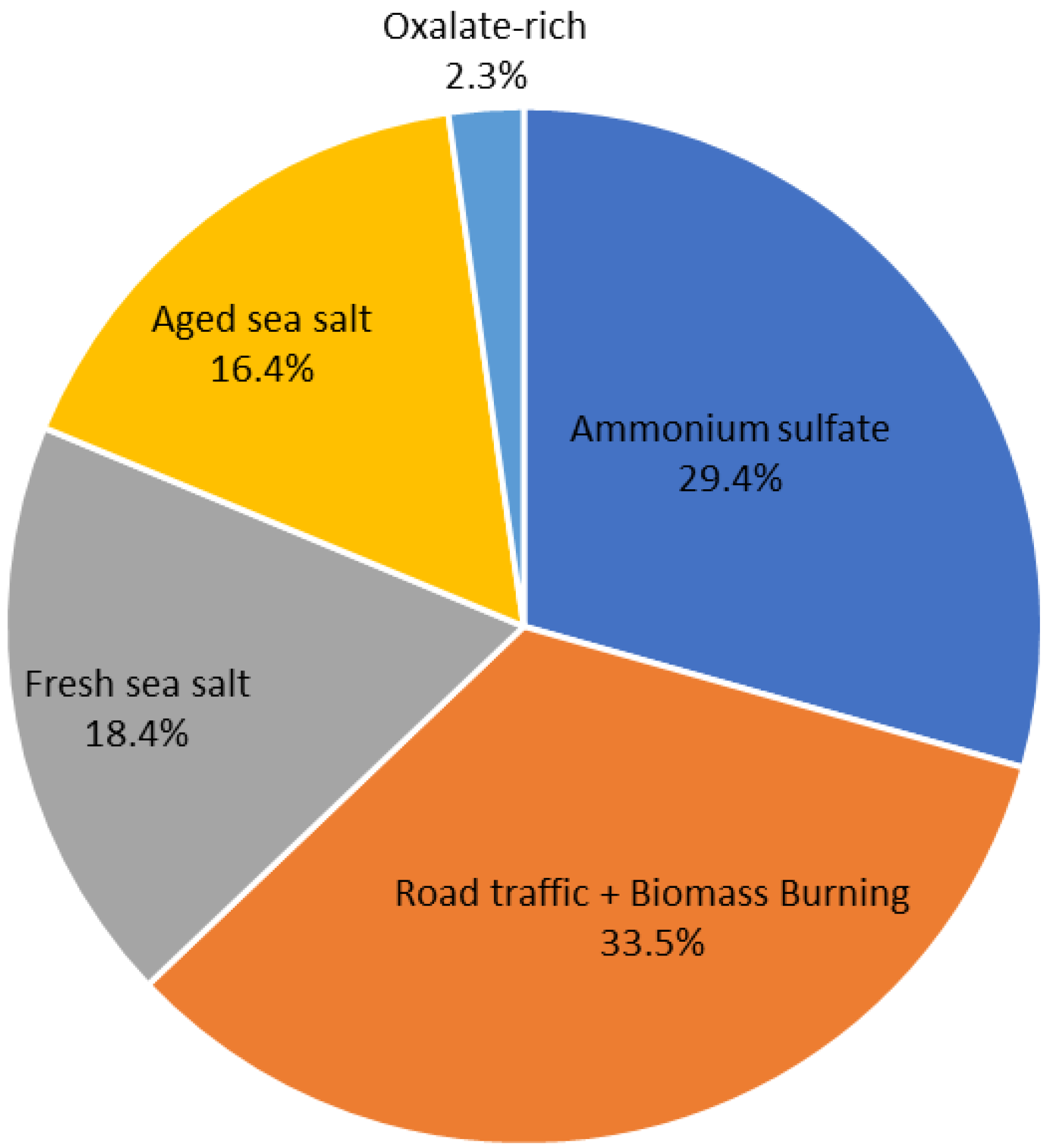
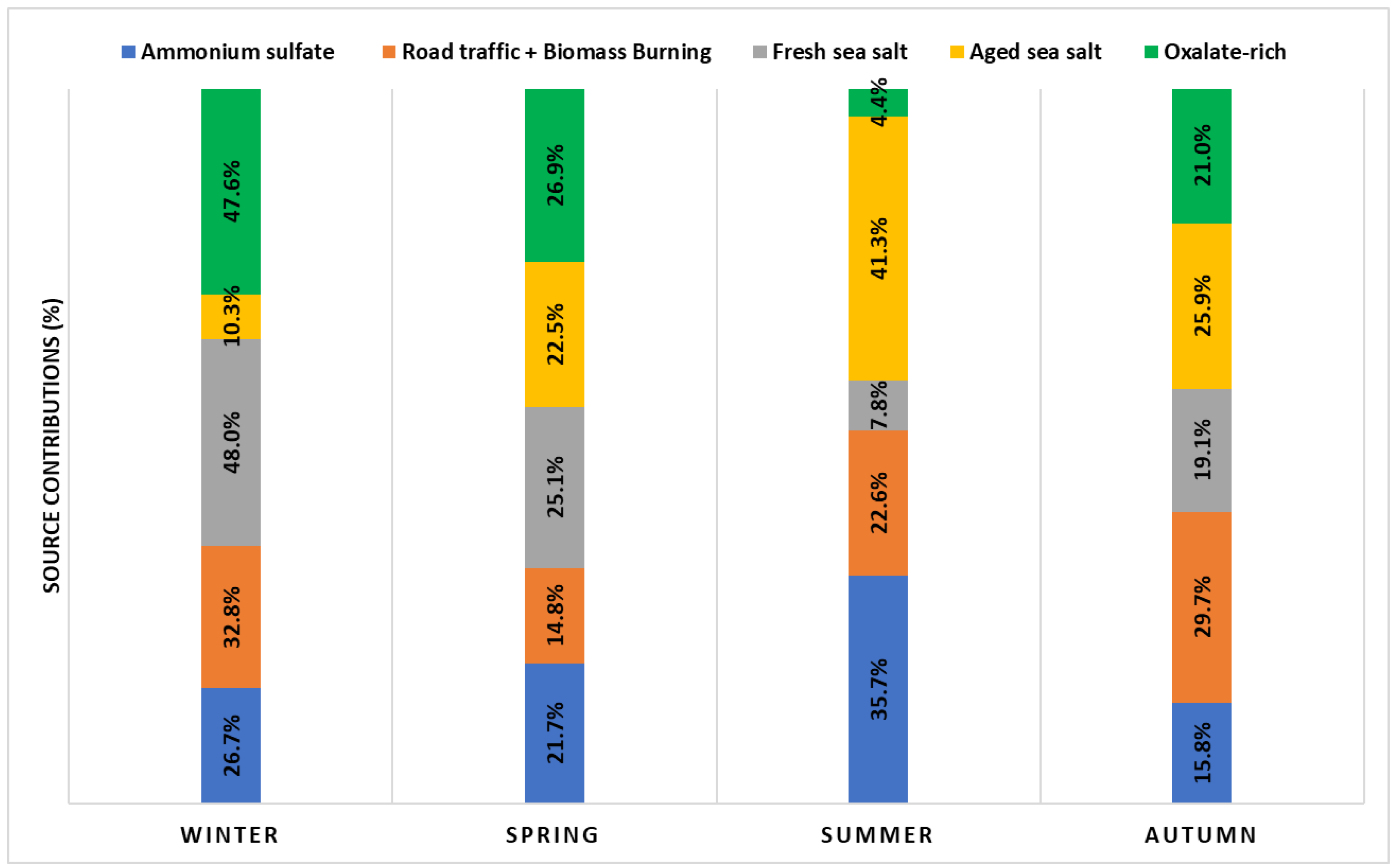
3.2. Source Apportionment of PM2.5: Source Profiles and Seasonal Variation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boucher, O.; Randall, D.; Artaxo, P.; Bretherton, C.; Feingold, G.; Forster, P.; Kerminen, V.-M.; Kondo, Y.; Liao, H.; Lohmann, U.; et al. Clouds and Aerosols. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013. [Google Scholar]
- Terzi, E.; Argyropoulos, G.; Bougatioti, A.; Mihalopoulos, N.; Nikolaou, K.; Samara, C. Chemical composition and mass closure of ambient PM10 at urban sites. Atmos. Environ. 2010, 44, 2231–2239. [Google Scholar] [CrossRef]
- Niessner, R. Chemical characterization of aerosols. Fresenius J. Anal. Chem. 1990, 337, 565–576. [Google Scholar] [CrossRef]
- Pey, J.; Querol, X.; Alastuey, A. Variations of levels and composition of PM10 and PM2.5 at an insular site in the Western Mediterranean. Atmos. Res. 2009, 94, 285–299. [Google Scholar] [CrossRef]
- Rogula-Kozłowska, W.; Klejnowski, K.; Rogula-Kopiec, P.; Ośródka, L.; Krajny, E.; Błaszczak, B.; Mathews, B. Spatial and seasonal variability of the mass concentration and chemical composition of PM2.5 in Poland. Air Qual. Atmos. Health 2013, 7, 41–58. [Google Scholar] [CrossRef] [Green Version]
- Perrino, C.; Catrambone, M.; Dalla Torre, S.; Rantica, E.; Sargolini, T.; Canepari, S. Seasonal variations in the chemical composition of particulate matter: A case study in the Po Valley. Part I: Macro-components and mass closure. Environ. Sci. Pollut. Res. 2013, 21, 3999–4009. [Google Scholar] [CrossRef]
- Samara, C.; Kantiranis, N.; Kollias, P.S.; Planou, S.; Kouras, A.; Besis, A.; Manoli, E.; Voutsa, D. Spatial and seasonal variations of the chemical, mineralogical and morphological features of quasi-ultrafine particles (PM0.49) at urban sites. Sci. Total Environ. 2016, 553, 392–403. [Google Scholar] [CrossRef]
- Almeida, S.M.; Pio, C.; Freitas, M.C.; Reis, M.A.; Trancoso, M.A. Approaching PM2.5 and PM2.5—10 source apportionment by mass balance analysis, principal component analysis and particle size distribution. Sci. Total Environ. 2006, 368, 663–674. [Google Scholar] [CrossRef]
- Bardouki, H.; Liakakou, H.; Economou, C.; Sciare, J.; Smolík, J.; Zdimal, V.; Eleftheriadis, K.; Lazaridis, M.; Dye, C.; Mihalopoulos, N. Chemical composition of size-resolved atmospheric aerosols in the eastern Mediterranean during summer and winter. Atmos. Environ. 2003, 37, 195–208. [Google Scholar] [CrossRef]
- Clemente, Á.; Yubero, E.; Galindo, N.; Crespo, J.; Nicolás, J.F.; Santacatalina, M.; Carratalá, A. Quantification of the impact of port activities on PM10 levels at the port-city boundary of a mediterranean city. J. Environ. Manag. 2020, 281, 111842. [Google Scholar] [CrossRef]
- Lemou, A.; Rabhi, L.; Merabet, H.; Ladji, R.; Nicolas, J.B.; Bonnaire, N.; Mustapha, M.A.; Dilmi, R.; Sciare, J.; Mihalopoulos, N.; et al. Chemical characterization of fine particles (PM2.5) at a coastal site in the South Western Mediterranean during the ChArMex experiment. Environ. Sci. Pollut. Res. 2020, 27, 20427–20445. [Google Scholar] [CrossRef]
- Perrino, C.; Catrambone, M.; Farao, C.; Canepari, S. Assessing the contribution of water to the mass closure of PM10. Atmos. Environ. 2016, 140, 555–564. [Google Scholar] [CrossRef]
- Sciare, J.; Cachier, H.; Oikonomou, K.; Ausset, P.; Sarda-Estève, R.; Mihalopoulos, N. Characterization of carbonaceous aerosols during the MINOS campaign in Crete, July–August 2001: A multi-analytical approach. Atmos. Chem. Phys. 2003, 3, 1743–1757. [Google Scholar] [CrossRef] [Green Version]
- Chow, J.C.; Lowenthal, D.H.; Chen, L.A.; Wang, X.; Watson, J.G. Mass reconstruction methods for PM2.5: A review. Air Qual. Atmos. Health 2015, 8, 243–263. [Google Scholar] [CrossRef] [Green Version]
- Masiol, M.; Squizzato, S.; Formenton, G.; Khan, M.B.; Hopke, P.K.; Nenes, A.; Pandis, S.N.; Tositti, L.; Benetello, F.; Visin, F.; et al. Hybrid multiple-site mass closure and source apportionment of PM2.5 and aerosol acidity at major cities in the Po Valley. Sci. Total Environ. 2020, 704, 135287. [Google Scholar] [CrossRef]
- Guinot, B.; Cachier, H.; Oikonomou, K. Geochemical perspectives from a new aerosol chemical mass closure. Atmos. Chem. Phys. 2006, 7, 1657–1670. [Google Scholar] [CrossRef] [Green Version]
- Naila, S.; Shahida, W. Source apportionment using reconstructed mass calculations. J. Environ. Sci. Health Part A Toxic/Hazard. Subst. Environ. Eng. 2014, 49, 463–477. [Google Scholar] [CrossRef]
- Belis, C.A.; Favez, O.; Mircea, M.; Diapouli, E.; Manousakas, M.-I.; Vratolis, S.; Gilardoni, S.; Paglione, M.; Decesari, S.; Mocnik, G.; et al. European Guide on Air Pollution Source Apportionment with Receptor Models—Revised Version 2019; EUR 29816 EN; Publications Office of the European Union: Luxembourg, 2019. [Google Scholar] [CrossRef]
- Diapouli, E.; Manousakas, M.I.; Vratolis, S.; Vasilatou, V.; Pateraki, S.; Bairachtari, K.A.; Querol, X.; Amato, F.; Alastuey, A.; Karanasiou, A.; et al. AIRUSE-LIFE +: Estimation of natural source contributions to urban ambient air PM 10 and PM 2. 5 concentrations in southern Europe—Implications to compliance with limit values. Atmos. Chem. Phys. 2016, 17, 3673–3685. [Google Scholar] [CrossRef] [Green Version]
- Paatero, P.; Eberly, S.I.; Brown, S.; Norris, G.A. Methods for estimating uncertainty in factor analytic solutions. Atmos. Meas. Technol. 2014, 7, 781–797. [Google Scholar] [CrossRef] [Green Version]
- Souto-Oliveira, C.E.; Kamigauti, L.Y.; Andrade, M.D.; Babinski, M. Improving Source Apportionment of Urban Aerosol Using Multi-Isotopic Fingerprints (MIF) and Positive Matrix Factorization (PMF): Cross-Validation and New Insights. Front. Environ. Sci. 2021, 9, 623915. [Google Scholar] [CrossRef]
- Bove, M.C.; Massabò, D.; Prati, P. PMF5.0 vs. CMB8.2: An inter-comparison study based on the new European SPECIEUROPE database. Atmos. Res. 2018, 201, 181–188. [Google Scholar] [CrossRef]
- Merabet, H.; Kerbachi, R.; Mihalopoulos, N.; Stavroulas, I.; Kanakidou, M.; Yassaa, N. Measurement of atmospheric black carbon in some South Mediterranean cities. Clean Air J. 2019, 29, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Dammak, R.; Chabbi, I.; Bahloul, M.; Azri, C. PM10 temporal variation and multi-scale contributions of sources and meteorology in Sfax, Tunisia. Air Qual. Atmos. Health 2020, 13, 617–628. [Google Scholar] [CrossRef]
- Abu-Allaban, M.M.; Lowenthal, D.H.; Gertler, A.W.; Labib, M. Sources of PM10 and PM2.5 in Cairo’s ambient air. Environ. Monit. Assess. 2007, 133, 417–425. [Google Scholar] [CrossRef]
- Khedidji, S.; Müller, K.; Rabhi, L.; Spindler, G.; Fomba, K.W.; Pinxteren, D.V.; Yassaa, N.; Herrmann, H. Chemical Characterization of Marine Aerosols in a South Mediterranean Coastal Area Located in Bou Ismaïl, Algeria. Aerosol Air Qual. Res. 2020, 20, 2448–2473. [Google Scholar] [CrossRef]
- Saad, M.H.; Masmoudi, M.M.; Chevaillier, S.; Laurent, B.; Lafon, S.; Alfaro, S.C. Variability of the elemental composition of airborne mineral dust along the coast of Central Tunisia. Atmos. Res. 2018, 209, 170–178. [Google Scholar] [CrossRef]
- Benchrif, A.; Guinot, B.; Bounakhla, M.; Cachier, H.; Damnati, B.; Baghdad, B. Aerosols in Northern Morocco: Input pathways and their chemical fingerprint. Atmos. Environ. 2018, 174, 140–147. [Google Scholar] [CrossRef]
- Metternich, P.; Georgii, H.; Groeneveld, K.O. Long range transport of atmospheric aerosol particles over the Mediterranean and Atlantic Ocean. Nucl. Instrum. Methods Phys. Res. Sect. B-Beam Interact. Mater. At. 1984, 3, 475–478. [Google Scholar] [CrossRef]
- Al-Momani, I.; Güllü, G.; Ölmez, I.; Eler, Ü.; Örtel, E.; Şirin, G.; Tuncel, G. Chemical composition of eastern Mediterranean aerosol and precipitation: Indications of long-range transport. Pure Appl. Chem. 1997, 69, 41–46. [Google Scholar] [CrossRef]
- Ancellet, G.; Pelon, J.; Totems, J.; Chazette, P.; Bazureau, A.; Sicard, M.; Iorio, T.D.; Dulac, F.; Mallet, M.D. Long range transport and mixing of aerosol sources during the 2013 North American biomass burning episode: Analysis of multiple lidar observations in the Western Mediterranean basin. Atmos. Chem. Phys. 2015, 16, 4725–4742. [Google Scholar] [CrossRef]
- Cachier, H.; Bremond, M.P.; Buat-Ménard, P. Determination of atmospheric soot carbon with a simple thermal method. Tellus B 1989, 41, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Genga, A.; Ielpo, P.; Siciliano, T.; Siciliano, M. Carbonaceous particles and aerosol mass closure in PM2.5 collected in a port city. Atmos. Res. 2017, 183, 245–254. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change; Wiley: Hoboken, NJ, USA, 1997. [Google Scholar] [CrossRef]
- Cesari, D.; Genga, A.; Ielpo, P.; Siciliano, M.; Mascolo, G.; Grasso, F.M.; Contini, D. Source apportionment of PM(2.5) in the harbour-industrial area of Brindisi (Italy): Identification and estimation of the contribution of in-port ship emissions. Sci. Total Environ. 2014, 497–498, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.; Daher, N.; Kam, W.; Shafer, M.M.; Ning, Z.; Schauer, J.J.; Sioutas, C. Spatial and temporal variation of chemical composition and mass closure of ambient coarse particulate matter (PM10-2.5) in the Los Angeles area. Atmos. Environ. 2011, 45, 2651–2662. [Google Scholar] [CrossRef]
- Zhang, Y.; Sartelet, K.; Zhu, S.; Wang, W.; Wu, S.Y.; Zhang, X.; Wang, K.; Tran, P.; Seigneur, C.; Wang, Z. Application of WRF/Chem-MADRID and WRF/Polyphemus in Europe—Part 2: Evaluation of chemical concentrations and sensitivity simulations. Atmos. Chem. Phys. 2013, 13, 6845–6875. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.G.; Chow, J.C.; Chen, L.A. Summary of Organic and Elemental Carbon/Black Carbon Analysis Methods and Intercomparisons. Aerosol Air Qual. Res. 2005, 5, 65–102. [Google Scholar] [CrossRef] [Green Version]
- Sciare, J.; Oikonomou, K.; Cachier, H.; Mihalopoulos, N.; Andreae, M.O.; Maenhaut, W.; Sarda-Estève, R. Aerosol mass closure and reconstruction of the light scattering coefficient over the Eastern Mediterranean Sea during the MINOS campaign. Atmos. Chem. Phys. 2005, 5, 2253–2265. [Google Scholar] [CrossRef] [Green Version]
- Turpin, B.J.; Lim, H. Species Contributions to PM2.5 Mass Concentrations: Revisiting Common Assumptions for Estimating Organic Mass. Aerosol Sci. Technol. 2001, 35, 602–610. [Google Scholar] [CrossRef]
- Guieu, C.; Loÿe-Pilot, M.; Ridame, C.; Thomas, C. Chemical characterization of the Saharan dust end-member: Some biogeochemical implications for the western Mediterranean Sea. J. Geophys. Res. 2002, 107, 4258. [Google Scholar] [CrossRef]
- Malm, W.C.; Sisler, J.F.; Huffman, D.; Eldred, R.A.; Cahill, T.A. Spatial and seasonal trends in particle concentration and optical extinction in the United States. J. Geophys. Res. 1994, 99, 1347–1370. [Google Scholar] [CrossRef]
- Chow, J.C.; Watson, J.G.; Fujita, E.M.; Lu, Z.; Lawson, D.R.; Ashbaugh, L.L. Temporal and spatial variations of PM2.5 and PM10 aerosol in the Southern California air quality study. Atmos. Environ. 1994, 28, 2061–2080. [Google Scholar] [CrossRef]
- Malm, W.C.; Schichtel, B.A.; Pitchford, M.L. Uncertainties in PM2.5 Gravimetric and Speciation Measurements and What We Can Learn from Them. J. Air Waste Manag. Assoc. 2011, 61, 1131–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiapello, I.; Bergametti, G.; Châtenet, B.; Bousquet, P.; Dulac, F.; Soares, E.S. Origins of African dust transported over the northeastern tropical Atlantic. J. Geophys. Res. 1997, 102, 13701–13709. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.E.; Kotchenruther, R.; Kuprov, R.; Silcox, G.D. Receptor model source attributions for Utah’s Salt Lake City airshed and the impacts of wintertime secondary ammonium nitrate and ammonium chloride aerosol. J. Air Waste Manag. Assoc. 2013, 63, 575–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Shi, G.; Han, B.; Wang, W.; Zhou, X.; Wang, J.; Li, X.; Feng, Y. The accuracy of two- and three-way positive matrix factorization models: Applying simulated multisite data sets. J. Air Waste Manag. Assoc. 2014, 64, 1122–1129. [Google Scholar] [CrossRef] [Green Version]
- Paatero, P.; Tapper, U. Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values. Environmetrics 1994, 5, 111–126. [Google Scholar] [CrossRef]
- Sara, C.; Luisa, C.; Bernd, G. Positive Matrix Factorisation (PMF)—An Introduction to the Chemometric Evaluation of Environmental Monitoring Data Using PMF; Joint Research Centre: Brussels, Belgium, 2009. [Google Scholar] [CrossRef]
- Kim, E.; Hopke, P.K. Improving source identification of fine particles in a rural northeastern U.S. area utilizing temperature-resolved carbon fractions. J. Geophys. Res. 2004, 109, D09204. [Google Scholar] [CrossRef]
- Norris, G.; Duvall, R.; Brown, S.; Bai, S. EPA Positive Matrix Factorization (PMF) 5.0 Fundamentals and User Guide; Technical Report; U.S. Environmental Protection Agency, National Exposure Research Laboratory: Washington, DC, USA, 2014.
- Brown, S.G.; Eberly, S.I.; Paatero, P.; Norris, G.A. Methods for estimating uncertainty in PMF solutions: Examples with ambient air and water quality data and guidance on reporting PMF results. Sci. Total Environ. 2015, 518–519, 626–635. [Google Scholar] [CrossRef]
- Farahmandkia, Z.; Moattar, F.; Zayeri, F.; Sadegh Sekhavatjou, M.; Mansouri, N. Contribution of point and small-scaled sources to the PM10 emission using positive matrix factorization model. J. Environ. Health Sci. Eng. 2017, 15, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, S.; Salameh, D.; Albinet, A.; Alleman, L.Y.; Waked, A.; Besombes, J.L.; Jacob, V.; Guillaud, G.; Meshbah, B.; Rocq, B.; et al. Comparison of PM10 Sources Profiles at 15 French Sites Using a Harmonized Constrained Positive Matrix Factorization Approach. Atmosphere 2019, 10, 310. [Google Scholar] [CrossRef] [Green Version]
- Veld, M.I.; Alastuey, A.; Pandolfi, M.; Amato, F.; Pérez, N.; Reche, C.; Via, M.; Minguillón, M.C.; Escudero, M.; Querol, X. Compositional changes of PM2.5 in NE Spain during 2009–2018: A trend analysis of the chemical composition and source apportionment. Sci. Total Environ. 2021, 795, 148728. [Google Scholar] [CrossRef] [PubMed]
- Salameh, D.; Detournay, A.; Pey, J.; Pérez, N.; Liguori, F.; Saraga, D.; Bove, M.C.; Brotto, P.; Cassola, F.; Massabò, D.; et al. PM2.5 chemical composition in five European Mediterranean cities: A 1-year study. Atmos. Res. 2015, 155, 102–117. [Google Scholar] [CrossRef]
- Pérez, N.; Pey, J.; Querol, X.; Alastuey, A.; López, J.M.; Viana, M. Partitioning of major and trace components in PM10–PM2.5–PM1 at an urban site in Southern Europe. Atmos. Environ. 2008, 42, 1677–1691. [Google Scholar] [CrossRef]
- Putaud, J.; Dingenen, R.V.; Alastuey, A.; Bauer, H.; Birmili, W.; Cyrys, J.; Flentje, H.E.; Fuzzi, S.; Gehrig, R.; Hansson, H.; et al. A European aerosol phenomenology 3: Physical and chemical characteristics of particulate matter from 60 rural, urban, and kerbside sites across Europe. Atmos. Environ. 2010, 44, 1308–1320. [Google Scholar] [CrossRef]
- Adon, A.J.; Liousse, C.; Doumbia, E.H.; Baeza-Squiban, A.; Cachier, H.; Léon, J.F.; Yoboué, V.; Akpo, A.B.; Galy-Lacaux, C.; Guinot, B.; et al. Physico-chemical characterization of urban aerosols from specific combustion sources in West Africa at Abidjan in Côte d’Ivoire and Cotonou in Benin in the frame of the DACCIWA program. Atmos. Chem. Phys. 2020, 20, 5327–5354. [Google Scholar] [CrossRef]
- Remoundaki, E.; Kassomenos, P.A.; Mantas, E.; Mihalopoulos, N.; Tsezos, M. Composition and Mass Closure of PM2.5 in Urban Environment (Athens, Greece). Aerosol Air Qual. Res. 2013, 13, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Rogula-Kozłowska, W.; Klejnowski, K.; Rogula-Kopiec, P.; Mathews, B.; Szopa, S. A Study on the Seasonal Mass Closure of Ambient Fine and Coarse Dusts in Zabrze, Poland. Bull. Environ. Contam. Toxicol. 2012, 88, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Putaud, J.P.; Raes, F.; Van Dingenen, R.; Brüggemann, E.; Facchini, M.C.; Decesari, S.; Fuzzi, S.; Gehrig, R.; Hüglin, C.; Laj, P.; et al. A european aerosol phenomenology—2: Chemical characteristics of particulate matter at kerbside, urban, rural and background sites in Europe. Atmos. Environ. 2004, 38, 2579–2595. [Google Scholar] [CrossRef]
- Myhre, G.; Myhre, C.E.L.; Samset, B.H.; Storelvmo, T. Aerosols and their Relation to Global Climate and Climate Sensitivity. Nat. Educ. Knowl. 2013, 4, 7. [Google Scholar]
- Hidy, G.M. Chapter 7—Atmospheric Aerosols. In Aerosols; Academic Press: Cambridge, MA, USA, 1984. [Google Scholar] [CrossRef]
- Babar, Z.B.; Park, J.; Lim, H. Influence of NH 3 on secondary organic aerosols from the ozonolysis and photooxidation of α-pinene in a flow reactor. Atmos. Environ. 2017, 164, 71–84. [Google Scholar] [CrossRef]
- Dasgupta, P.K.; Campbell, S.W.; Al-Horr, R.S.; Ullah, S.M.; Li, J.; Amalfitano, C.; Poor, N.D. Conversion of sea salt aerosol to NaNO3 and the production of HCl: Analysis of temporal behavior of aerosol chloride/nitrate and gaseous HCl/HNO3 concentrations with AIM. Atmos. Environ. 2007, 41, 4242–4257. [Google Scholar] [CrossRef]
- Vecchi, R.; Chiari, M.; D’Alessandro, A.; Fermo, P.; Lucarelli, F.; Mazzei, F.; Nava, S.; Piazzalunga, A.; Prati, P.; Silvani, F.; et al. A mass closure and PMF source apportionment study on the sub-micron sized aerosol fraction at urban sites in Italy. Atmos. Environ. 2008, 42, 2240–2253. [Google Scholar] [CrossRef]
- Tian, S.; Pan, Y.; Wang, Y. Size-resolved source apportionment of particulate matter in urban Beijing during haze and non-haze episodes. Atmos. Chem. Phys. 2016, 16, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Speer, R.E.; Edney, E.O.; Kleindienst, T.E. Impact of organic compounds on the concentrations of liquid water in ambient PM2.5. J. Aerosol Sci. 2003, 34, 63–77. [Google Scholar] [CrossRef]
- Dingenen, R.V.; Raes, F.; Putaud, J.; Baltensperger, U.; Charron, A.; Facchini, M.C.; Decesari, S.; Fuzzi, S.; Gehrig, R.; Hansson, H.; et al. A European aerosol phenomenology-1: Physical characteristics of particulate matter at kerbside, urban, rural and background sites in Europe. Atmos. Environ. 2004, 38, 2561–2577. [Google Scholar] [CrossRef]
- Scerri, M.; Kandler, K.; Weinbruch, S.; Yubero, E.; Galindo, N.; Prati, P.; Caponi, L.; Massabò, D. Estimation of the contributions of the sources driving PM2.5 levels in a Central Mediterranean coastal town. Chemosphere 2018, 211, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Manousakas, M.; Papaefthymiou, H.; Diapouli, E.; Migliori, A.; Karydas, A.G.; Bogdanović-Radović, I.; Eleftheriadis, K. Assessment of PM2.5 sources and their corresponding level of uncertainty in a coastal urban area using EPA PMF 5.0 enhanced diagnostics. Sci. Total Environ. 2017, 574, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nava, S.; Calzolai, G.; Chiari, M.; Giannoni, M.; Giardi, F.; Becagli, S.; Severi, M.; Traversi, R.; Lucarelli, F. Source Apportionment of PM2.5 in Florence (Italy) by PMF Analysis of Aerosol Composition Records. Atmosphere 2020, 11, 484. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, C.; Tao, J.; Zhang, L.; Liang, X.; Ma, J.; Gao, H.; Huang, T.; Zhang, K. Chemical characterization and source apportionment of PM2.5 in a semi-arid and petrochemical-industrialized city, Northwest China. Sci. Total Environ. 2016, 573, 1031–1040. [Google Scholar] [CrossRef]
- Tan, J.; Zhang, L.; Zhou, X.; Duan, J.; Li, Y.; Hu, J.; He, K. Chemical characteristics and source apportionment of PM2.5 in Lanzhou, China. Sci. Total Environ. 2017, 601–602, 1743–1752. [Google Scholar] [CrossRef]
- Lonati, G.; Giugliano, M.; Butelli, P.; Romele, L.; Tardivo, R. Major chemical components of PM2.5 in Milan (Italy). Atmos. Environ. 2005, 39, 1925–1934. [Google Scholar] [CrossRef]
- Petit, J.; Pallarès, C.; Favez, O.; Alleman, L.Y.; Bonnaire, N.; Rivière, E.D. Sources and Geographical Origins of PM10 in Metz (France) Using Oxalate as a Marker of Secondary Organic Aerosols by Positive Matrix Factorization Analysis. Atmosphere 2019, 10, 370. [Google Scholar] [CrossRef] [Green Version]
- Samara, C.; Voutsa, D.; Kouras, A.; Eleftheriadis, K.; Maggos, T.; Saraga, D.E.; Petrakakis, M.J. Organic and elemental carbon associated to PM10 and PM2.5 at urban sites of northern Greece. Environ. Sci. Pollut. Res. 2013, 21, 1769–1785. [Google Scholar] [CrossRef] [PubMed]
- Manousakas, M.I.; Florou, K.; Pandis, S.N. Source Apportionment of Fine Organic and Inorganic Atmospheric Aerosol in an Urban Background Area in Greece. Atmosphere 2020, 11, 330. [Google Scholar] [CrossRef] [Green Version]
- Jeong, C.; Herod, D.; Dabek-Złotorzyńska, E.; Ding, L.C.; McGuire, M.L.; Evans, G.J. Identification of the sources and geographic origins of black carbon using factor analysis at paired rural and urban sites. Environ. Sci. Technol. 2013, 47, 8462–8470. [Google Scholar] [CrossRef] [PubMed]
- Borlaza, L.J.; Weber, S.; Uzu, G.; Jacob, V.; Cañete, T.; Micallef, S.; Trébuchon, C.; Slama, R.; Favez, O.; Jaffrezo, J.L. Disparities in particulate matter (PM10) origins and oxidative potential at a city scale (Grenoble, France)—Part 1: Source apportionment at three neighbouring sites. Atmos. Chem. Phys. 2021, 21, 5415–5437. [Google Scholar] [CrossRef]
- Hien, P.D.; Bac, V.T.; Thinh, N.T.; Anh, H.L.; Thang, D.D.; Nghia, N.T. A Comparison Study of Chemical Compositions and Sources of PM1.0 and PM2.5 in Hanoi. Aerosol Air Qual. Res. 2021, 21, 210056. [Google Scholar] [CrossRef]
- Pathak, R.; Yao, X.; Lau, A.K.H.; Chan, C.K. Acidity and concentrations of ionic species of PM2.5 in Hong Kong. Atmos. Environ. 2003, 37, 1113–1124. [Google Scholar] [CrossRef]
- Crilley, L.R.; Lucarelli, F.; Bloss, W.J.; Harrison, R.M.; Beddows, D.C.; Calzolai, G.; Nava, S.; Valli, G.; Bernardoni, V.; Vecchi, R. Source apportionment of fine and coarse particles at a roadside and urban background site in London during the 2012 summer ClearfLo campaign. Environ. Pollut. 2017, 220, 766–778. [Google Scholar] [CrossRef] [Green Version]
- Pérez, N.; Pey, J.; Reche, C.; Cortés, J.; Alastuey, A.; Querol, X. Impact of harbour emissions on ambient PM10 and PM2.5 in Barcelona (Spain): Evidences of secondary aerosol formation within the urban area. Sci. Total Environ. 2016, 571, 237–250. [Google Scholar] [CrossRef]
- Martinelango, P.; Dasgupta, P.K.; Al-Horr, R.S. Atmospheric production of oxalic acid/oxalate and nitric acid/nitrate in the Tampa Bay airshed: Parallel pathways. Atmos. Environ. 2007, 41, 4258–4269. [Google Scholar] [CrossRef]
- Xing, L.; Fu, T.; Cao, J.J.; Lee, S.; Wang, G.; Ho, K.F.; Cheng, M.; You, C.; Wang, T. Seasonal and spatial variability of the OM/OC mass ratios and high regional correlation between oxalic acid and zinc in Chinese urban organic aerosols. Atmos. Chem. Phys. 2013, 13, 4307–4318. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Yu, J. Is vehicle exhaust a significant primary source of oxalic acid in ambient aerosols? Geophys. Res. Lett. 2007, 34, L02808. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhuang, G.; Wang, Q.; Liu, T.; Huang, K.; Fu, J.S.; Li, J.; Lin, Y.; Zhang, R.; Deng, C. Characteristics, sources and formation of aerosol oxalate in an Eastern Asia megacity and its implication to haze pollution. Atmos. Chem. Phys. Discuss. 2011, 11, 22075–22112. [Google Scholar] [CrossRef]
- Feng, J.; Guo, Z.; Zhang, T.; Yao, X.; Chan, C.K.; Fang, M. Source and formation of secondary particulate matter in PM2.5 in Asian continental outflow. J. Geophys. Res. 2012, 117, D03302. [Google Scholar] [CrossRef] [Green Version]
- Yamasoe, M.A.; Artaxo, P.; Miguel, A.H.; Allen, A.G. Chemical composition of aerosol particles from direct emissions of vegetation fires in the Amazon Basin: Water-soluble species and trace elements. Atmos. Environ. 2000, 34, 1641–1653. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbonaceous Species | Inorganic Ions | Organic Markers | |||
|---|---|---|---|---|---|
| Species | BC | EC | Na+, NH4+, K+, Mg2+, Ca2+, Cl−, NO3−, SO42− | Oxalate | Levoglucosan, Glucose |
| Uncertainties (µg/m3) | 0.15 | 0.40 | 0.05 | 0.01 | <0.001 |
| Present Study | 14 Southern European Sites [58] | Barcelona (Spain) [57] | Bou-Ismail (Algeria) [11] | Montseny (Spain) [55] | Brindisi Harbor (Italy) [33] | Crete Island (Greece) [39] | Lisbon (Portugal) [8] | Marseille (France) [56] | |
|---|---|---|---|---|---|---|---|---|---|
| May 2011–Apr 2012 | 1996–2007 | 2003–2006 | 2012–2013 | 2018 | Jun–Oct 2012 | 26 Jul–23 Aug 2001 | 2001 | Jul 2011–Jul 2012 | |
| Urban | Urban | Urban | Rural (Coastal) | Rural | Urban industrial | Background (150 m a.s.l) | Suburban | Urban background | |
| PM2.5 | 17.96 | 3–35 | 29 | 12.3 | 5.55 | 16 | 17.41 | 14 | 19.6 |
| Sea salt | 6 (1.14) | 6 | 2 (0.5) | 10 (1.36–2.53) | n.a. | 3(0.6) | (0.22) | 5.4 | 2.3 (0.33) |
| Ammonium | 6 (1.09) | n.a. | 5 (1.4) | 5 (1.10) | 9 (0.50) | 16(2.8) | n.a. | 5.7 | 7.5 (1.5) |
| Nitrate | 6 (1.04) | 7 | 9 (2.7) | 4 (1.02) | 10 (n.a.) | 2(0.3) | (0.07) | 6.4 | 8.5 (1.7) |
| Sulfate | 17 (2.99) | 15 | 16 (4.6) | 25 (3.09) * | 19 (1.11) ** | 20(3.6) ** | n.a. | 18 | 10.9 (2.2) * |
| SIA | 28 (5.05) | n.a. | 30 (8.7) | 34 (5.03) | 38 (n.a.) | 38 (6.7) | (8.06) | 30.1 | 27 (5.4) |
| POM | 34 (6.04) | 23 | (9) | 50 (1.74) | 50 (2.83) | 33 (6) | (6.13) *** | 30 | 42 (8.6) |
| Mineral dust | 9 (1.65) | 11 | 16 (4.8) | 7 (1.52) | 7 (0.38) | 22 (4) | (0.54) | 8.7 | 19 (n.a.) |
| BC | 18 (3.24) | 8 | (2.3) | 6 (0.83) | 3 (0.15) | 3 (0.5) | (1.18) | 6.8–18 | 10 (1.8) |
| n.d. | 4 (0.75) | - | 18 (5.3) | - | - | 1 (0.2) | (1.02) | n.a. | n.a. |
| Location | Type | Study Period | Ca2+-to-Dust Conversion Factor, f | OC-to-POM Conversion Factor, k | Reference | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| f | Intercept, b | r2 | n | % Coarse Closure * | r2 | n | k | % Fine Closure ** | r2 | n | ||||
| Tetouan, Morocco | Urban | May 2011–Apr 2012 | 0.229 | +1.04 | 0.52 | 41 | 111.8 | 0.96 | 51 | 1.20 | 95.6 | 0.97 | 84 | This work |
| Paris, France | Urban | Jun 2004–Jul 2005 | 0.150 | +0.39 | 0.67 | 20 | 99.7 | 0.78 | 20 | 1.40 | 99.1 | 0.89 | 25 | [16] |
| Florence, Italy | Urban | Jul 2002–Jun 2003 | 0.120 | +0.33 | 0.56 | 44 | 99.6 | 0.73 | 46 | 1.50 | 97.9 | 0.85 | 41 | [16] |
| Gonesse, France | Peri-urban | Sept 2004–Jul 2005 | 0.072 | +0.19 | 0.90 | 26 | 99.3 | 0.77 | 26 | 1.60 | 98.7 | 0.86 | 30 | [16] |
| Beijing, China | Urban | 9–27 Aug 2004 | 0.069–0.085 | +0.007–+0.77 | 0.79–0.98 | 10–27 | 99.4–106.7 | 0.88–0.96 | 11–24 | 1.50–1.70 | 99.0–99.9 | 0.87–0.99 | 12–27 | [16] |
| Beijing, China | Urban | 10–31 Jan 2003 | 0.055–0.082 | +0.43–+1.07 | 0.78–0.94 | 14–29 | 97.5–99.8 | 0.85–0.95 | 20–28 | 1.55–1.85 | 99.2–99.6 | 0.85–0.96 | 19–28 | [16] |
| Abidjan, Côte d’Ivoire | Urban | Wet season (2015, 2016) | 0.015–0.15 | 0.9 | - | - | - | - | 1.8 *** | - | - | - | [59] | |
| Abidjan, Côte d’Ivoire | Urban | Dry season (2016, 2017) | 0.006–0.07 | - | 0.9 | - | 1.8 *** | [59] | ||||||
| Athens, Greece | Urban | 16 Mar–19 Apr 2010 | - | - | - | - | - | - | - | 1.8 | 73 | - | 15 | [60] |
| Factor | Identified Factors | Specific Tracers |
|---|---|---|
| 1 | Ammonium sulfate | SO42−, NH4+, K+, NO3− |
| 2 | Road traffic and biomass burning | OC, BC |
| 3 | Fresh sea salt | Cl−, K+, NO3− |
| 4 | Aged sea salt | Mg2+, Na+, Ca2+ |
| 5 | Oxalate-rich factors | Oxalate, NO3− |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benchrif, A.; Tahri, M.; Guinot, B.; Chakir, E.M.; Zahry, F.; Bagdhad, B.; Bounakhla, M.; Cachier, H.; Costabile, F. Aerosols in Northern Morocco-2: Chemical Characterization and PMF Source Apportionment of Ambient PM2.5. Atmosphere 2022, 13, 1701. https://doi.org/10.3390/atmos13101701
Benchrif A, Tahri M, Guinot B, Chakir EM, Zahry F, Bagdhad B, Bounakhla M, Cachier H, Costabile F. Aerosols in Northern Morocco-2: Chemical Characterization and PMF Source Apportionment of Ambient PM2.5. Atmosphere. 2022; 13(10):1701. https://doi.org/10.3390/atmos13101701
Chicago/Turabian StyleBenchrif, Abdelfettah, Mounia Tahri, Benjamin Guinot, El Mahjoub Chakir, Fatiha Zahry, Bouamar Bagdhad, Moussa Bounakhla, Hélène Cachier, and Francesca Costabile. 2022. "Aerosols in Northern Morocco-2: Chemical Characterization and PMF Source Apportionment of Ambient PM2.5" Atmosphere 13, no. 10: 1701. https://doi.org/10.3390/atmos13101701