
Genome-Wide Association Mapping for Yield and Yield-Related Traits in Rice (Oryza Sativa L.) Using SNPs Markers
 ,
,
Abstract
:
1. Introduction
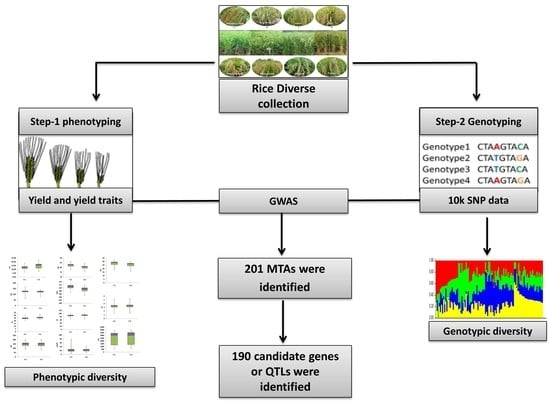
2. Materials and Methods
2.1. Plant Material
2.2. Field Cultivation and Management
2.3. Phenotyping
2.4. DNA Isolation and Genotyping
2.5. Genotypic Data Analysis
2.6. Population Structure and GWAS Analysis
2.7. Gene Annotation
2.8. Principal Component Analysis
3. Results
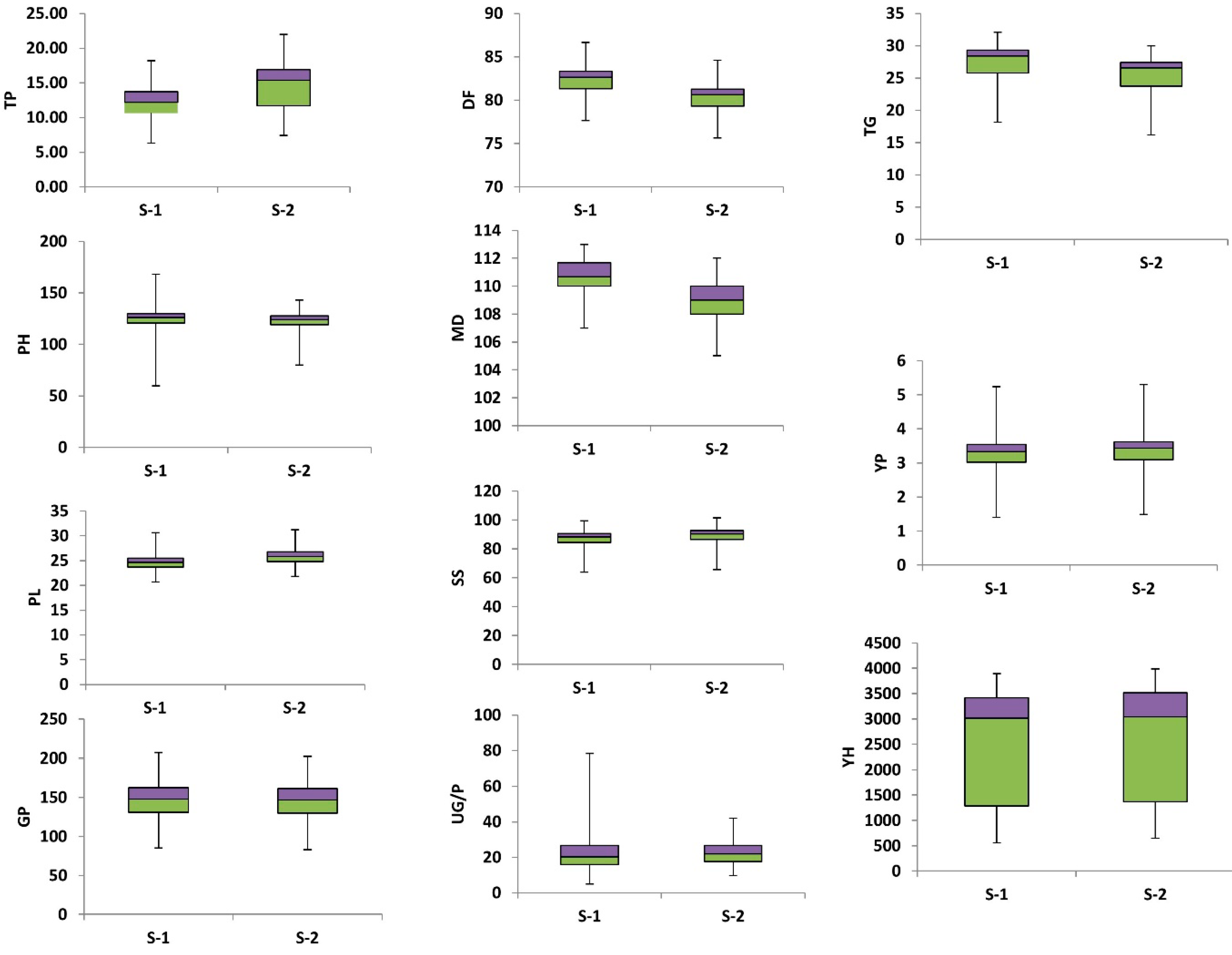
3.1. Phenotypic Diversity
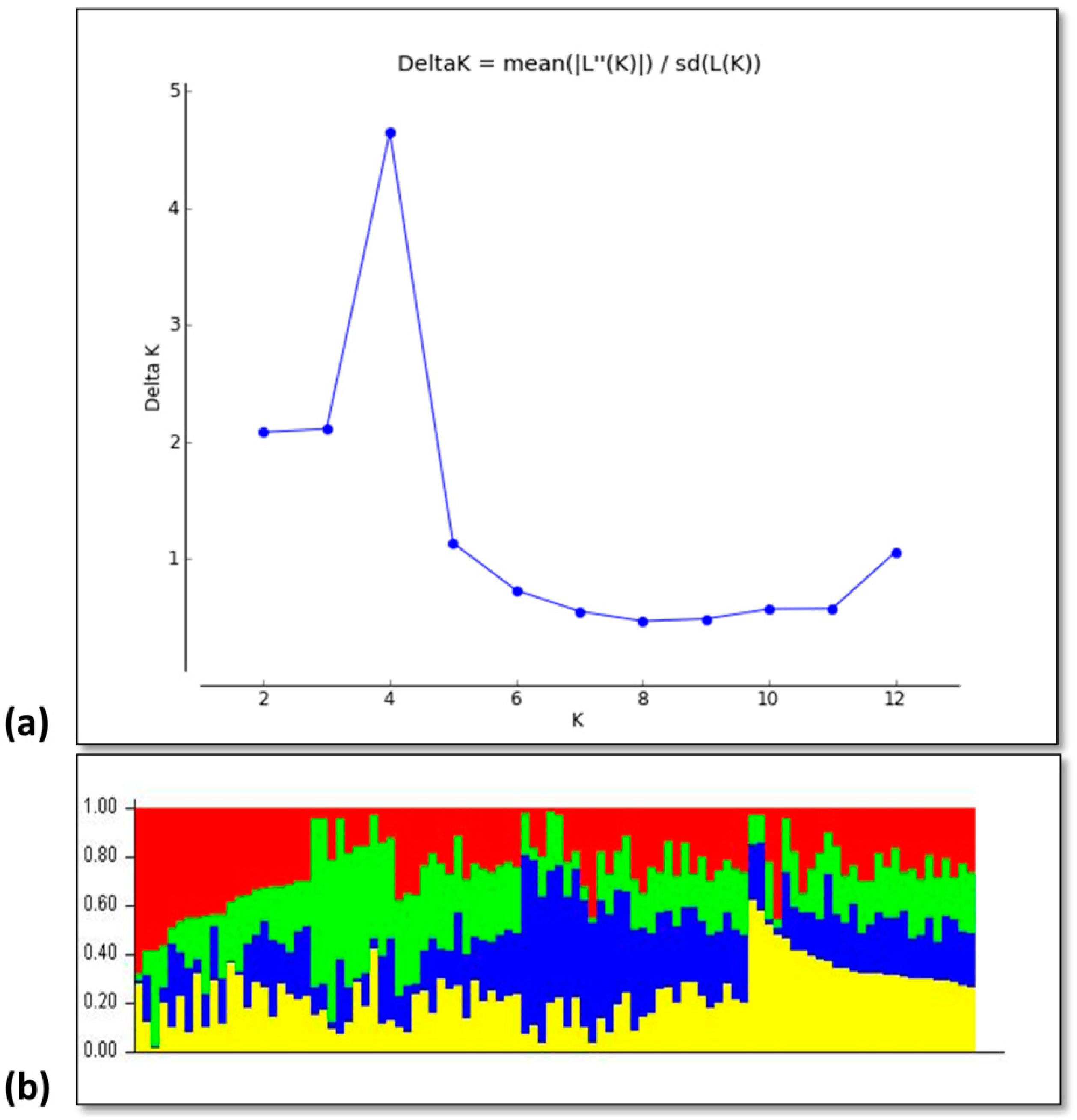
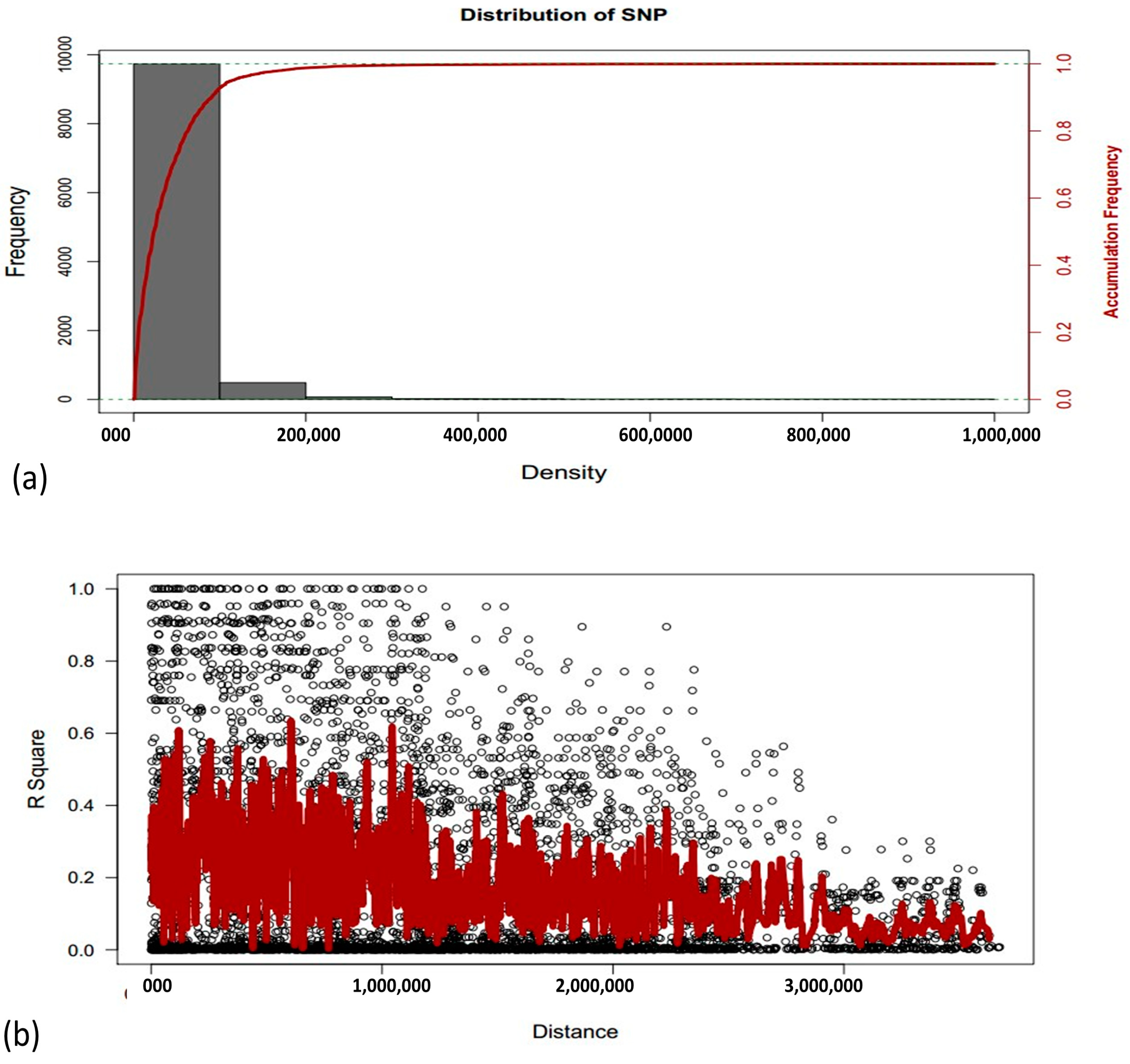
3.2. Genotypic Diversity
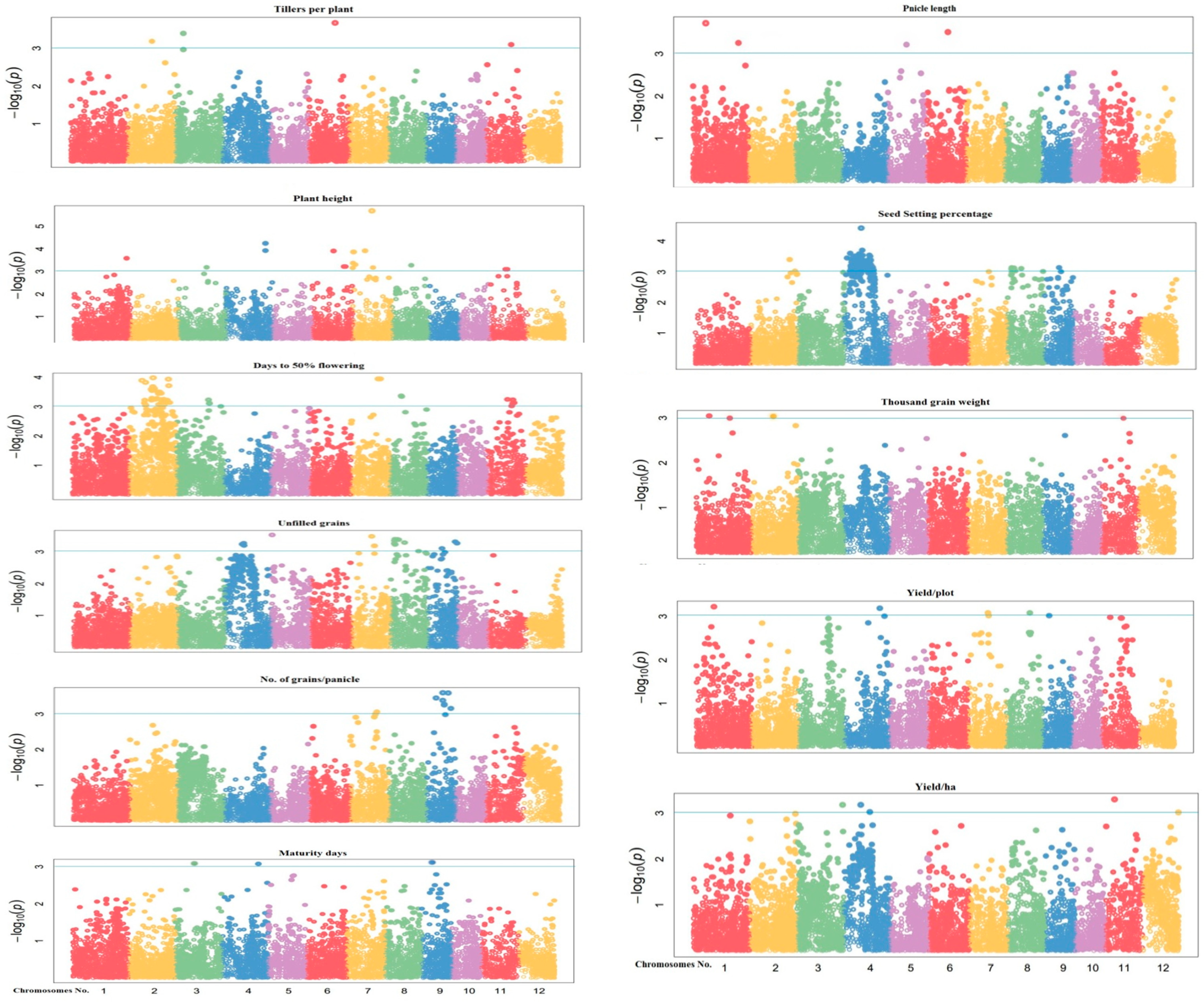
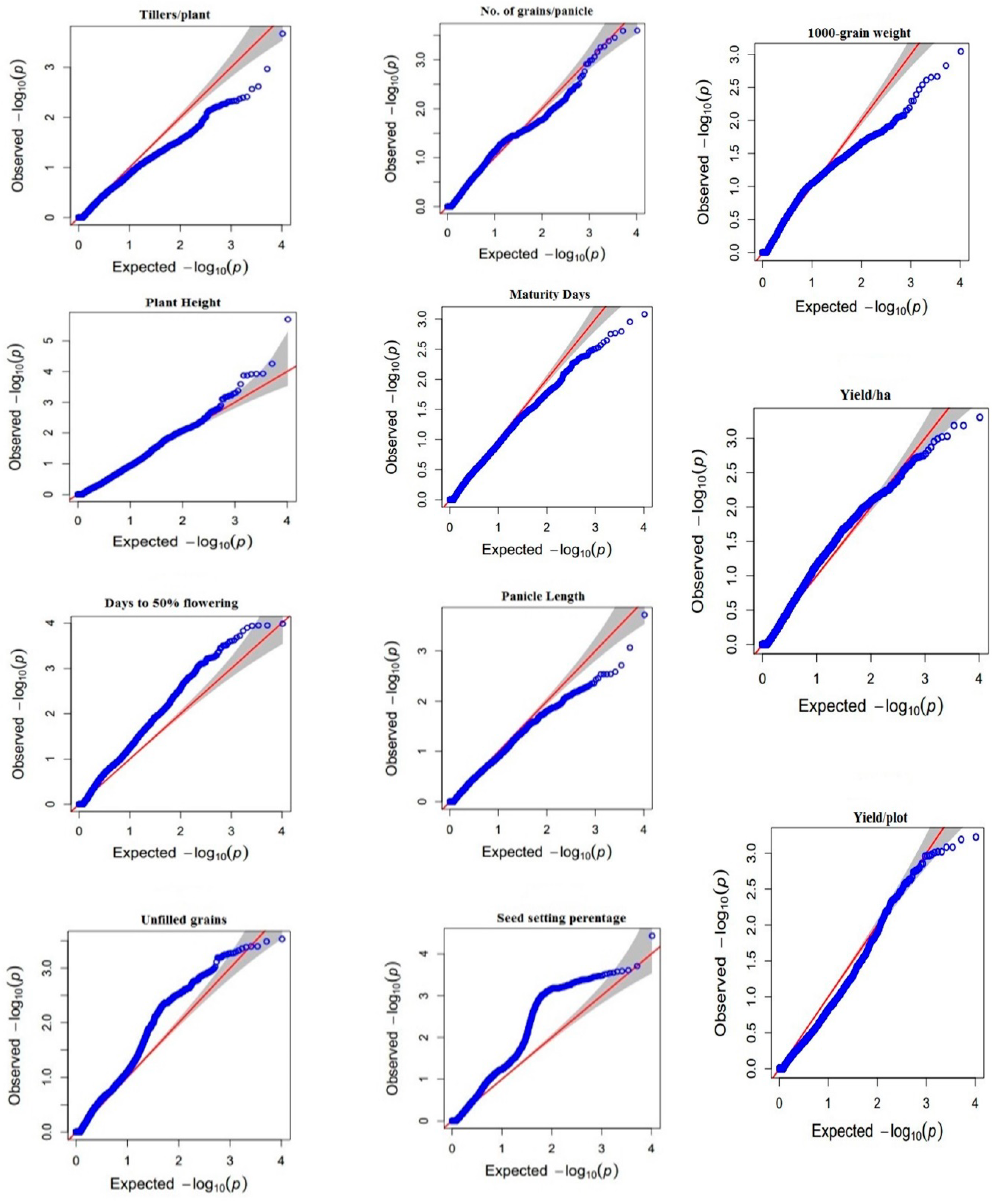
3.3. Genome-Wide Association Studies Using 10k SNP Array
3.3.1. Tillers Per Plant (TP)
3.3.2. Plant Height (PH)
3.3.3. Days to 50 Percent Flowering (DF)
3.3.4. Unfilled Grains per Panicle (UG/P)
3.3.5. Number of Grains per Panicle (G/P)
3.3.6. Days to Maturity (DM)
3.3.7. Panicle Length (PL)
3.3.8. Seed Setting Percentage (SS)
3.3.9. 1000 Grain Weight (TGW)
3.3.10. Yield per Plot (Y/P)
3.3.11. Yield per Hectare (Y/H)
3.4. Gene Annotation of the Identified SNP Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stallworth, S.; Shrestha, S.; Schumaker, B.; Roma-Burgos, N.; Tseng, T.-M. Screening Diverse Weedy Rice (Oryza sativa spp.) Mini Germplasm for Tolerance to Heat and Complete Submergence Stress During Seedling Stage. Front. Agron. 2021, 3, 642335. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.-C.; Lee, J.; Yu, Y.; Yang, K.; Choi, B.-S.; Koh, H.-J.; Waminal, N.E.; Choi, H.-I.; Kim, N.-H.; et al. Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci. Rep. 2015, 5, 15655. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, K.; Yuan, Q.; Liu, X.; Liu, Z.; Lin, X.; Zeng, R.; Zhu, H.; Dong, G.; Qian, Q.; et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 2012, 44, 950–954. [Google Scholar] [CrossRef]
- Zeng, D.; Tian, Z.; Rao, Y.; Dong, G.; Yang, Y.; Huang, L.; Leng, Y.; Xu, J.; Sun, C.; Zhang, G.; et al. Rational design of high-yield and superior-quality rice. Nat. Plants 2017, 3, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, H.; Ma, B.; Liu, G.; Wang, J.; Wang, J.; Gao, R.; Li, J.; Liu, J.; Xu, J.; et al. A natural tandem array alleviates epigenetic repression of IPA1 and leads to superior yielding rice. Nat. Commun. 2017, 8, 14789. [Google Scholar] [CrossRef]
- Sakamoto, T.; Matsuoka, M. Identifying and exploiting grain yield genes in rice. Curr. Opin. Plant Biol. 2008, 11, 209–214. [Google Scholar] [CrossRef]
- Huang, R.; Jiang, L.; Zheng, J.; Wang, T.; Wang, H.; Huang, Y.; Hong, Z. Genetic bases of rice grain shape: So many genes, so little known. Trends Plant Sci. 2013, 18, 218–226. [Google Scholar] [CrossRef]
- Varshney, R.K.; Terauchi, R.; McCouch, S.R. Harvesting the Promising Fruits of Genomics: Applying Genome Sequencing Technologies to Crop Breeding. PLoS Biol. 2014, 12, e1001883. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, M.; Ashraf, U.; Liu, S.; Zhang, J. Exploring the Relationships Between Yield and Yield-Related Traits for Rice Varieties Released in China From 1978 to 2017. Front. Plant Sci. 2019, 10, 543. [Google Scholar] [CrossRef]
- Kim, M.-S.; Ko, S.-R.; Le, V.T.; Jee, M.-G.; Jung, Y.J.; Kang, K.-K.; Cho, Y.-G. Development of SNP Markers from GWAS for Selecting Seed Coat and Aleurone Layers in Brown Rice (Oryza sativa L.). Genes 2022, 13, 1805. [Google Scholar] [CrossRef] [PubMed]
- Morales, K.Y.; Singh, N.; Perez, F.A.; Ignacio, J.C.; Thapa, R.; Arbelaez, J.D.; Tabien, R.E.; Famoso, A.; Wang, D.R.; Septiningsih, E.M.; et al. An improved 7K SNP array, the C7AIR, provides a wealth of validated SNP markers for rice breeding and genetics studies. PLoS ONE 2020, 15, e0232479. [Google Scholar] [CrossRef] [PubMed]
- Fujino, K.; Obara, M.; Shimizu, T.; Koyanagi, K.O.; Ikegaya, T. Genome-wide association mapping focusing on a rice population derived from rice breeding programs in a region. Breed. Sci. 2015, 65, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Zhang, F.; Chong, X.; Song, A.; Guan, Z.; Fang, W.; Chen, F. Genome-wide association study identifies favorable SNP alleles and candidate genes for waterlogging tolerance in chrysanthemums. Hortic. Res. 2019, 6, 21. [Google Scholar] [CrossRef]
- Zhang, K.; Qin, Z.S.; Liu, J.S.; Chen, T.; Waterman, M.S.; Sun, F. Haplotype Block Partitioning and Tag SNP Selection Using Genotype Data and Their Applications to Association Studies. Genome Res. 2004, 14, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, W.; Jiang, C.; Wang, X.; Xiong, H.; Todorovska, E.G.; Yin, Z.; Chen, Y.; Wang, X.; Xie, J.; et al. Genetic Architecture and Candidate Genes for Deep-Sowing Tolerance in Rice Revealed by Non-syn GWAS. Front. Plant Sci. 2018, 9, 332. [Google Scholar] [CrossRef]
- Neang, S.; de Ocampo, M.; A Egdane, J.; Platten, J.D.; Ismail, A.M.; Seki, M.; Suzuki, Y.; Skoulding, N.S.; Kano-Nakata, M.; Yamauchi, A.; et al. A GWAS approach to find SNPs associated with salt removal in rice leaf sheath. Ann. Bot. 2020, 126, 1193–1202. [Google Scholar] [CrossRef]
- Pantalião, G.F.; Narciso, M.; Guimarães, C.; Castro, A.; Colombari, J.M.; Breseghello, F.; Rodrigues, L.; Vianello, R.P.; Borba, T.O.; Brondani, C. Genome wide association study (GWAS) for grain yield in rice cultivated under water deficit. Genetica 2016, 144, 651–664. [Google Scholar] [CrossRef]
- Wu, J.; Feng, F.; Lian, X.; Teng, X.; Wei, H.; Yu, H.; Xie, W.; Yan, M.; Fan, P.; Li, Y.; et al. Genome-wide Association Study (GWAS) of mesocotyl elongation based on re-sequencing approach in rice. BMC Plant Biol. 2015, 15, 218. [Google Scholar] [CrossRef]
- Gawel, N.J.; Jarret, R.L. A modified CTAB DNA extraction procedure forMusa andIpomoea. Plant Mol. Biol. Rep. 1991, 9, 262–266. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Wen, X.; Falush, D. Documentation for Structure Software: Version 2.3; University of Chicago: Chicago, IL, USA, 2010; pp. 1–37. [Google Scholar]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.; Dreisigacker, S.; Lopes, M.; Chavez, P.; Reynolds, M.P. Genome-wide association study for grain yield and related traits in an elite spring wheat population grown in temperate irrigated environments. Theor. Appl. Genet. 2015, 128, 353–363. [Google Scholar] [CrossRef]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Blake, T.K.; Budde, A.D.; Chao, S.; Hayes, P.M.; Horsley, R.D.; Obert, D.E.; Ullrich, S.E.; Smith, K.P. A genome-wide association study of malting quality across eight U.S. barley breeding programs. Theor. Appl. Genet. 2015, 128, 705–721. [Google Scholar] [CrossRef]
- Kwon, Y.; Ham, T.-H.; Kim, J.; Lee, G.; Lee, Y.; Lee, J. Genome Wide Association study of rice (Oryza sativa L.) during heading stage under a high temperature. Plant Breed. Biotechnol. 2021, 9, 104–111. [Google Scholar] [CrossRef]
- Arbuckle, J.L. IBM SPSS Amos 20 User’s Guide; SPSS Inc.: Chicago, IL, USA, 2011; pp. 226–229. [Google Scholar]
- Bekele, B.D.; Rakhi, S.; Naveen, G.; Kundur, P.; Shashidhar, H. Estimation of genetic variability and correlation studies for grain zinc concentrations and yield related traits in selected rice (Oryza sativa L.) genotypes. Asian J. Exp. Biol. Sci. 2013, 4, 345–351. [Google Scholar]
- Kamana, B.; Ankur, P.; Subarna, S.; Prasad, K.B.; Koshraj, U. Genetic variability, correlation and path analysis of rice genotypes in rainfed condition at Lamjung, Nepal. Russ. J. Agric. Socio-Econ. Sci. 2019, 92, 274–280. [Google Scholar]
- Reig-Valiente, J.L.; Viruel, J.; Sales, E.; Marqués, L.; Terol, J.; Gut, M.; Derdak, S.; Talón, M.; Domingo, C. Genetic Diversity and Population Structure of Rice Varieties Cultivated in Temperate Regions. Rice 2016, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Girma, B.T.; Kitil, M.A.; Banje, D.G.; Biru, H.M.; Serbessa, T.B. Genetic Variability Study of Yield and Yield Related Traits in Rice (Oryza sativa L.) Genotypes. Adv. Crop. Sci. Technol. 2018, 6, 4. [Google Scholar] [CrossRef]
- Paswan, S.; Sharma, V.; Singh, V.; Ahmad, A. Genetic variability studies for yield and related attributes in rice genotypes (Oryza sativa L.). Res. J. Agric. Sci. 2014, 5, 750–752. [Google Scholar]
- Yao, J.; Wang, L.; Liu, L.; Zhao, C.; Zheng, Y. Association mapping of agronomic traits on chromosome 2A of wheat. Genetica 2009, 137, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.G.M.-D.; Khan, A.S.; Li, M.-J.; Khan, S.H.; Kashif, M. Early selection of bread wheat genotypes using morphological and photosynthetic attributes conferring drought tolerance. J. Integr. Agric. 2019, 18, 2483–2491. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Phung, N.T.P.; Mai, C.D.; Mournet, P.; Frouin, J.; Droc, G.; Ta, N.K.; Jouannic, S.; Lê, L.T.; Do, V.N.; Gantet, P.; et al. Characterization of a panel of Vietnamese rice varieties using DArT and SNP markers for association mapping purposes. BMC Plant Biol. 2014, 14, 371. [Google Scholar] [CrossRef]
- Swamy, B.P.M.; Shamsudin, N.A.A.; Rahman, S.N.A.; Mauleon, R.; Ratnam, W.; Cruz, M.T.S.; Kumar, A. Association Mapping of Yield and Yield-related Traits Under Reproductive Stage Drought Stress in Rice (Oryza sativa L.). Rice 2017, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Reig-Valiente, J.L.; Marqués, L.; Talón, M.; Domingo, C. Genome-wide association study of agronomic traits in rice cultivated in temperate regions. BMC Genom. 2018, 19, 706. [Google Scholar] [CrossRef]
- Liu, C.; Chen, K.; Zhao, X.; Wang, X.; Shen, C.; Zhu, Y.; Dai, M.; Qiu, X.; Yang, R.; Xing, D.; et al. Identification of genes for salt tolerance and yield-related traits in rice plants grown hydroponically and under saline field conditions by genome-wide association study. Rice 2019, 12, 88. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kenney-Hunt, J.P.; Pavlicev, M.; Peck, J.R.; Waxman, D.; Cheverud, J.M. Pleiotropic scaling of gene effects and the ‘cost of complexity’. Nature 2008, 452, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.H. Pleiotropic models of quantitative variation. Genetics 1990, 124, 773–782. [Google Scholar] [CrossRef]
- Pavlicev, M.; Wagner, G.P. A model of developmental evolution: Selection, pleiotropy and compensation. Trends Ecol. Evol. 2012, 27, 316–322. [Google Scholar] [CrossRef]
- Noon, E.P.-B.; Sabarís, G.; Ortiz, D.M.; Sager, J.; Liebowitz, A.; Stern, D.L.; Frankel, N. Comprehensive analysis of a cis-regulatory region reveals pleiotropy in enhancer function. Cell Rep. 2018, 22, 3021–3031. [Google Scholar] [CrossRef]
- Atwell, S.; Huang, Y.S.; Vilhjálmsson, B.J.; Willems, G.; Horton, M.; Li, Y.; Meng, D.; Platt, A.; Tarone, A.M.; Hu, T.T.; et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 2010, 465, 627–631. [Google Scholar] [CrossRef]
- Hu, J.; Xiao, C.; He, Y. Recent progress on the genetics and molecular breeding of brown planthopper resistance in rice. Rice 2016, 9, 30. [Google Scholar] [CrossRef]
- Biscarini, F.; Cozzi, P.; Casella, L.; Riccardi, P.; Vattari, A.; Orasen, G.; Perrini, R.; Tacconi, G.; Tondelli, A.; Biselli, C.; et al. Genome-Wide Association Study for Traits Related to Plant and Grain Morphology, and Root Architecture in Temperate Rice Accessions. PLoS ONE 2016, 11, e0155425. [Google Scholar] [CrossRef]
- Mogga, M.; Sibiya, J.; Shimelis, H.; Mbogo, D.; Muzhingi, T.; Lamo, J.; Yao, N. Correction: Diversity analysis and genome-wide association studies of grain shape and eating quality traits in rice (Oryza sativa L.) using DArT markers. PLoS ONE 2019, 14, e0212078. [Google Scholar] [CrossRef] [PubMed]
- Begum, H.; Spindel, J.E.; Lalusin, A.; Borromeo, T.; Gregorio, G.; Hernandez, J.; Virk, P.; Collard, B.; McCouch, S.R. Genome-Wide Association Mapping for Yield and Other Agronomic Traits in an Elite Breeding Population of Tropical Rice (Oryza sativa). PLoS ONE 2015, 10, e0119873. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, Y.; Wei, X.; Li, C.; Wang, A.; Zhao, Q.; Li, W.; Guo, Y.; Deng, L.; Zhu, C.; et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 2012, 44, 32–39. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Group | Number of Genotypes | Name of Genotypes |
|---|---|---|---|
| 1 | Group I | 12 | Lemont?, Bond, IR-36, Delvex, Teqing, Nira, Cica, Koshihikari, IR-64, Bellmont, Taducan, Dee Geo Woo |
| 2 | Group II | 21 | Yangzi-95, Gui-99, L-203, CE-65, Zao-40, LA-110, Rando, Jakson, WC-4644, Tsai Yuan Chung, Cica-6, IR-456-3-2-1, Newbonnet, Newrex, Taichung Native-1, Sinum Paga Selection, Stg-663228, Lebonnet, Starbonnet, Della, Toro-2 |
| 3 | Group III | 39 | Delitus, Dellrose, CDR-448, CDR-201, B5-Xiequizao, Roxero regue, H-256-76-1-1-1, Palman, Jasmine-85, A-301, L-202, VE GOLD, L-203, IR-6, Sathi basmati, shaheen basmati, basmati-198, basmati-370, basmati-Pak, Basmati-385, Basmati-515, R- 456, CB-5, CB-10, CB-11, CB-12, CB-13, L-203, VeGold, TP-49, Hill Long Grain, L-202, A-301, L-202, V-203, PALMAN, 87-1-550, 79, 923 |
| 4 | Group IV | 28 | CB-14, CB-15, CB-16, CB-17, CB-19, CB-20, CB-209, CB-21, CB-22, CB-26, CB-27, CB-28, CB-29, CB-30, CB-31, CB-32, CB-33, CB-34, CB-36, CB-38, CB-39, CB-40, CB-41, CB-43, CB-44, KSK-282, KSK-133, Roxero regue |
| Sr. No | Traits | SNP | Chro | Position | Gene ID | Region | p Value | R2 | Strand | MTAs |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PH | OsGRb14446 | 7 | 14594194 | Os07g0436100|13882; Os07g0436350|3483; Os07g0437000|24064 | Intergenic | 2.02 × 10−6 | 25.55 | − | 16 |
| 2 | DF | OsGRb30080 | 2 | 18212087 | Os02g0508500|45221; Os02g0510100|19402; Os02g0510300|27182 | Intergenic | 1.03 × 10−4 | 19.84 | + | 49 |
| 3 | DM | OsGRb09564 | 4 | 27939281 | Os04g0557500 | CDS | 8.27 × 10−4 | 12.97 | − | 3 |
| 4 | T/P | OsGRb13190 | 6 | 20245648 | Os06g0538900|20956; Os06g0539100|13974; Os06g0539500|11602; Os06g0540050|35029; Os06g0540200|36824 | Intergenic | 2.11 × 10−4 | 17.57 | − | 4 |
| 5 | PL | OsGRb23906 | 1 | 10116371 | Os01g0283000; Os01g0283000 | Intron | 1.91 × 10−4 | 18.77 | − | 4 |
| 6 | G/P | OsGRb28603 | 9 | 12952275 | Os09g0381600|37451 | Intergenic | 2.52 × 10−4 | 20.79 | + | 8 |
| 7 | UG/P | OsGRg07442 | 5 | 258353 | Os05g0104700 | 3UTR | 2.92 × 10−4 | 15.74 | + | 20 |
| 8 | SS | OsGRb30591 | 4 | 12914840 | Os04g0294401|3101; Os04g0294812|20440; Os04g0295100|40925 | Intergenic | 3.70 × 10−5 | 22.69 | − | 81 |
| 9 | TGW | OsGRb23906 | 1 | 10116371 | Os01g0283000; Os01g0283000 | Intron | 9.02 × 10−4 | 15.63 | − | 4 |
| 10 | Y/Plot | OsGRb01011 | 1 | 13770374 | Os01g0346700|8390; Os01g0347100|29581; Os01g0347200|37151 | Intergenic | 5.94 × 10−4 | 16.03 | − | 7 |
| 11 | Y/H | OsGRb20658 | 11 | 7220561 | Os11g0235250|23265; Os11g0235700|1114 | Intergenic | 4.95 × 10−4 | 18.71 | + | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashfaq, M.; Rasheed, A.; Zhu, R.; Ali, M.; Javed, M.A.; Anwar, A.; Tabassum, J.; Shaheen, S.; Wu, X. Genome-Wide Association Mapping for Yield and Yield-Related Traits in Rice (Oryza Sativa L.) Using SNPs Markers. Genes 2023, 14, 1089. https://doi.org/10.3390/genes14051089
Ashfaq M, Rasheed A, Zhu R, Ali M, Javed MA, Anwar A, Tabassum J, Shaheen S, Wu X. Genome-Wide Association Mapping for Yield and Yield-Related Traits in Rice (Oryza Sativa L.) Using SNPs Markers. Genes. 2023; 14(5):1089. https://doi.org/10.3390/genes14051089
Chicago/Turabian StyleAshfaq, Muhammad, Abdul Rasheed, Renshan Zhu, Muhammad Ali, Muhammad Arshad Javed, Alia Anwar, Javaria Tabassum, Shabnum Shaheen, and Xianting Wu. 2023. "Genome-Wide Association Mapping for Yield and Yield-Related Traits in Rice (Oryza Sativa L.) Using SNPs Markers" Genes 14, no. 5: 1089. https://doi.org/10.3390/genes14051089