
FDXR-Associated Oculopathy: Congenital Amaurosis and Early-Onset Severe Retinal Dystrophy as Common Presenting Features in a Chinese Population

Abstract
:1. Introduction
2. Materials and Methods
2.1. Probands and Pedigrees
2.2. Identification of FDXR Variants
2.3. Characterizing the Phenotypes of Patients with FDXR Variants
3. Results
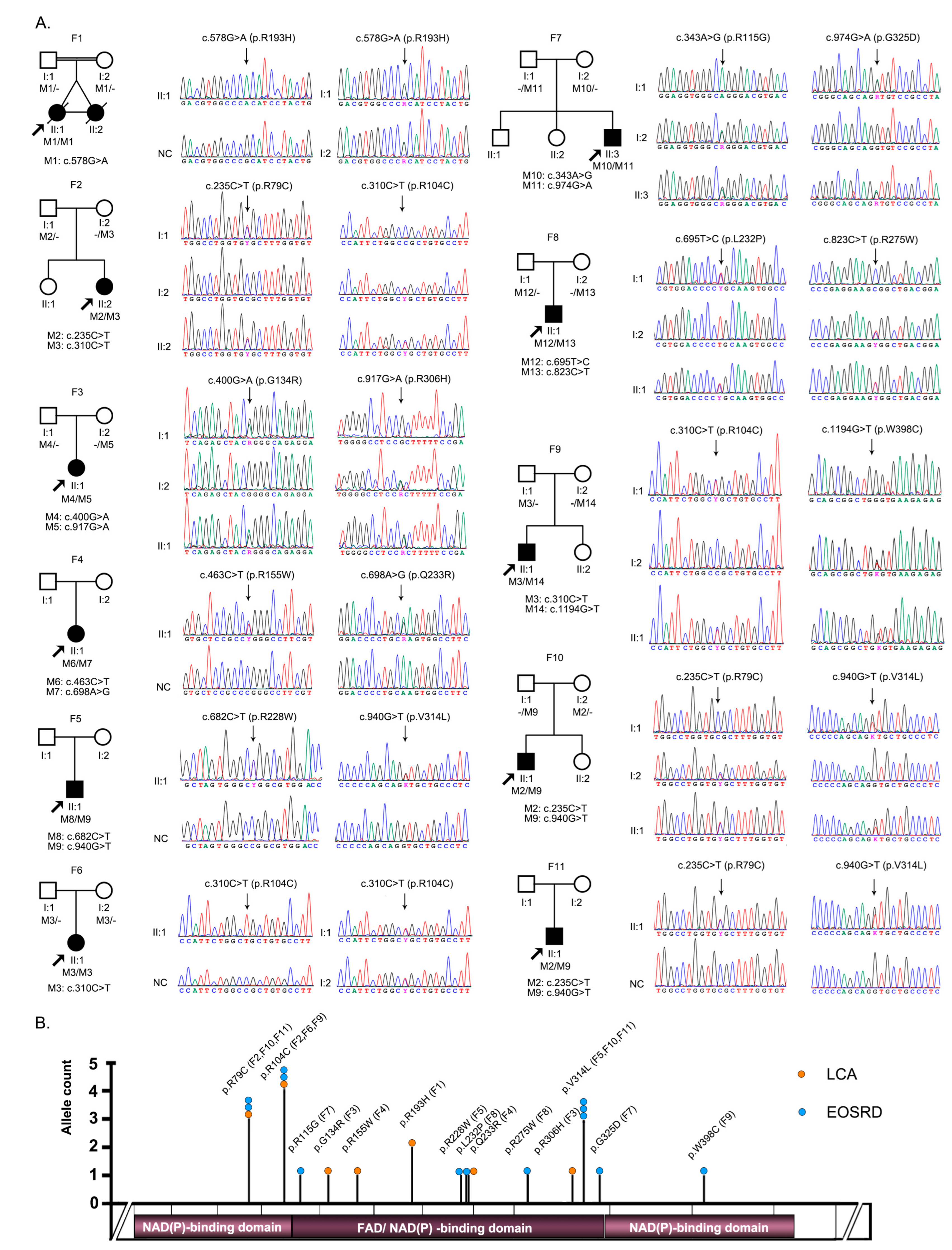
3.1. Identification of Pathogenic or Likely Pathogenic Variants in FDXR
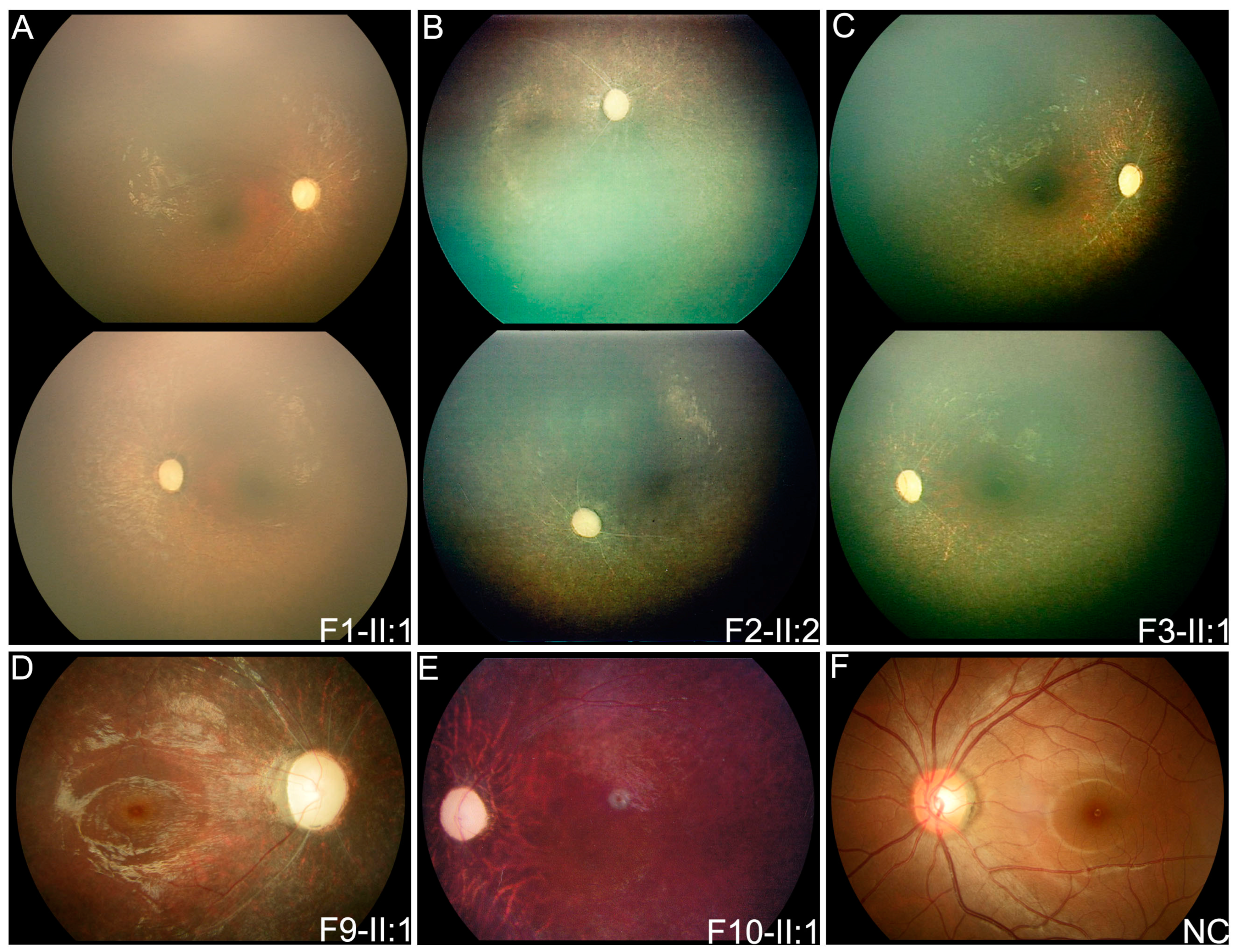
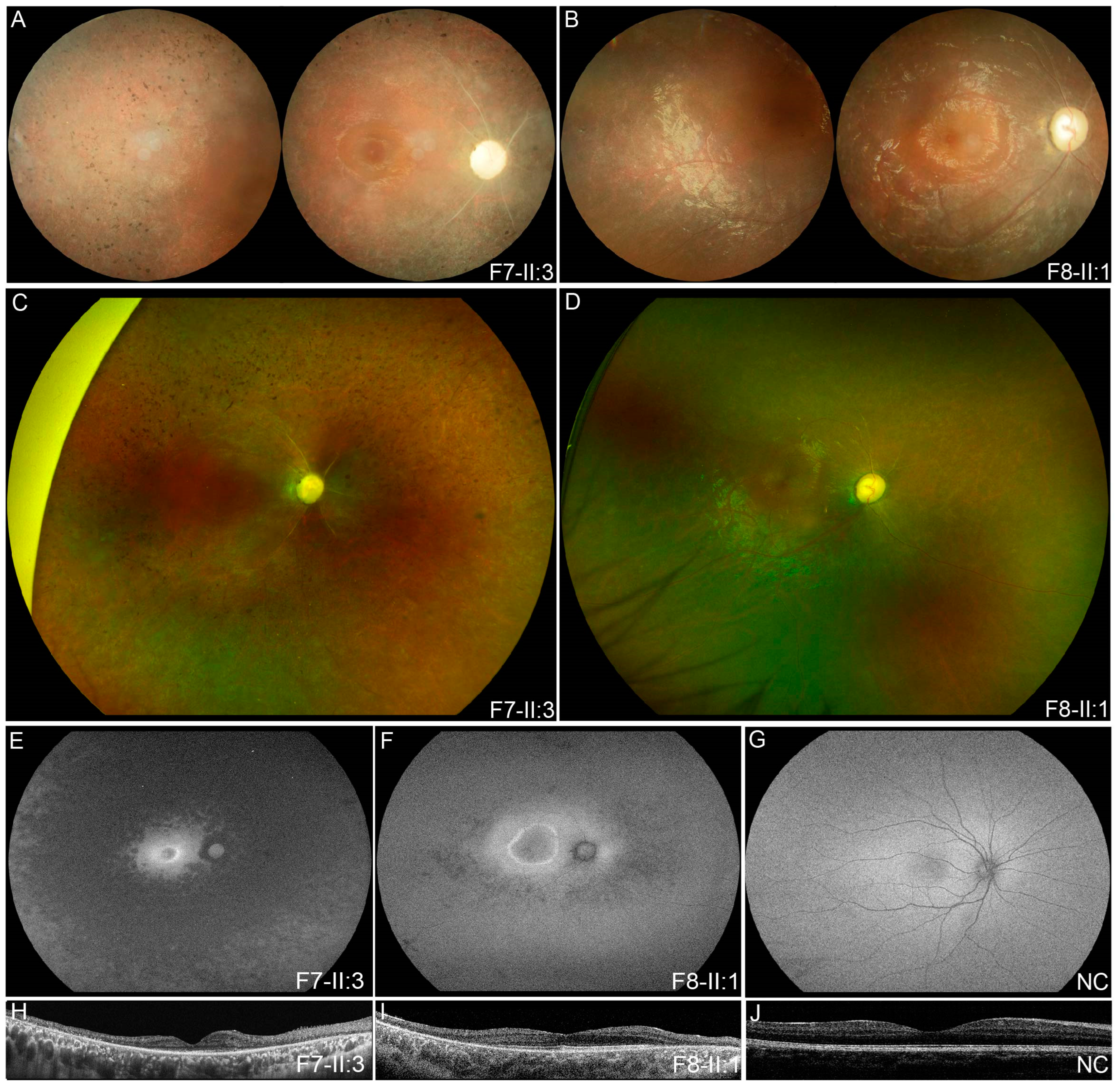
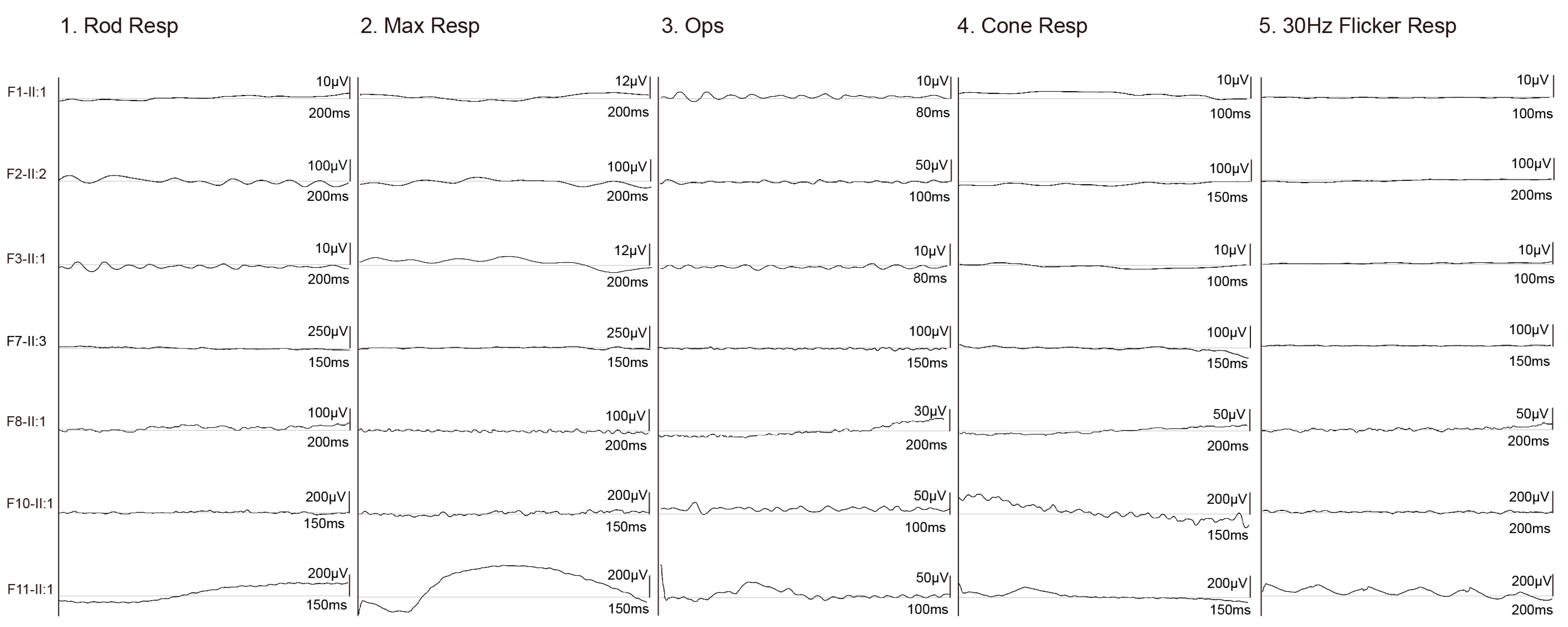
3.2. Clinical Phenotypes of Eight Families with FDXR Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paul, A.; Drecourt, A.; Petit, F.; Deguine, D.D.; Vasnier, C.; Oufadem, M.; Masson, C.; Bonnet, C.; Masmoudi, S.; Mosnier, I.; et al. FDXR Mutations Cause Sensorial Neuropathies and Expand the Spectrum of Mitochondrial Fe-S-Synthesis Diseases. Am. J. Hum. Genet. 2017, 101, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Hanukoglu, I. Conservation of the Enzyme-Coenzyme Interfaces in FAD and NADP Binding Adrenodoxin Reductase-A Ubiquitous Enzyme. J. Mol. Evol. 2017, 85, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Solish, S.B.; Picado-Leonard, J.; Morel, Y.; Kuhn, R.W.; Mohandas, T.K.; Hanukoglu, I.; Miller, W.L. Human adrenodoxin reductase: Two mRNAs encoded by a single gene on chromosome 17cen----q25 are expressed in steroidogenic tissues. Proc. Natl. Acad. Sci. USA 1988, 85, 7104–7108. [Google Scholar] [CrossRef]
- Yang, L.; Slone, J.; Zou, W.; Queme, L.F.; Jankowski, M.P.; Yin, F.; Huang, T. Systemic Delivery of AAV-Fdxr Mitigates the Phenotypes of Mitochondrial Disorders in Fdxr Mutant Mice. Mol. Ther. Methods Clin. Dev. 2020, 18, 84–97. [Google Scholar] [CrossRef]
- Jurkute, N.; Shanmugarajah, P.D.; Hadjivassiliou, M.; Higgs, J.; Vojcic, M.; Horrocks, I.; Nadjar, Y.; Touitou, V.; Lenaers, G.; Poh, R.; et al. Expanding the FDXR-Associated Disease Phenotype: Retinal Dystrophy Is a Recurrent Ocular Feature. Investig. Opthalmol. Vis. Sci. 2021, 62, 2. [Google Scholar] [CrossRef]
- Rocatcher, A.; Desquiret-Dumas, V.; Charif, M.; Ferré, M.; Gohier, P.; Mirebeau-Prunier, D.; Verny, C.; Milea, D.; Lenaers, G.; Bonneau, D.; et al. The top 10 most frequently involved genes in hereditary optic neuropathies in 2186 probands. Brain 2023, 146, 455–460. [Google Scholar] [CrossRef]
- Stenton, S.L.; Piekutowska-Abramczuk, D.; Kulterer, L.; Kopajtich, R.; Claeys, K.G.; Ciara, E.; Eisen, J.; Ploski, R.; Pronicka, E.; Malczyk, K.; et al. Expanding the clinical and genetic spectrum of FDXR deficiency by functional validation of variants of uncertain significance. Hum. Mutat. 2021, 42, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, Y.; Li, J.; Song, Z.; Yi, Z.; Li, F.; Xue, J.; Zhang, W.; Wang, C. Report of a case with ferredoxin reductase (FDXR) gene variants in a Chinese boy exhibiting hearing loss, visual impairment, and motor retardation. Int. J. Dev. Neurosci. 2021, 81, 364–369. [Google Scholar] [CrossRef]
- Keller, N.; Paketci, C.; Altmueller, J.; Fuhrmann, N.; Wunderlich, G.; Schrank, B.; Unver, O.; Yilmaz, S.; Boostani, R.; Karimiani, E.G.; et al. Genomic variants causing mitochondrial dysfunction are common in hereditary lower motor neuron disease. Hum. Mutat. 2021, 42, 460–472. [Google Scholar] [CrossRef]
- Slone, J.; Peng, Y.; Chamberlin, A.; Harris, B.; Kaylor, J.; McDonald, M.T.; Lemmon, M.; El-Dairi, M.A.; Tchapyjnikov, D.; Gonzalez-Krellwitz, L.A.; et al. Biallelic mutations in FDXR cause neurodegeneration associated with inflammation. J. Hum. Genet. 2018, 63, 1211–1222. [Google Scholar] [CrossRef]
- Peng, Y.; Shinde, D.N.; Valencia, C.A.; Mo, J.S.; Rosenfeld, J.; Truitt Cho, M.; Chamberlin, A.; Li, Z.; Liu, J.; Gui, B.; et al. Biallelic mutations in the ferredoxin reductase gene cause novel mitochondriopathy with optic atrophy. Hum. Mol. Genet. 2017, 26, 4937–4950. [Google Scholar] [CrossRef]
- Liu, P.; Meng, L.; Normand, E.A.; Xia, F.; Song, X.; Ghazi, A.; Rosenfeld, J.; Magoulas, P.L.; Braxton, A.; Ward, P.; et al. Reanalysis of Clinical Exome Sequencing Data. N. Engl. J. Med. 2019, 380, 2478–2480. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Hong, Y.; Xu, K.; Zhang, C. Novel biallelic compound heterozygous mutations in FDXR cause optic atrophy in a young female patient: A case report. Int. J. Ophthalmol. 2021, 14, 1796–1798. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Q.; Shen, T.; Xiao, X.; Li, S.; Guan, L.; Zhang, J.; Zhu, Z.; Yin, Y.; Wang, P.; et al. Comprehensive mutation analysis by whole-exome sequencing in 41 Chinese families with Leber congenital amaurosis. Investig. Opthalmol. Vis. Sci. 2013, 54, 4351–4357. [Google Scholar] [CrossRef] [PubMed]
- Slone, J.D.; Yang, L.; Peng, Y.; Queme, L.F.; Harris, B.; Rizzo, S.J.S.; Green, T.; Ryan, J.L.; Jankowski, M.P.; Reinholdt, L.G.; et al. Integrated analysis of the molecular pathogenesis of FDXR-associated disease. Cell Death Dis. 2020, 11, 423. [Google Scholar] [CrossRef]
- Falk, M.J.; Zhang, Q.; Nakamaru-Ogiso, E.; Kannabiran, C.; Fonseca-Kelly, Z.; Chakarova, C.; Audo, I.; Mackay, D.S.; Zeitz, C.; Borman, A.D.; et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 2012, 44, 1040–1045. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, M.J.; Kim, S.Y.; Lim, B.C.; Kim, K.J.; Seong, M.W.; Lee, J.S.; Chae, J.H. Novel compound heterozygous ACO2 mutations in an infant with progressive encephalopathy: A newly identified neurometabolic syndrome. Brain Dev. 2020, 42, 680–685. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, Q. A novel variant in IDH3A identified in a case with Leber congenital amaurosis accompanied by macular pseudocoloboma. Ophthalmic Genet. 2018, 39, 662–663. [Google Scholar] [CrossRef]
- Yi, Z.; Xiao, X.; Li, S.; Sun, W.; Zhang, Q. Pathogenicity discrimination and genetic test reference for CRX variants based on genotype-phenotype analysis. Exp. Eye Res. 2019, 189, 107846. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, W.; Zhou, J.; Li, X.; Jiang, Y.; Li, S.; Jia, X.; Xiao, X.; Ouyang, J.; Wang, Y.; et al. Different Phenotypes Represent Advancing Stages of ABCA4-Associated Retinopathy: A Longitudinal Study of 212 Chinese Families From a Tertiary Center. Investig. Opthalmol. Vis. Sci. 2022, 63, 28. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, W.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Zhang, Q. Clinical and Genetic Analysis of 63 Families Demonstrating Early and Advanced Characteristic Fundus as the Signature of CRB1 Mutations. Am. J. Ophthalmol. 2021, 223, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.S.; Holzbaur, E.L. Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. Elife 2020, 9, e50260. [Google Scholar] [CrossRef]
- Welch, B.M.; Keil, A.P.; van ‘t Erve, T.J.; Deterding, L.J.; Williams, J.G.; Lih, F.B.; Cantonwine, D.E.; McElrath, T.F.; Ferguson, K.K. Longitudinal profiles of plasma eicosanoids during pregnancy and size for gestational age at delivery: A nested case-control study. PLoS Med. 2020, 17, e1003271. [Google Scholar] [CrossRef] [PubMed]
- Stimpfel, M.; Jancar, N.; Virant-Klun, I. New Challenge: Mitochondrial Epigenetics? Stem Cell Rev. Rep. 2018, 14, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Leber, T. Ueber Retinitis pigmentosa und angeborene Amaurose. Graefes Arch. Clin. Exp. Ophthalmol. 1869, 15, 25. [Google Scholar] [CrossRef]
- Franceschetti, A.; Dieterle, P. Diagnostic and prognostic importance of the electroretinogram in tapetoretinal degeneration with reduction of the visual field and hemeralopia. Confin. Neurol. 1954, 14, 184–186. [Google Scholar] [CrossRef]
- Yi, Z.; Sun, W.; Xiao, X.; Li, S.; Jia, X.; Li, X.; Yu, B.; Wang, P.; Zhang, Q. Novel variants in GUCY2D causing retinopathy and the genotype-phenotype correlation. Exp. Eye Res. 2021, 208, 108637. [Google Scholar] [CrossRef]
- Xiao, X.; Sun, W.; Li, S.; Jia, X.; Zhang, Q. Spectrum, frequency, and genotype-phenotype of mutations in SPATA7. Mol. Vis. 2019, 25, 821–833. [Google Scholar]
- Li, S.; Xiao, X.; Yi, Z.; Sun, W.; Wang, P.; Zhang, Q. RPE65 mutation frequency and phenotypic variation according to exome sequencing in a tertiary centre for genetic eye diseases in China. Acta Ophthalmol. 2020, 98, e181–e190. [Google Scholar] [CrossRef]
- Yi, Z.; Li, S.; Wang, S.; Xiao, X.; Sun, W.; Zhang, Q. Clinical features and genetic spectrum of NMNAT1-associated retinal degeneration. Eye 2021, 36, 2279–2285. [Google Scholar] [CrossRef]
- Dharmaraj, S.; Leroy, B.P.; Sohocki, M.M.; Koenekoop, R.K.; Perrault, I.; Anwar, K.; Khaliq, S.; Devi, R.S.; Birch, D.G.; De Pool, E.; et al. The phenotype of Leber congenital amaurosis in patients with AIPL1 mutations. Arch. Ophthalmol. 2004, 122, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, S.; Jiang, Y.; Wang, Y.; Ouyang, J.; Yi, Z.; Sun, W.; Jia, X.; Xiao, X.; Wang, P.; et al. Pathogenic variants in CEP290 or IQCB1 cause earlier onset retinopathy in Senior-Loken syndrome compared to those in INVS, NPHP3, or NPHP4. Am. J. Ophthalmol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Ouyang, J.; Sun, W.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Zhang, Q. Biallelic mutations in USP45, encoding a deubiquitinating enzyme, are associated with Leber congenital amaurosis. J. Med. Genet. 2019, 56, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.A.; Hancock, J.; Ghanekar, S.D.; Kuo, S.H.; Dohse, C.A.; Vega, J. Emerging therapies in Friedreich’s Ataxia. Expert Rev. Neurother. 2020, 20, 1215–1228. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Nucleotide | Effect | Bioinformatic Prediction Tools | GnomAD | Previously Reported | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| ID | Change | REVEL | CADD | SIFT | Polyphen2 | PROVEAN | Fathmm-MKL Coding | Frequency | ||
| M1 | c.578G > A | p.R193H | 0.896 | 31 | DA | PRD | DE | DE | 4/248218 | Slone J, et al. [6] |
| M2 | c.235C > T | p.R79C | 0.653 | 29 | DA | PRD | DE | DE | 2/250924 | / |
| M3 | c.310C > T | p.R104C | 0.517 | 31 | DA | PRD | DE | DE | 16/281296 | / |
| M4 | c.400G > A | p.G134R | 0.818 | 29.7 | DA | PRD | DE | DE | 1/249762 | / |
| M5 | c.917G > A | p.R306H | 0.196 | 22.8 | T | B | DE | DE | 2/242172 | / |
| M6 | c.463C > T | p.R115G | 0.545 | 24.4 | DA | PRD | DE | DE | 4/282390 | Slone J, et al. [6] |
| M7 | c.698A > G | p.Q233R | 0.604 | 26.5 | DA | POD | DE | DE | 2/251196 | / |
| M8 | c.682C > T | p.R228W | 0.793 | 32 | DA | PRD | DE | DE | 4/251200 | / |
| M9 | c.940G > T | p.V314L | 0.13 | 22.5 | T | B | N | DE | 18/274502 | Song, S, et al. [11] |
| M10 | c.343A > G | p.R115G | 0.409 | 23.6 | T | B | DE | DE | / | / |
| M11 | c.974G > A | p.G325D | 0.213 | 24.1 | DA | B | DE | DE | / | / |
| M12 | c.695T > C | p.L232P | 0.776 | 29.8 | DA | POD | DE | DE | / | / |
| M13 | c.823C > T | p.R275W | 0.336 | 24.3 | DA | PRD | DE | DE | 2/221422 | Jurkute N, et al. [9] |
| M14 | c.1194G > T | p.W398C | 0.533 | 31 | DA | PRD | DE | DE | / | / |
| Family | Patient | Nucleotide Change | Gender | Age (Years) At | First | BCVA | Fundus Changes | Others | ||
|---|---|---|---|---|---|---|---|---|---|---|
| ID | ID | Onset | Exam | Symptom | Right | Left | ||||
| F1 | F1-II:1 | c.[578G > A]; [578G > A] | F | FMB | 0.8 | Nystagmus | PVT | PVT | ODP, SRV, SPR | DD |
| F2 | F2-II:2 | c.[235C > T]; [310C > T] | F | FMB | 2 | Nystagmus | PVT | PVT | ODP, SRV, SPR | MR |
| F3 | F3-II:1 | c.[400G > A]; [c.917G > A] | F | FMB | 1 | Nystagmus | PVT | PVT | ODP, SRV, SPR | FC, DD, MW |
| F4 | F4-II:1 | c.[463C > T]; [698A > G] | F | FMB | 6 | Nystagmus | 0.05 | LP | ODP, SRV, SPR | MW |
| F5 | F5-II:1 | c.[682C > T]; [c.940G > T] | M | EC | 16 | Nyctalopia | 0.1 | 0.025 | ODP, SRV, TP | / |
| F6 | F6-II:1 | c.[310C > T]; [310C > T] | M | EC | 19 | Nyctalopia | HM | HM | ODP, SRV, TP | / |
| F7 | F7-II:3 | c.[343A > G]; [974G > A] | M | EC | 12 | Nyctalopia | 0.30 | 0.30 | ODP, SRV, SPR | / |
| F8 | F8-II:1 | c.[695T > C]; [823C > T] | M | 5 | 6 | Poor vision | 0.03 | LP | ODP, SRV, TP | / |
| F9 | F9-II:1 | c.[310C > T]; [1194G > T] | F | 3 | 4 | Nyctalopia | 0.2 | 0.25 | ODP, SRV, SPR | / |
| F10 | F10-II:1 | c.[235C > T]; [940G > T] | M | EC | 7 | Poor vision | HM | 0.04 | ODP, SARV, TP | / |
| F11 | F11-II:1 | c.[235C > T]; [940G > T] | F | 3 | 5 | Poor vision | 0.08 | 0.06 | ODP, SARV, TP | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, S.; Zheng, Y.; Yi, Z.; Wang, Y.; Jiang, Y.; Ouyang, J.; Li, S.; Xiao, X.; Sun, W.; Wang, P.; et al. FDXR-Associated Oculopathy: Congenital Amaurosis and Early-Onset Severe Retinal Dystrophy as Common Presenting Features in a Chinese Population. Genes 2023, 14, 952. https://doi.org/10.3390/genes14040952
Yi S, Zheng Y, Yi Z, Wang Y, Jiang Y, Ouyang J, Li S, Xiao X, Sun W, Wang P, et al. FDXR-Associated Oculopathy: Congenital Amaurosis and Early-Onset Severe Retinal Dystrophy as Common Presenting Features in a Chinese Population. Genes. 2023; 14(4):952. https://doi.org/10.3390/genes14040952
Chicago/Turabian StyleYi, Shutong, Yuxi Zheng, Zhen Yi, Yingwei Wang, Yi Jiang, Jiamin Ouyang, Shiqiang Li, Xueshan Xiao, Wenmin Sun, Panfeng Wang, and et al. 2023. "FDXR-Associated Oculopathy: Congenital Amaurosis and Early-Onset Severe Retinal Dystrophy as Common Presenting Features in a Chinese Population" Genes 14, no. 4: 952. https://doi.org/10.3390/genes14040952