
Functional Analysis of a Novel, Non-Canonical RPGR Splice Variant Causing X-Linked Retinitis Pigmentosa
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
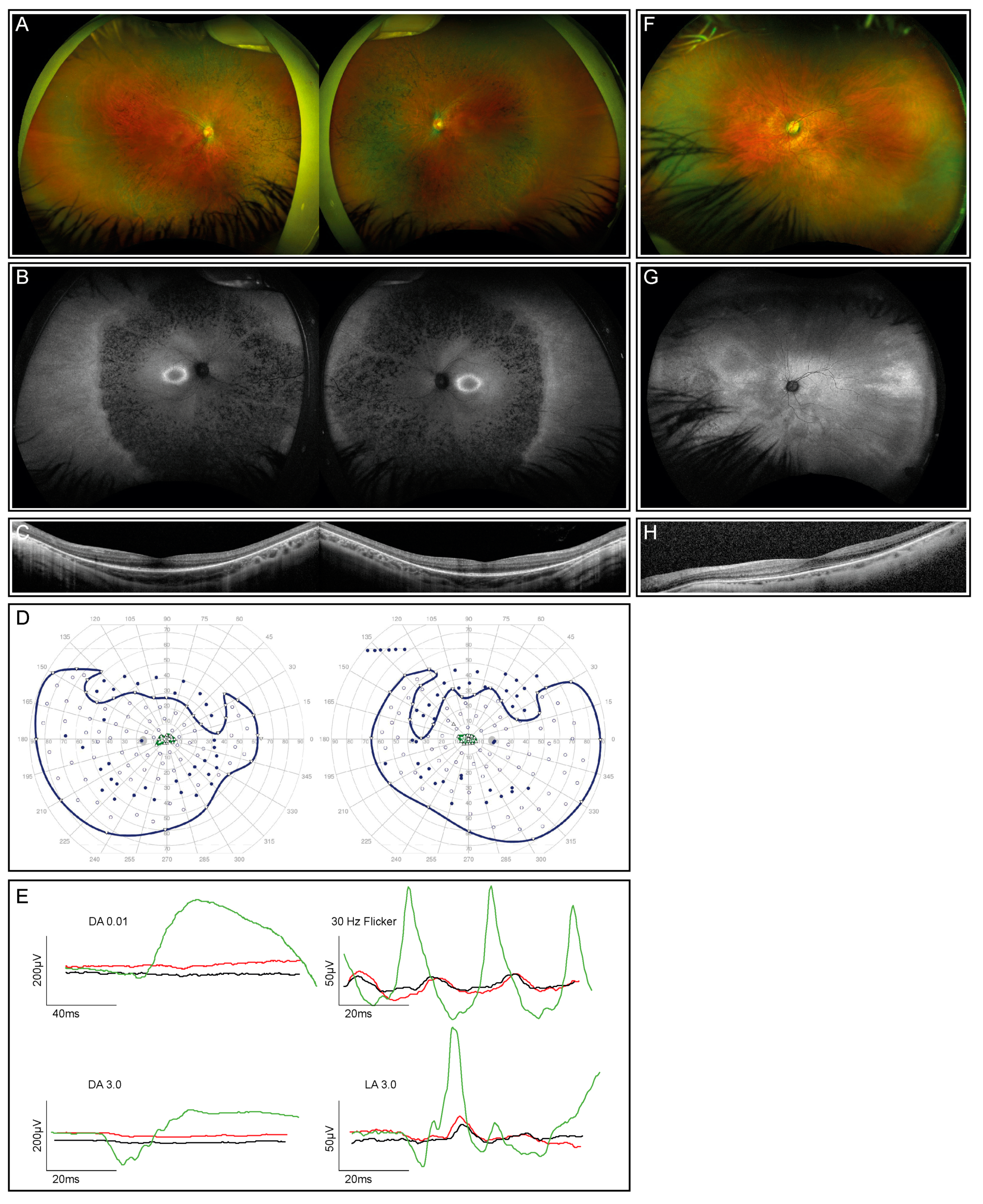
2.1. Clinical Examinations
2.2. Genetic Testing
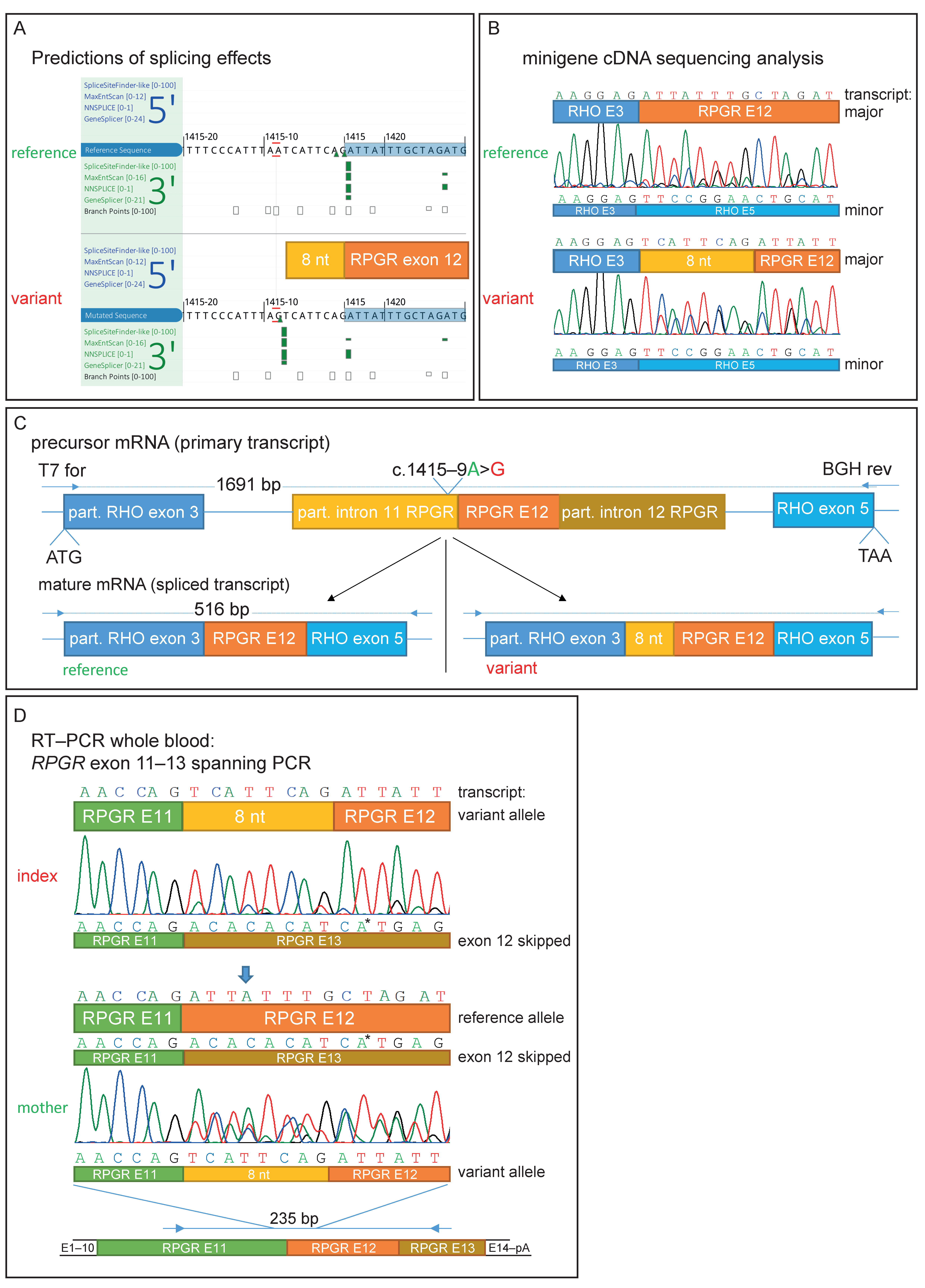
2.3. Functional Analyses
3. Results
3.1. Clinical Presentation
3.2. Genetic Testing of the Patient and His Mother
3.3. Functional Characterization of an Intronic Sequence Variant
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ayuso, C.; Millan, J.M. Retinitis pigmentosa and allied conditions today: A paradigm of translational research. Genome Med. 2010, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef]
- Perea-Romero, I.; Gordo, G.; Iancu, I.F.; Del Pozo-Valero, M.; Almoguera, B.; Blanco-Kelly, F.; Carreño, E.; Jimenez-Rolando, B.; Lopez-Rodriguez, R.; Lorda-Sanchez, I.; et al. Author Correction: Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci. Rep. 2021, 11, 10340. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, V.; Karali, M.; Melillo, P.; Testa, F.; Brunetti-Pierri, R.; Musacchia, F.; Condroyer, C.; Neidhardt, J.; Audo, I.; Zeitz, C.; et al. Spectrum of Disease Severity in Patients with X-Linked Retinitis Pigmentosa due to RPGR Mutations. Investig. Ophthalmol. Vis. Sci. 2020, 61, 36. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.A.; Rosner, B.; Weigel-DiFranco, C.; Dryja, T.P.; Berson, E.L. Disease course of patients with X-linked retinitis pigmentosa due to RPGR gene mutations. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Hosch, J.; Lorenz, B.; Stieger, K. RPGR: Role in the photoreceptor cilium, human retinal disease, and gene therapy. Ophthalmic Genet. 2011, 32, 1–11. [Google Scholar] [CrossRef]
- Iftikhar, M.; Kherani, S.; Kaur, R.; Lemus, M.; Nefalar, A.; Usmani, B.; Junaid, N.; Campochiaro, P.A.; Scholl, H.P.; Shah, S.M. Progression of Retinitis Pigmentosa as Measured on Microperimetry: The PREP-1 Study. Ophthalmol. Retin. 2018, 2, 502–507. [Google Scholar] [CrossRef]
- Bird, A.C. X-linked retinitis pigmentosa. Br. J. Ophthalmol. 1975, 59, 177–199. [Google Scholar] [CrossRef]
- Nanda, A.; Salvetti, A.P.; Clouston, P.; Downes, S.M.; MacLaren, R.E. Exploring the Variable Phenotypes of RPGR Carrier Females in Assessing their Potential for Retinal Gene Therapy. Genes 2018, 9, 643. [Google Scholar] [CrossRef]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Schwahn, U.; Lenzner, S.; Dong, J.; Feil, S.; Hinzmann, B.; van Duijnhoven, G.; Kirschner-Schwabe, R.; Hemberger, M.; Bergen, A.A.; Rosenberg, T.; et al. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat. Genet. 1998, 19, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Lyraki, R.; Megaw, R.; Hurd, T. Disease mechanisms of X-linked retinitis pigmentosa due to RP2 and RPGR mutations. Biochem. Soc. Trans. 2016, 44, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Roepman, R.; Bauer, D.; Rosenberg, T.; van Duijnhoven, G.; van de Vosse, E.; Platzer, M.; Rosenthal, A.; Ropers, H.-H.; Cremers, F.P.M.; Berger, W. Identification of a gene disrupted by a microdeletion in a patient with X-linked retinitis pigmentosa (XLRP). Hum. Mol. Genet. 1996, 5, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Roepman, R.; van Duijnhoven, G.; Rosenberg, T.; Pinckers, A.J.; Bleeker-Wagemakers, L.M.; Bergen, A.A.; Post, J.; Beck, A.; Reinhardt, R.; Ropers, H.-H.; et al. Positional cloning of the gene for X-linked retinitis pigmentosa 3: Homology with the guanine-nucleotide-exchange factor RCC1. Hum. Mol. Genet. 1996, 5, 1035–1041. [Google Scholar] [CrossRef]
- Schmid, F.; Glaus, E.; Cremers, F.P.; Kloeckener-Gruissem, B.; Berger, W.; Neidhardt, J. Mutation- and tissue-specific alterations of RPGR transcripts. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1628–1635. [Google Scholar] [CrossRef]
- Kirschner, R.; Rosenberg, T.; Schultz-Heienbrok, R.; Lenzner, S.; Feil, S.; Roepman, R.; Cremers, F.P.M.; Ropers, H.-H.; Berger, W. RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum. Mol. Genet. 1999, 8, 1571–1578. [Google Scholar] [CrossRef]
- Megaw, R.D.; Soares, D.C.; Wright, A.F. RPGR: Its role in photoreceptor physiology, human disease, and future therapies. Exp. Eye Res. 2015, 138, 32–41. [Google Scholar] [CrossRef]
- Hong, D.H.; Pawlyk, B.; Sokolov, M.; Strissel, K.J.; Yang, J.; Tulloch, B.; Wright, A.F.; Arshavsky, V.Y.; Li, T. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2413–2421. [Google Scholar] [CrossRef]
- Mavlyutov, T.A.; Zhao, H.; Ferreira, P.A. Species-specific subcellular localization of RPGR and RPGRIP isoforms: Implications for the phenotypic variability of congenital retinopathies among species. Hum. Mol. Genet. 2002, 11, 1899–1907. [Google Scholar] [CrossRef]
- Iannaccone, A.; Breuer, D.K.; Wang, X.F.; Kuo, S.F.; Normando, E.M.; Filippova, E.; Baldi, A.; Hiriyanna, S.; Macdonald, C.B.; Baldi, F.; et al. Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation. J. Med. Genet. 2003, 40, e118. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Black, G.C.; Rice, J.M.; Hart-Holden, N.; Jones, A.; O’Grady, A.; Ramsden, S.; Wright, A.F. RPGR mutation analysis and disease: An update. Hum. Mutat. 2007, 28, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Maggi, J.; Koller, S.; Bahr, L.; Feil, S.; Kivrak Pfiffner, F.; Hanson, J.V.M.; Maspoli, A.; Gerth-Kahlert, C.; Berger, W. Long-Range PCR-Based NGS Applications to Diagnose Mendelian Retinal Diseases. Int. J. Mol. Sci. 2021, 22, 1508. [Google Scholar] [CrossRef]
- Maggi, J.; Roberts, L.; Koller, S.; Rebello, G.; Berger, W.; Ramesar, R. De Novo Assembly-Based Analysis of RPGR Exon ORF15 in an Indigenous African Cohort Overcomes Limitations of a Standard Next-Generation Sequencing (NGS) Data Analysis Pipeline. Genes 2020, 11, 800. [Google Scholar] [CrossRef]
- Robson, A.G.; Nilsson, J.; Li, S.; Jalali, S.; Fulton, A.B.; Tormene, A.P.; Holder, G.E.; Brodie, S.E. ISCEV guide to visual electrodiagnostic procedures. Doc. Ophthalmol. 2018, 136, 1–26. [Google Scholar] [CrossRef]
- Available online: https://gatk.broadinstitute.org/hc/en-us/articles/360035535932-Germline-short-variant-discovery-SNPs-Indels- (accessed on 17 March 2023).
- UZH Institut für Medizinische Molekulargenetik. Gen-Panel für Untersuchungen Mittels Hochdurchsatzsequezierung 2021 [Updated 09.09.2021]. Available online: https://www.medmolgen.uzh.ch/de/services/Hauptgene.html (accessed on 17 March 2023).
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Haug, P.; Koller, S.; Maggi, J.; Lang, E.; Feil, S.; Wlodarczyk, A.; Bähr, L.; Steindl, K.; Rohrbach, M.; Gerth-Kahlert, C.; et al. Whole Exome Sequencing in Coloboma/Microphthalmia: Identification of Novel and Recurrent Variants in Seven Genes. Genes 2021, 12, 65. [Google Scholar] [CrossRef]
- Rechsteiner, D.; Issler, L.; Koller, S.; Lang, E.; Bahr, L.; Feil, S.; Rüegger, C.M.; Kottke, R.; Toelle, S.P.; Zweifel, N.; et al. Genetic Analysis in a Swiss Cohort of Bilateral Congenital Cataract. JAMA Ophthalmol. 2021, 139, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Koller, S.; Bahr, L.; Toteberg-Harms, M.; Atac, D.; Roulez, F.; Bahr, A.; Steindl, K.; Feil, S.; Berger, W.; et al. Exome Sequencing in a Swiss Childhood Glaucoma Cohort Reveals CYP1B1 and FOXC1 Variants as Most Frequent Causes. Transl. Vis. Sci. Technol. 2020, 9, 47. [Google Scholar] [CrossRef]
- Kilgore, D.A.; Kilgore, T.A.; Sukpraprut-Braaten, S.; Schaefer, G.B.; Uwaydat, S.H. Multimodal imaging of an RPGR carrier female. Ophthalmic Genet. 2021, 42, 312–316. [Google Scholar] [CrossRef]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; van Genderen, M.; Neidhardt, J.; Luhmann, U.F.; Hoeben, F.; Forster, U.; Wycisk, K.; Mátyás, G.; Hoyng, C.B.; Riemslag, F.; et al. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4328–4335. [Google Scholar] [CrossRef] [PubMed]
- Baala, L.; Romano, S.; Khaddour, R.; Saunier, S.; Smith, U.M.; Audollent, S.; Ozilou, C.; Faivre, L.; Laurent, N.; Foliguet, B.; et al. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am. J. Hum. Genet. 2007, 80, 186–194. [Google Scholar] [CrossRef]
- Bessant, D.A.; Payne, A.M.; Mitton, K.P.; Wang, Q.L.; Swain, P.K.; Plant, C.; Bird, A.C.; Zack, D.J.; Swaroop, A.; Bhattacharya, S.S. A mutation in NRL is associated with autosomal dominant retinitis pigmentosa. Nat. Genet. 1999, 21, 355–356. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Available online: https://www.gtexportal.org/home/ (accessed on 17 March 2023).
- de Heer, A.M.; Collin, R.W.; Huygen, P.L.; Schraders, M.; Oostrik, J.; Rouwette, M.; Kunst, H.P.; Kremer, H.; Cremers, C.W. Progressive sensorineural hearing loss and normal vestibular function in a Dutch DFNB7/11 family with a novel mutation in TMC1. Audiol. Neurootol. 2011, 16, 93–105. [Google Scholar] [CrossRef]
- Kortum, F.; Kieninger, S.; Mazzola, P.; Kohl, S.; Wissinger, B.; Prokisch, H.; Stingl, K.; Weisschuh, N. X-Linked Retinitis Pigmentosa Caused by Non-Canonical Splice Site Variants in RPGR. Int. J. Mol. Sci. 2021, 22, 850. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.; Gallone, G.; Short, P.J.; McRae, J.F.; Ironfield, H.; Wynn, E.H.; Gerety, S.S.; He, L.; Kerr, B.; Johnson, D.S.; et al. Pathogenicity and selective constraint on variation near splice sites. Genome Res. 2019, 29, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Sangermano, R.; Khan, M.; Cornelis, S.S.; Richelle, V.; Albert, S.; Garanto, A.; Elmelik, D.; Qamar, R.; Lugtenberg, D.; van den Born, L.I.; et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018, 28, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Reurink, J.; Oostrik, J.; Aben, M.; Ramos, M.G.; van Berkel, E.; Oldak, M.; van Wijk, E.; Kremer, H.; Roosing, S.; Cremers, F.P.M. Minigene-Based Splice Assays Reveal the Effect of Non-Canonical Splice Site Variants in USH2A. Int. J. Mol. Sci. 2022, 23, 13343. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Munoz, A.; Liquori, A.; Garcia-Bohorquez, B.; Jaijo, T.; Aller, E.; Millan, J.M.; García-García, G. Functional assays of non-canonical splice-site variants in inherited retinal dystrophies genes. Sci. Rep. 2022, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Toulis, V.; Cortes-Gonzalez, V.; Castro-Miro, M.; Sallum, J.F.; Catala-Mora, J.; Villanueva-Mendoza, C.; Ciccioli, M.; Gonzàlez-Duarte, R.; Valero, R.; Marfany, G. Increasing the Genetic Diagnosis Yield in Inherited Retinal Dystrophies: Assigning Pathogenicity to Novel Non-canonical Splice Site Variants. Genes 2020, 11, 378. [Google Scholar] [CrossRef]
- Weisschuh, N.; Mazzola, P.; Bertrand, M.; Haack, T.B.; Wissinger, B.; Kohl, S.; Stingl, K. Clinical Characteristics of POC1B-Associated Retinopathy and Assignment of Pathogenicity to Novel Deep Intronic and Non-Canonical Splice Site Variants. Int. J. Mol. Sci. 2021, 22, 5396. [Google Scholar] [CrossRef]
- Khan, M.; Cornelis, S.S.; Pozo-Valero, M.D.; Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTalbishi, A.; De Baere, E.; et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet. Med. 2020, 22, 1235–1246. [Google Scholar] [CrossRef]
- Wang, Z.; Xiao, X.; Van Nostrand, E.; Burge, C.B. General and specific functions of exonic splicing silencers in splicing control. Mol. Cell 2006, 23, 61–70. [Google Scholar] [CrossRef]
- Blencowe, B.J. Exonic splicing enhancers: Mechanism of action, diversity and role in human genetic diseases. Trends Biochem. Sci. 2000, 25, 106–110. [Google Scholar] [CrossRef]
- Hong, D.H.; Li, T. Complex expression pattern of RPGR reveals a role for purine-rich exonic splicing enhancers. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3373–3382. [Google Scholar]
- Glaus, E.; Schmid, F.; Da Costa, R.; Berger, W.; Neidhardt, J. Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR splice defect in patient-derived cells. Mol. Ther. 2011, 19, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Vossing, C.; Owczarek-Lipska, M.; Nagel-Wolfrum, K.; Reiff, C.; Juschke, C.; Neidhardt, J. Translational Read-Through Therapy of RPGR Nonsense Mutations. Int. J. Mol. Sci. 2020, 21, 8418. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | cNomen | gnomAD (%) | CADD PHRED | Dist. NSS | LSE | Splice AI (Score) | HGMD | ACMG | Zyg |
|---|---|---|---|---|---|---|---|---|---|
| CC2D2A | NM_001080522.2: c.1017C>T | 0.0008 | 0.0 | −1 | DL (0.05) | - | LB | het | |
| GRM6 | NM_000843.4: c.138G>A | 0.0822 | 12.1 | 154 | NAS | AG (0.17) | - | LB | het |
| TMEM67 | NM_153704.6: c.2848G>A | 0.0008 | 28.3 | −60 | all (0.00) | DM? | VUS | het | |
| INPP5E | NM_019892.6: c.1159+8C>T | 0.4092 | 0.4 | 8 | DL (0.02) | - | B | het | |
| NRL | NM_006177.3: c.14C>G | 0.0004 | 23.7 | 41 | all (0.00) | - | VUS | het | |
| RPGR | NM_001034853.2: c.1415-9A>G | - | 23.9 | −9 | NAS | AG/AL (0.98)/(0.95) | - | VUS | hem |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koller, S.; Beltraminelli, T.; Maggi, J.; Wlodarczyk, A.; Feil, S.; Baehr, L.; Gerth-Kahlert, C.; Menghini, M.; Berger, W. Functional Analysis of a Novel, Non-Canonical RPGR Splice Variant Causing X-Linked Retinitis Pigmentosa. Genes 2023, 14, 934. https://doi.org/10.3390/genes14040934
Koller S, Beltraminelli T, Maggi J, Wlodarczyk A, Feil S, Baehr L, Gerth-Kahlert C, Menghini M, Berger W. Functional Analysis of a Novel, Non-Canonical RPGR Splice Variant Causing X-Linked Retinitis Pigmentosa. Genes. 2023; 14(4):934. https://doi.org/10.3390/genes14040934
Chicago/Turabian StyleKoller, Samuel, Tim Beltraminelli, Jordi Maggi, Agnès Wlodarczyk, Silke Feil, Luzy Baehr, Christina Gerth-Kahlert, Moreno Menghini, and Wolfgang Berger. 2023. "Functional Analysis of a Novel, Non-Canonical RPGR Splice Variant Causing X-Linked Retinitis Pigmentosa" Genes 14, no. 4: 934. https://doi.org/10.3390/genes14040934