
Optimizing Sparse Testing for Genomic Prediction of Plant Breeding Crops

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Data Sets
2.1.1. Wheat Data
2.1.2. Maize Data
2.2. Statistical Model
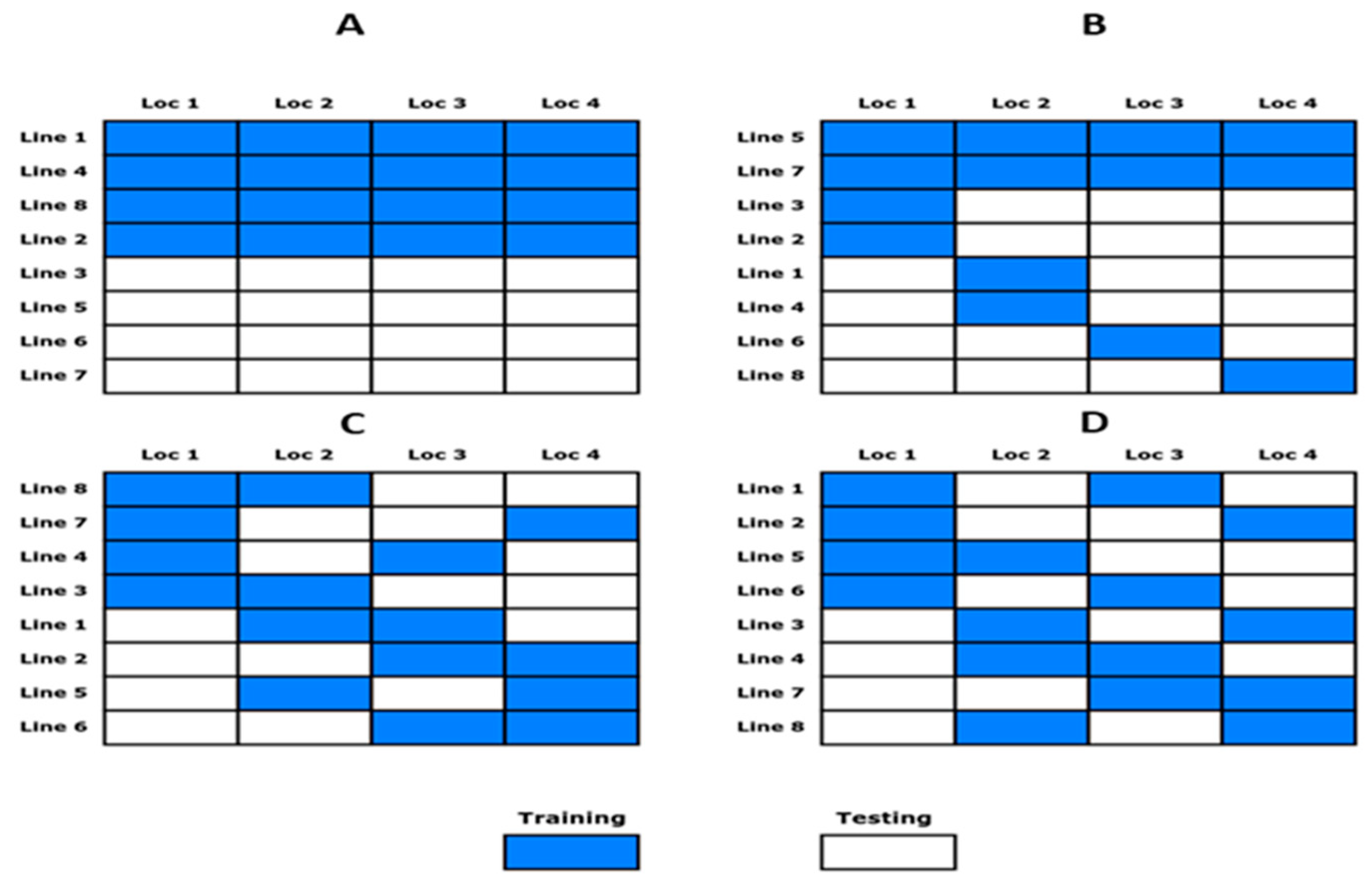
2.3. Sparse Testing Methods for the Allocation of Lines to Environments
2.3.1. Method 1 (M1)-Allocation of Fraction of Lines in All Locations
2.3.2. Method 2 (M2)-Allocation of Fraction of Lines with Some Shared Lines in Locations
2.3.3. Method 3 (M3)-Random Allocation of Fraction of Lines to Locations under Incomplete Locations
2.3.4. Method 4 (M4)-Allocation of Lines to Locations Using the IBD Principle
2.4. Cross-Validation Strategy
3. Results
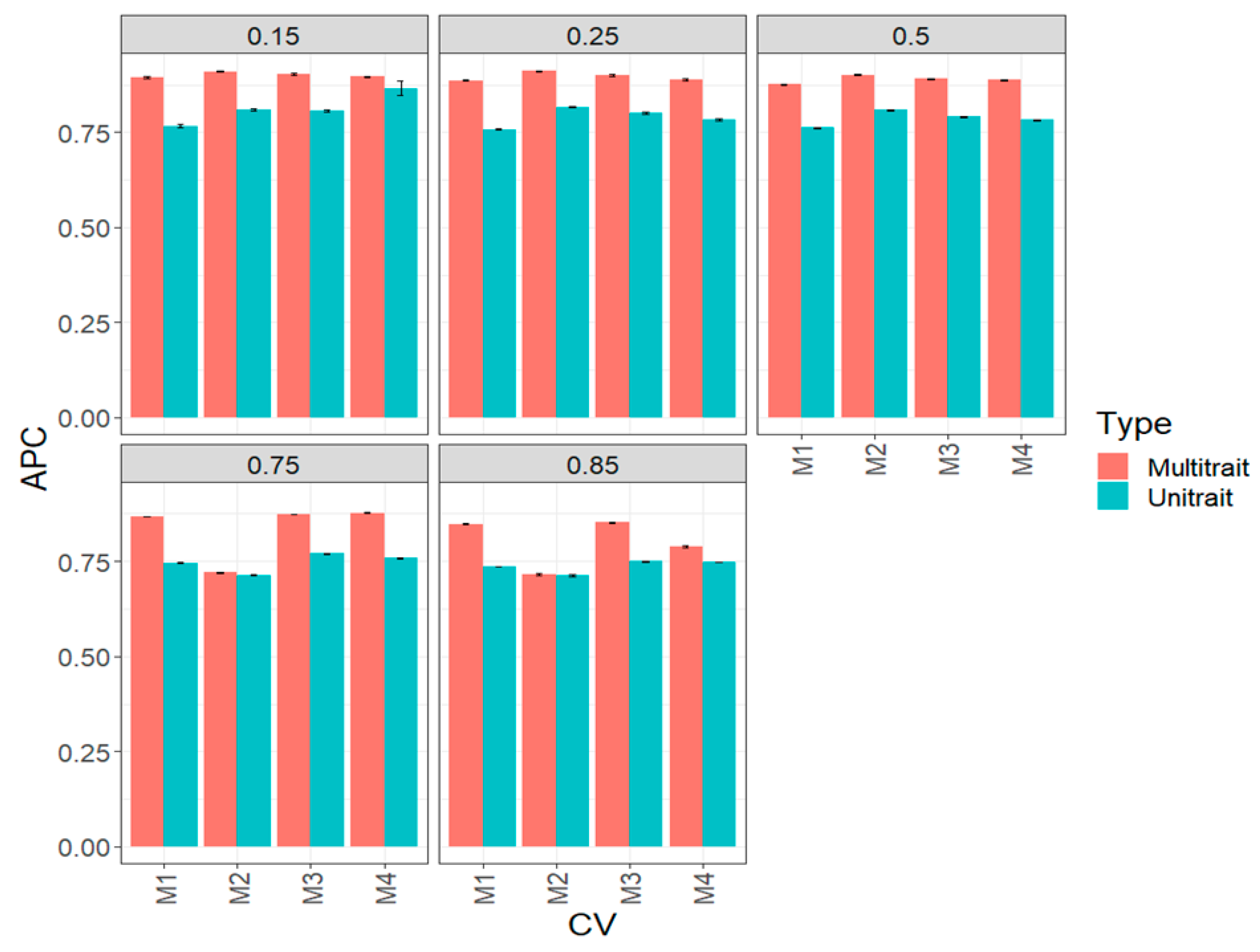
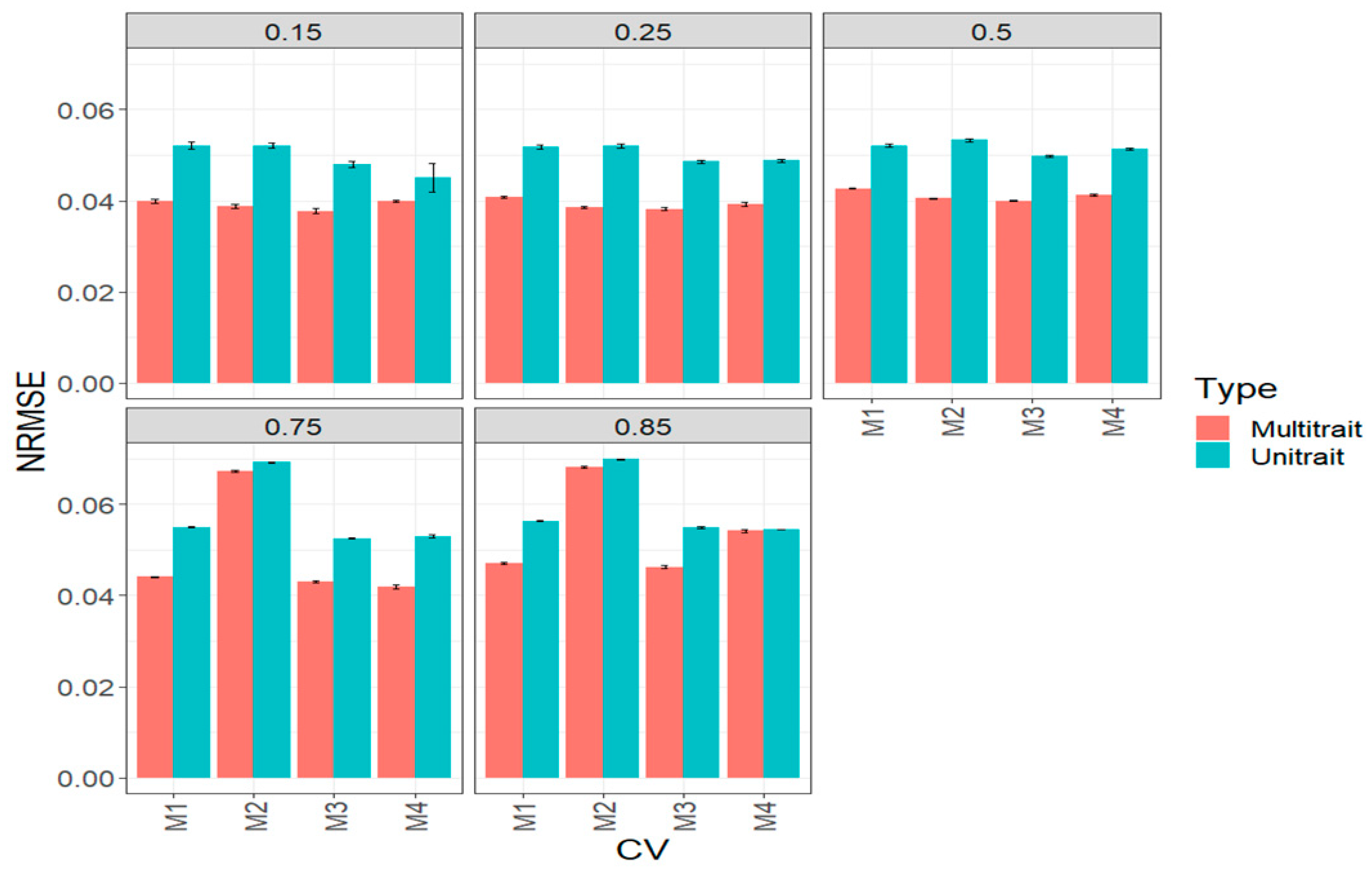
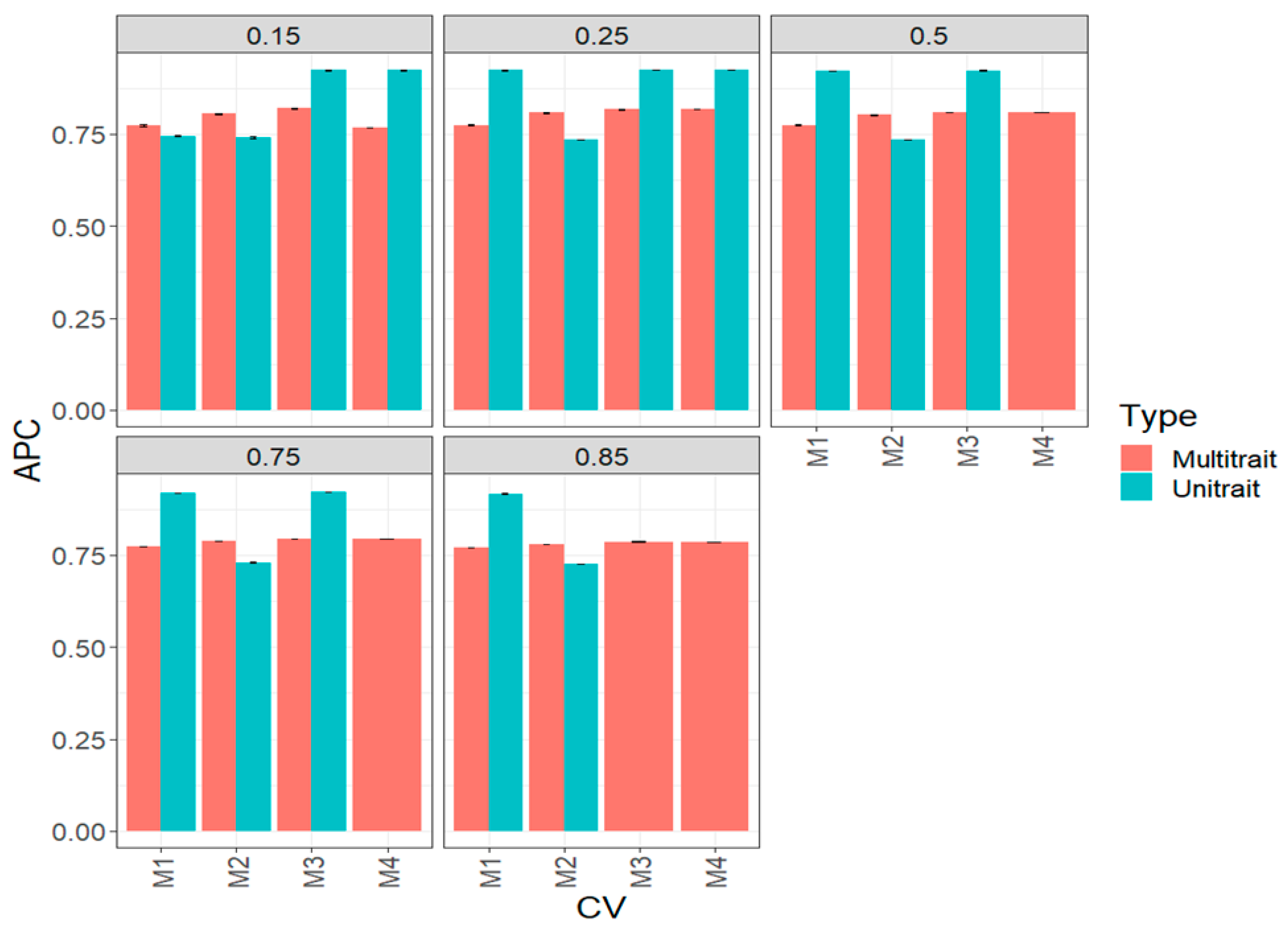
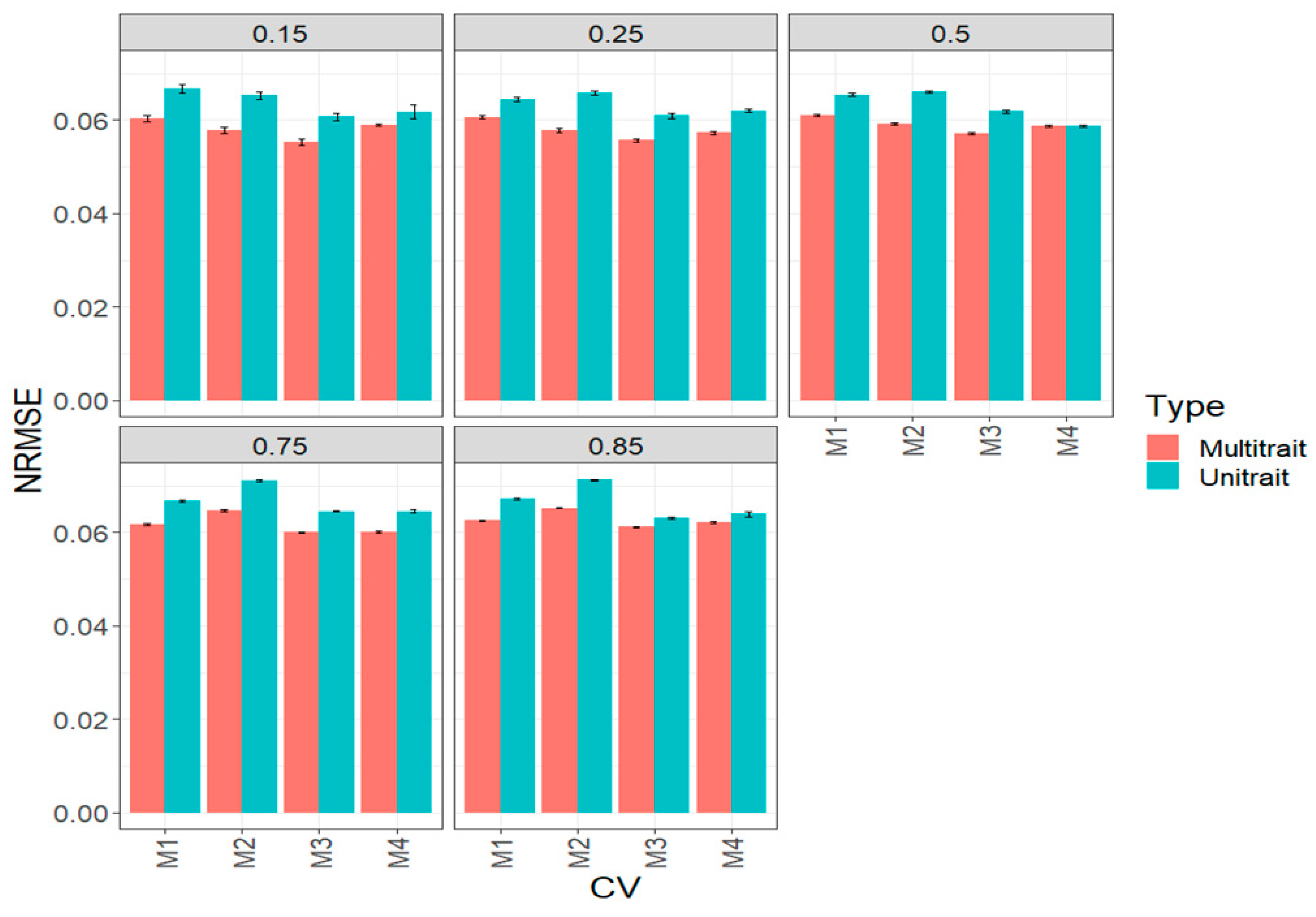
3.1. Complete Maize Data Set (Big Maize Data Set)
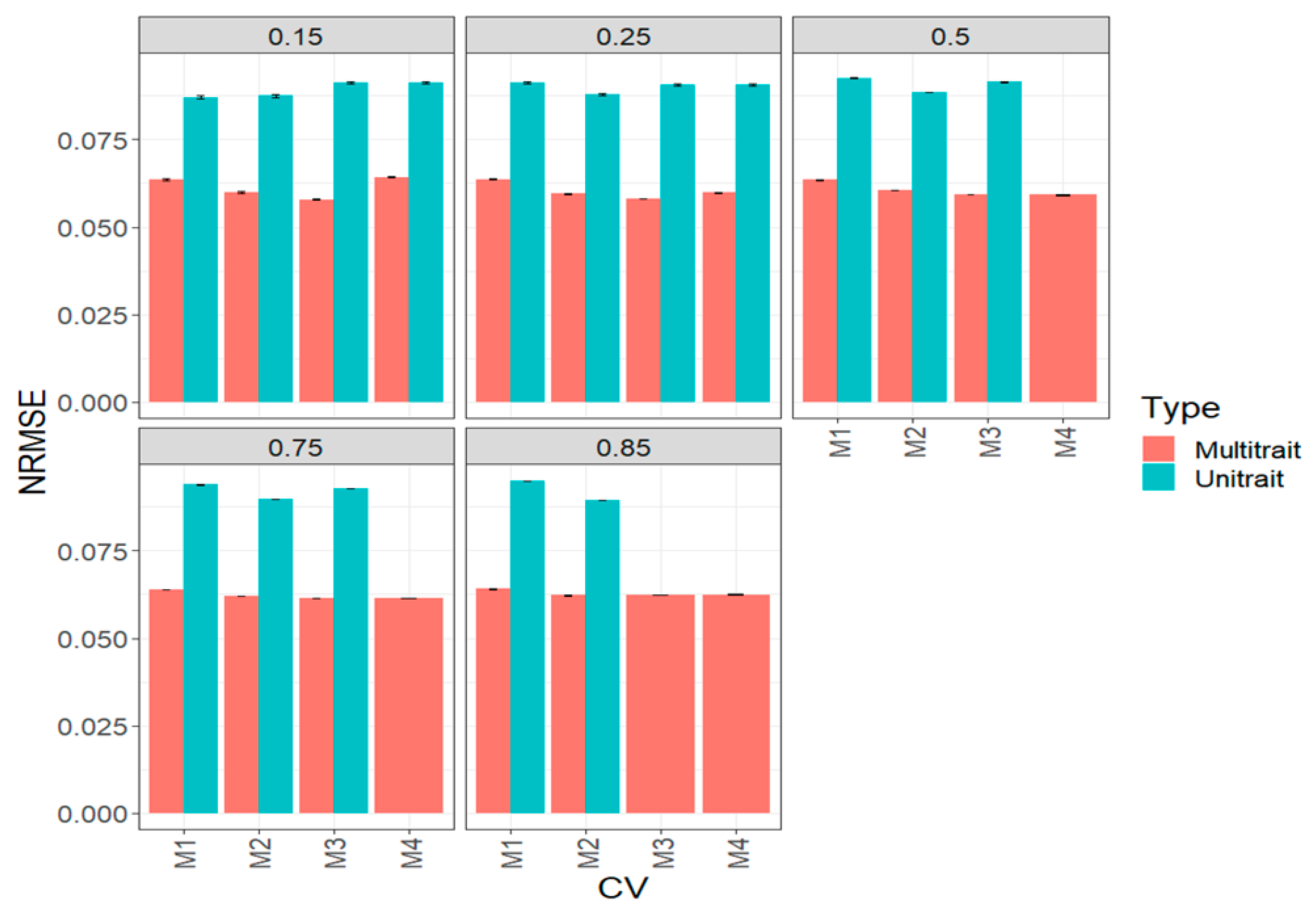
3.2. Complete Wheat Data Set (Big Wheat Data Set)
3.3. Across Data Sets
3.4. Assessing the Benefits of Sparse Testing Methods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | CV | Prop_Testing | Trait Type | NRMSE | NRMSE_SE | APC | APC_SE |
|---|---|---|---|---|---|---|---|
| Maize | M1 | 0.15 | Multi | 0.040 | 0.001 | 0.894 | 0.007 |
| Maize | M1 | 0.15 | Uni | 0.052 | 0.002 | 0.767 | 0.009 |
| Maize | M1 | 0.25 | Multi | 0.041 | 0.000 | 0.886 | 0.003 |
| Maize | M1 | 0.25 | Uni | 0.052 | 0.001 | 0.758 | 0.004 |
| Maize | M1 | 0.50 | Multi | 0.043 | 0.000 | 0.876 | 0.002 |
| Maize | M1 | 0.50 | Uni | 0.052 | 0.001 | 0.761 | 0.002 |
| Maize | M1 | 0.75 | Multi | 0.044 | 0.000 | 0.867 | 0.001 |
| Maize | M1 | 0.75 | Uni | 0.055 | 0.000 | 0.746 | 0.002 |
| Maize | M1 | 0.85 | Multi | 0.047 | 0.000 | 0.848 | 0.003 |
| Maize | M1 | 0.85 | Uni | 0.056 | 0.000 | 0.736 | 0.002 |
| Maize | M2 | 0.15 | Multi | 0.039 | 0.001 | 0.910 | 0.004 |
| Maize | M2 | 0.15 | Uni | 0.052 | 0.001 | 0.808 | 0.007 |
| Maize | M2 | 0.25 | Multi | 0.039 | 0.001 | 0.910 | 0.002 |
| Maize | M2 | 0.25 | Uni | 0.052 | 0.001 | 0.816 | 0.004 |
| Maize | M2 | 0.50 | Multi | 0.041 | 0.000 | 0.901 | 0.002 |
| Maize | M2 | 0.50 | Uni | 0.053 | 0.001 | 0.809 | 0.003 |
| Maize | M2 | 0.75 | Multi | 0.067 | 0.000 | 0.720 | 0.005 |
| Maize | M2 | 0.75 | Uni | 0.069 | 0.000 | 0.714 | 0.003 |
| Maize | M2 | 0.85 | Multi | 0.068 | 0.000 | 0.716 | 0.004 |
| Maize | M2 | 0.85 | Uni | 0.070 | 0.000 | 0.713 | 0.004 |
| Maize | M3 | 0.15 | Multi | 0.038 | 0.001 | 0.902 | 0.006 |
| Maize | M3 | 0.15 | Uni | 0.048 | 0.001 | 0.807 | 0.007 |
| Maize | M3 | 0.25 | Multi | 0.038 | 0.001 | 0.900 | 0.004 |
| Maize | M3 | 0.25 | Uni | 0.049 | 0.001 | 0.801 | 0.003 |
| Maize | M3 | 0.50 | Multi | 0.040 | 0.000 | 0.891 | 0.002 |
| Maize | M3 | 0.50 | Uni | 0.050 | 0.000 | 0.791 | 0.003 |
| Maize | M3 | 0.75 | Multi | 0.043 | 0.000 | 0.874 | 0.002 |
| Maize | M3 | 0.75 | Uni | 0.053 | 0.000 | 0.771 | 0.002 |
| Maize | M3 | 0.85 | Multi | 0.046 | 0.001 | 0.853 | 0.003 |
| Maize | M3 | 0.85 | Uni | 0.055 | 0.000 | 0.750 | 0.003 |
| Maize | M4 | 0.15 | Multi | 0.040 | 0.000 | 0.896 | 0.003 |
| Maize | M4 | 0.15 | Uni | 0.045 | 0.006 | 0.866 | 0.038 |
| Maize | M4 | 0.25 | Multi | 0.039 | 0.001 | 0.889 | 0.005 |
| Maize | M4 | 0.25 | Uni | 0.049 | 0.001 | 0.783 | 0.004 |
| Maize | M4 | 0.50 | Multi | 0.041 | 0.001 | 0.887 | 0.004 |
| Maize | M4 | 0.50 | Uni | 0.051 | 0.001 | 0.782 | 0.002 |
| Maize | M4 | 0.75 | Multi | 0.042 | 0.001 | 0.877 | 0.003 |
| Maize | M4 | 0.75 | Uni | 0.053 | 0.001 | 0.759 | 0.003 |
| Maize | M4 | 0.85 | Multi | 0.054 | 0.001 | 0.789 | 0.007 |
| Maize | M4 | 0.85 | Uni | 0.054 | 0.000 | 0.748 | 0.001 |
| Data Set | CV | Prop_Testing | Trait Type | NRMSE | NRMSE_SE | APC | APC_SE |
|---|---|---|---|---|---|---|---|
| Wheat | M1 | 0.15 | Multi | 0.064 | 0.001 | 0.774 | 0.004 |
| Wheat | M1 | 0.15 | Uni | 0.087 | 0.001 | 0.744 | 0.004 |
| Wheat | M1 | 0.25 | Multi | 0.064 | 0.000 | 0.775 | 0.002 |
| Wheat | M1 | 0.25 | Uni | 0.091 | 0.001 | 0.924 | 0.001 |
| Wheat | M1 | 0.50 | Multi | 0.064 | 0.000 | 0.775 | 0.001 |
| Wheat | M1 | 0.50 | Uni | 0.093 | 0.000 | 0.922 | 0.000 |
| Wheat | M1 | 0.75 | Multi | 0.064 | 0.000 | 0.774 | 0.001 |
| Wheat | M1 | 0.75 | Uni | 0.094 | 0.000 | 0.919 | 0.000 |
| Wheat | M1 | 0.85 | Multi | 0.064 | 0.000 | 0.772 | 0.001 |
| Wheat | M1 | 0.85 | Uni | 0.095 | 0.000 | 0.917 | 0.000 |
| Wheat | M2 | 0.15 | Multi | 0.060 | 0.001 | 0.805 | 0.004 |
| Wheat | M2 | 0.15 | Uni | 0.088 | 0.001 | 0.740 | 0.005 |
| Wheat | M2 | 0.25 | Multi | 0.059 | 0.000 | 0.809 | 0.002 |
| Wheat | M2 | 0.25 | Uni | 0.088 | 0.001 | 0.735 | 0.002 |
| Wheat | M2 | 0.50 | Multi | 0.060 | 0.000 | 0.802 | 0.001 |
| Wheat | M2 | 0.50 | Uni | 0.089 | 0.000 | 0.734 | 0.001 |
| Wheat | M2 | 0.75 | Multi | 0.062 | 0.000 | 0.787 | 0.001 |
| Wheat | M2 | 0.75 | Uni | 0.090 | 0.000 | 0.730 | 0.001 |
| Wheat | M2 | 0.85 | Multi | 0.062 | 0.000 | 0.780 | 0.001 |
| Wheat | M2 | 0.85 | Uni | 0.089 | 0.000 | 0.727 | 0.000 |
| Wheat | M3 | 0.15 | Multi | 0.058 | 0.001 | 0.820 | 0.003 |
| Wheat | M3 | 0.15 | Uni | 0.091 | 0.001 | 0.924 | 0.001 |
| Wheat | M3 | 0.25 | Multi | 0.058 | 0.000 | 0.817 | 0.001 |
| Wheat | M3 | 0.25 | Uni | 0.091 | 0.001 | 0.925 | 0.001 |
| Wheat | M3 | 0.50 | Multi | 0.059 | 0.000 | 0.809 | 0.001 |
| Wheat | M3 | 0.50 | Uni | 0.092 | 0.000 | 0.923 | 0.000 |
| Wheat | M3 | 0.75 | Multi | 0.061 | 0.000 | 0.794 | 0.001 |
| Wheat | M3 | 0.75 | Uni | 0.093 | 0.000 | 0.921 | 0.000 |
| Wheat | M3 | 0.85 | Multi | 0.062 | 0.000 | 0.787 | 0.001 |
| Wheat | M4 | 0.15 | Multi | 0.064 | 0.000 | 0.768 | 0.001 |
| Wheat | M4 | 0.15 | Uni | 0.091 | 0.001 | 0.924 | 0.001 |
| Wheat | M4 | 0.25 | Multi | 0.060 | 0.000 | 0.818 | 0.001 |
| Wheat | M4 | 0.25 | Uni | 0.091 | 0.001 | 0.925 | 0.001 |
| Wheat | M4 | 0.50 | Multi | 0.059 | 0.000 | 0.810 | 0.001 |
| Wheat | M4 | 0.75 | Multi | 0.061 | 0.000 | 0.794 | 0.001 |
| Wheat | M4 | 0.85 | Multi | 0.062 | 0.000 | 0.785 | 0.001 |
| CV | Prop_Testing | Trait Type | NRMSE | NRMSE_SE | APC | APC_SE |
|---|---|---|---|---|---|---|
| M1 | 0.15 | Multi | 0.060 | 0.001 | 0.773 | 0.010 |
| M2 | 0.15 | Multi | 0.058 | 0.001 | 0.808 | 0.009 |
| M3 | 0.15 | Multi | 0.055 | 0.001 | 0.818 | 0.008 |
| M4 | 0.15 | Multi | 0.059 | 0.001 | 0.787 | 0.005 |
| M1 | 0.15 | Uni | 0.067 | 0.002 | 0.737 | 0.012 |
| M2 | 0.15 | Uni | 0.065 | 0.002 | 0.770 | 0.011 |
| M3 | 0.15 | Uni | 0.061 | 0.002 | 0.803 | 0.009 |
| M4 | 0.15 | Uni | 0.062 | 0.003 | 0.800 | 0.020 |
| M1 | 0.25 | Multi | 0.061 | 0.001 | 0.773 | 0.005 |
| M2 | 0.25 | Multi | 0.058 | 0.001 | 0.806 | 0.004 |
| M3 | 0.25 | Multi | 0.056 | 0.001 | 0.811 | 0.004 |
| M4 | 0.25 | Multi | 0.057 | 0.001 | 0.808 | 0.004 |
| M1 | 0.25 | Uni | 0.064 | 0.001 | 0.757 | 0.007 |
| M2 | 0.25 | Uni | 0.066 | 0.001 | 0.766 | 0.006 |
| M3 | 0.25 | Uni | 0.061 | 0.001 | 0.791 | 0.005 |
| M4 | 0.25 | Uni | 0.062 | 0.001 | 0.785 | 0.004 |
| M1 | 0.50 | Multi | 0.061 | 0.000 | 0.770 | 0.003 |
| M2 | 0.50 | Multi | 0.059 | 0.000 | 0.799 | 0.002 |
| M3 | 0.50 | Multi | 0.057 | 0.000 | 0.803 | 0.003 |
| M4 | 0.50 | Multi | 0.059 | 0.000 | 0.794 | 0.003 |
| M1 | 0.50 | Uni | 0.065 | 0.001 | 0.752 | 0.004 |
| M2 | 0.50 | Uni | 0.066 | 0.001 | 0.767 | 0.003 |
| M3 | 0.50 | Uni | 0.062 | 0.001 | 0.786 | 0.004 |
| M4 | 0.50 | Uni | 0.059 | 0.000 | 0.773 | 0.004 |
| M1 | 0.75 | Multi | 0.062 | 0.000 | 0.766 | 0.002 |
| M2 | 0.75 | Multi | 0.065 | 0.000 | 0.756 | 0.002 |
| M3 | 0.75 | Multi | 0.060 | 0.000 | 0.782 | 0.002 |
| M4 | 0.75 | Multi | 0.060 | 0.001 | 0.783 | 0.003 |
| M1 | 0.75 | Uni | 0.067 | 0.000 | 0.746 | 0.003 |
| M2 | 0.75 | Uni | 0.071 | 0.000 | 0.735 | 0.003 |
| M3 | 0.75 | Uni | 0.065 | 0.000 | 0.765 | 0.003 |
| M4 | 0.75 | Uni | 0.065 | 0.001 | 0.726 | 0.005 |
| M1 | 0.85 | Multi | 0.063 | 0.000 | 0.761 | 0.002 |
| M2 | 0.85 | Multi | 0.065 | 0.000 | 0.748 | 0.002 |
| M3 | 0.85 | Multi | 0.061 | 0.000 | 0.772 | 0.002 |
| M4 | 0.85 | Multi | 0.062 | 0.000 | 0.751 | 0.004 |
| M1 | 0.85 | Uni | 0.067 | 0.000 | 0.744 | 0.003 |
| M2 | 0.85 | Uni | 0.071 | 0.000 | 0.731 | 0.003 |
| M3 | 0.85 | Uni | 0.063 | 0.000 | 0.736 | 0.003 |
| M4 | 0.85 | Uni | 0.064 | 0.001 | 0.725 | 0.010 |
References
- Crespo-Herrera, L.; Howard, R.; Piepho, H.P.; Pérez-Rodríguez, P.; Montesinos-López, O.A.; Burgueño, J.; Singh, R.; Mondal, S.; Jarquín, D.; Crossa, J. Genome-enabled prediction for sparse testing in multi-environmental wheat trials. Plant Genome 2021, 14, e20151. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.A.; Jarquín, D.; de Los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic Selection in Plant Breeding: Methods, Models, and Perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef]
- Roorkiwal, M.; Rathore, A.; Das, R.R.; Singh, M.K.; Jain, A.; Srinivasan, S.; Gaur, P.M.; Chellapilla, B.; Tripathi, S.; Li, Y.; et al. Genome-enabled prediction models for yield related traits in Chickpea. Front. Plant Sci. 2016, 7, 1666. [Google Scholar] [CrossRef]
- Wolfe, M.D.; Del Carpio, D.P.; Alabi, O.; Ezenwaka, L.C.; Ikeogu, U.N.; Kayondo, I.S.; Lozano, R.; Okeke, U.G.; Ozimati, A.A.; Williams, E.; et al. Prospects for Genomic Selection in Cassava Breeding. Plant Genome 2017, 10, 15. [Google Scholar] [CrossRef]
- Huang, M.; Balimponya, E.G.; Mgonja, E.M.; McHale, L.K.; Luzi-Kihupi, A.; Wang, G.-L.; Sneller, C.H. Use of genomic selection in breeding rice (Oryza sativa L.) for resistance to rice blast (Magnaporthe oryzae). Mol. Breed. 2019, 39, 114. [Google Scholar] [CrossRef]
- Cordell, H.J. Epistasis: What it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum. Mol. Genet. 2002, 11, 2463–2468. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.H.; Williams, S.M. Epistasis and its implications for personal genetics. Am. J. Hum. Genet. 2009, 85, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Lehner, B. Molecular mechanisms of epistasis within and between genes. Trends Genet. 2011, 27, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Buil, A.; Brown, A.A.; Lappalainen, T.; Vinuela, A.; Davies, M.N.; Zheng, H.F.; Richerds, J.B.; Glass, D.; Small, K.S.; Durbin, R.; et al. Gene-gene and gene environments interaction detected by transcriptome sequence analyses in twins. Nat. Genet. 2015, 47, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B.; Butler, D.G.; Cavanagh, C.R.; Cullis, B.R. Multiphase variety trials using both composite and individual replicate samples: A model-based design approach. J. Agric. Sci. 2015, 153, 1017–1029. [Google Scholar] [CrossRef]
- Smith, A.B.; Ganesalingam, A.; Kuchel, H.; Cullis, B.R. Factor analytic mixed models for the provision of grower information from national crop variety testing programs. Theor. Appl. Genet. 2015, 128, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Jarquín, D.; Howard, R.; Crossa, J.; Beyene, Y.; Gowda, M.; Martini, J.W.R.; Pazaran, G.C.; Burgueño, J.; Pacheco, A.; Grondona, M.; et al. Genomic prediction enhanced sparse testing for multi-environment trials. G3 Genes Genomes Genet. 2020, 10, 2725–2739. [Google Scholar] [CrossRef] [PubMed]
- Federer, W.T.; Nair, R.C.; Raghavarao, D. Some augmented row-column designs. Biometrics 1975, 31, 361–373. [Google Scholar] [CrossRef]
- Piepho, H.-P.; Williams, E.R. Augmented Row–Column Designs for a Small Number of Checks. Agron. J. 2016, 108, 2256–2262. [Google Scholar] [CrossRef]
- Coombes, N.E. DiGGeR, a Spatial Design Program. Biometric Bulletin; NSW Department of Primary Industries: Orange, NSW, Australia, 2009. [Google Scholar]
- Butler, D.G.; Cullis, B.R.; Gilmour, A.R.; Gogel, B.G.; Thomson, R. ASRmel-R Reference Manual Version 4; VSN International Ltd.: Hemel Hempstead, UK, 2017. [Google Scholar]
- Van Raden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; de los Campos, G. Genome-Wide Regression and Prediction with the BGLR Statistical Package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Montesinos-Lopez, O.A.; Montesinos-Lopez, A.; Acosta, R.; Varshney, R.K.; Bentley, A.; Crossa, J. Using an incomplete block design to allocate lines to environments improves sparse genome-based prediction in plant breeding. Plant Genome 2022, 15, e20194. [Google Scholar] [CrossRef] [PubMed]
- Möhring, J.; Piepho, H.-P. Comparison of weighting in two-stage analysis of plant breeding trials. Crop Sci. 2009, 49, 1977–1988. [Google Scholar] [CrossRef]
- Damesa, T.M.; Möhring, J.; Worku, M.; Piepho, H.-P. One step at a time: Stage-wise analysis of a series of experiments. Agron. J. 2017, 109, 845–857. [Google Scholar] [CrossRef]
- Montesinos-López, O.A.; Montesinos-López, A.; Crossa, J. Multivariate Statistical Machine Learning Methods for Genomic Prediction; Montesinos López, O.A., Montesinos López, A., Crossa, J., Eds.; Springer International Publishing: Cham, Switzerland, 2022; ISBN 978-3-030-89010-0. [Google Scholar]







| Sparse Designs with Different % of trn Data | Gains (or Loss) of Sparse Designs for Each % of trn | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Concept | Standard | 85 | 75 | 50 | 25 | 15 | 85 | 75 | 50 | 25 | 15 |
| Scenario 1 | |||||||||||
| Total trts | 250 | 294 | 333 | 500 | 1000 | 1667 | 17.60 | 33.20 | 100.00 | 300.00 | 566.80 |
| New lines | 225 | 269 | 308 | 475 | 975 | 1642 | 19.56 | 36.89 | 111.11 | 333.33 | 629.78 |
| Checks | 25 | 25 | 25 | 25 | 25 | 25 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Reps | 1 | 0.85 | 0.75 | 0.5 | 0.25 | 0.15 | −15.00 | −25.00 | −50.00 | −75.00 | −85.00 |
| Locs | 4 | 4 | 4 | 4 | 4 | 4 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| R | 4 | 3.4 | 3 | 2 | 1 | 0.6 | −15.00 | −25.00 | −50.00 | −75.00 | −85.00 |
| Total_plots | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Total trts | 250 | 294 | 333 | 500 | 1000 | 1667 | 17.60 | 33.20 | 100.00 | 300.00 | 566.80 |
| Plots/trt | 4.44 | 3.72 | 3.25 | 2.11 | 1.03 | 0.61 | −16.36 | −26.95 | −52.63 | −76.92 | −86.30 |
| Scenario 2 | |||||||||||
| Total trts | 4500 | 5294 | 6000 | 9000 | 18000 | 30000 | 17.64 | 33.33 | 100.00 | 300.00 | 566.67 |
| New lines | 4450 | 5244 | 5950 | 8950 | 17950 | 29950 | 17.84 | 33.71 | 101.12 | 303.37 | 573.03 |
| Checks | 50 | 50 | 50 | 50 | 50 | 50 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Reps | 1 | 0.85 | 0.75 | 0.5 | 0.25 | 0.15 | −15.00 | −25.00 | −50.00 | −75.00 | −85.00 |
| Locs | 4 | 4 | 4 | 4 | 4 | 4 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| R | 4 | 3.4 | 3 | 2 | 1 | 0.6 | −15.00 | −25.00 | −50.00 | −75.00 | −85.00 |
| Tot_Plots | 18000 | 18000 | 18000 | 18000 | 18000 | 18000 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Total trts | 4500 | 5294 | 6000 | 9000 | 18000 | 30000 | 17.64 | 33.33 | 100.00 | 300.00 | 566.67 |
| Plots/trt | 4.04 | 3.43 | 3.03 | 2.01 | 1.00 | 0.60 | −15.14 | −25.21 | −50.28 | −75.21 | −85.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montesinos-López, O.A.; Saint Pierre, C.; Gezan, S.A.; Bentley, A.R.; Mosqueda-González, B.A.; Montesinos-López, A.; van Eeuwijk, F.; Beyene, Y.; Gowda, M.; Gardner, K.; et al. Optimizing Sparse Testing for Genomic Prediction of Plant Breeding Crops. Genes 2023, 14, 927. https://doi.org/10.3390/genes14040927
Montesinos-López OA, Saint Pierre C, Gezan SA, Bentley AR, Mosqueda-González BA, Montesinos-López A, van Eeuwijk F, Beyene Y, Gowda M, Gardner K, et al. Optimizing Sparse Testing for Genomic Prediction of Plant Breeding Crops. Genes. 2023; 14(4):927. https://doi.org/10.3390/genes14040927
Chicago/Turabian StyleMontesinos-López, Osval A., Carolina Saint Pierre, Salvador A. Gezan, Alison R. Bentley, Brandon A. Mosqueda-González, Abelardo Montesinos-López, Fred van Eeuwijk, Yoseph Beyene, Manje Gowda, Keith Gardner, and et al. 2023. "Optimizing Sparse Testing for Genomic Prediction of Plant Breeding Crops" Genes 14, no. 4: 927. https://doi.org/10.3390/genes14040927