

Retinal Phenotyping of a Murine Model of Lafora Disease

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Line
2.2. Electroretinogram Testing, Retinal Photography and Optical Coherence Tomography
2.3. Histological Analysis
3. Results
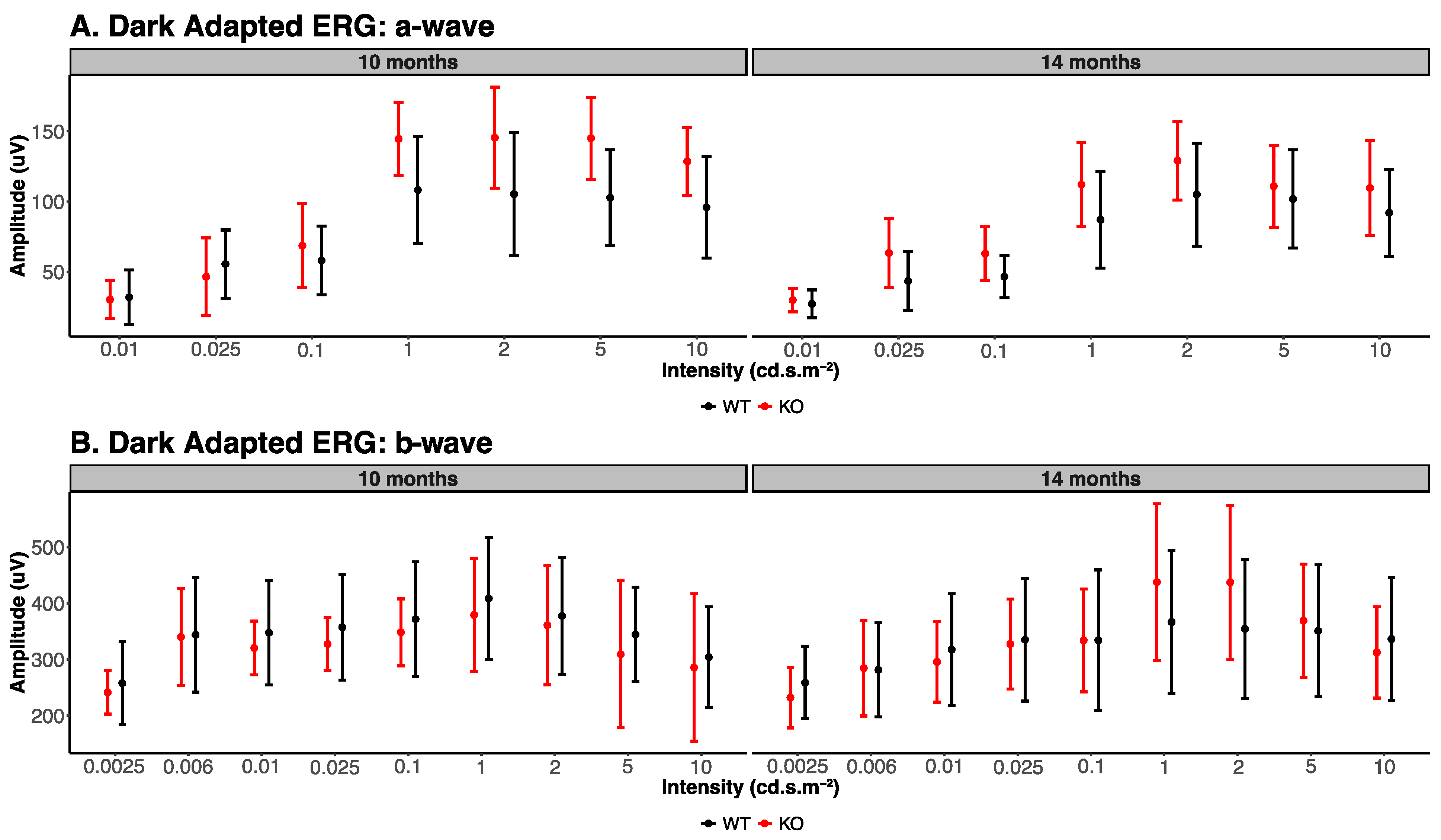
3.1. Electroretinogram Analysis
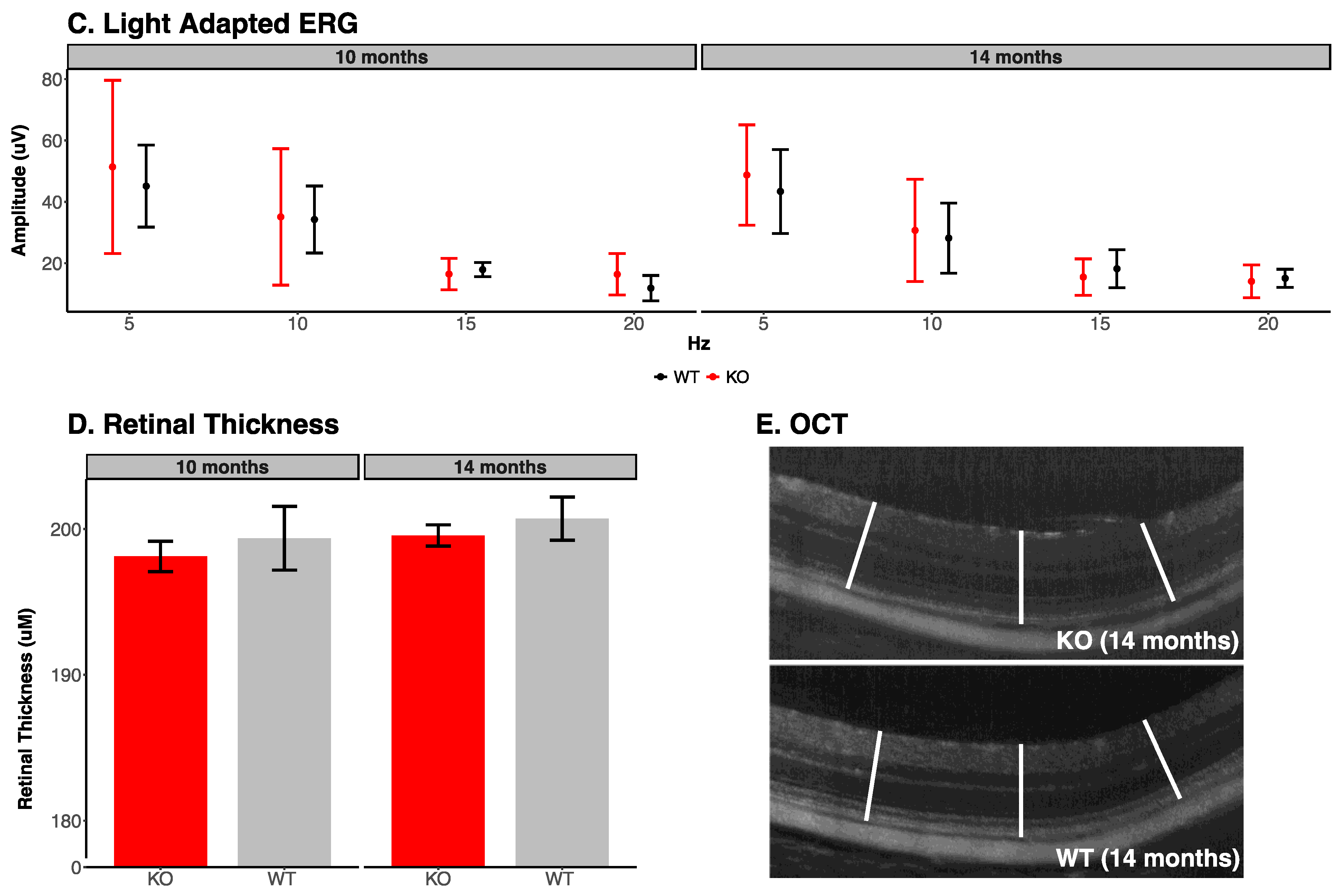
3.2. Retinal Photography and Optical Coherence Tomography
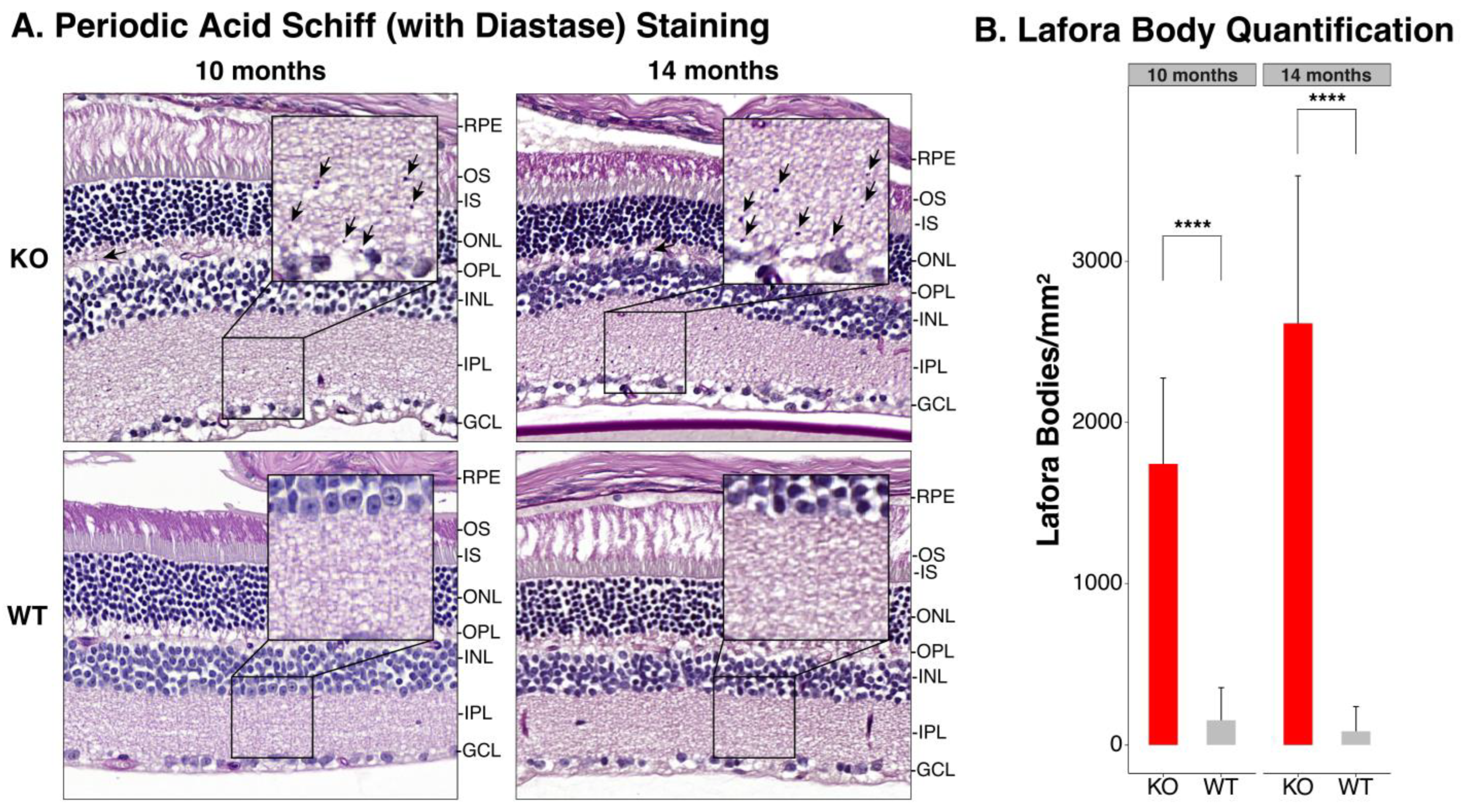
3.3. Periodic Acid Schiff-Diastase Staining of the Retina
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lafora, G.R.; Glueck, B. Beitrag zur Histopathologie der myoklonischen Epilepsie. Z. Gesamte Neurol. Psychiatr. 1911, 6, 1–14. [Google Scholar] [CrossRef]
- Minassian, B.A.; Lee, J.R.; Herbrick, J.A.; Huizenga, J.; Soder, S.; Mungall, A.J.; Dunham, I.; Gardner, R.; Fong, C.Y.; Carpenter, S.; et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 1998, 20, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Young, E.J.; Ianzano, L.; Munteanu, I.; Zhao, X.; Christopoulos, C.C.; Avanzini, G.; Elia, M.; Ackerley, C.A.; Jovic, N.J.; et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 2003, 35, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Puri, R.; Singh, S.; Mittal, S.; Dubey, D. Recent advances in the molecular basis of Lafora’s progressive myoclonus epilepsy. J. Hum. Genet. 2006, 51, 1–8. [Google Scholar] [CrossRef]
- Minassian, B.A. Lafora’s disease: Towards a clinical, pathologic, and molecular synthesis. Pediatr. Neurol. 2001, 25, 21–29. [Google Scholar] [CrossRef]
- Turnbull, J.; Tiberia, E.; Striano, P.; Genton, P.; Carpenter, S.; Ackerley, C.A.; Minassian, B.A. Lafora disease. Epileptic Disord. 2016, 18, 38–62. [Google Scholar] [CrossRef]
- Striano, P.; Zara, F.; Turnbull, J.; Girard, J.M.; Ackerley, C.A.; Cervasio, M.; De Rosa, G.; Del Basso-De Caro, M.L.; Striano, S.; Minassian, B.A. Typical progression of myoclonic epilepsy of the Lafora type: A case report. Nat. Clin. Pract. Neurol. 2008, 4, 106–111. [Google Scholar] [CrossRef]
- Franceschetti, S.; Gambardella, A.; Canafoglia, L.; Striano, P.; Lohi, H.; Gennaro, E.; Ianzano, L.; Veggiotti, P.; Sofia, V.; Biondi, R.; et al. Clinical and genetic findings in 26 Italian patients with Lafora disease. Epilepsia 2006, 47, 640–643. [Google Scholar] [CrossRef]
- Araya, N.; Takahashi, Y.; Shimono, M.; Fukuda, T.; Kato, M.; Nakashima, M.; Matsumoto, N.; Saitsu, H. A recurrent homozygous NHLRC1 variant in siblings with Lafora disease. Hum. Genome Var. 2018, 5, 16. [Google Scholar] [CrossRef]
- Acharya, J.N.; Satishchandra, P.; Asha, T.; Shankar, S.K. Lafora’s disease in south India: A clinical, electrophysiologic, and pathologic study. Epilepsia 1993, 34, 476–487. [Google Scholar] [CrossRef]
- Ferlazzo, E.; Canafoglia, L.; Michelucci, R.; Gambardella, A.; Gennaro, E.; Pasini, E.; Riguzzi, P.; Plasmati, R.; Volpi, L.; Labate, A.; et al. Mild Lafora disease: Clinical, neurophysiologic, and genetic findings. Epilepsia 2014, 55, e129–e133. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.; Beydoun, A.; Drury, I. Longitudinal EEG studies in a kindred with Lafora disease. Epilepsia 1991, 32, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Macri, A.; Tumber, A.; Koukas, N.; Ahonen, S.; Striano, P.; Minassian, B. Ocular phenotype and electroretinogram abnormalities in Lafora disease: A “window to the brain”. Neurology 2018, 91, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Orsini, A.; Ferrari, D.; Riva, A.; Santangelo, A.; Macri, A.; Freri, E.; Canafoglia, L.; D’Aniello, A.; Di Gennaro, G.; Massimetti, G.; et al. Ocular phenotype and electroretinogram abnormalities in Lafora disease and correlation with disease stage. J. Neurol. 2022, 269, 3597–3604. [Google Scholar] [CrossRef]
- Heitkotter, H.; Linderman, R.E.; Cava, J.A.; Woertz, E.N.; Mastey, R.R.; Summerfelt, P.; Chui, T.Y.; Rosen, R.B.; Patterson, E.J.; Vincent, A.; et al. Retinal alterations in patients with Lafora disease. Am. J. Ophthalmol. Case Rep. 2021, 23, 101146. [Google Scholar] [CrossRef]
- Holland, J.M.; Davis, W.C.; Prieur, D.J.; Collins, G.H. Lafora’s disease in the dog. A comparative study. Am. J. Pathol. 1970, 58, 509–530. [Google Scholar]
- Kaiser, E.; Krauser, K.; Schwartz-Porsche, D. Lafora disease (progressive myoclonic epilepsy) in the Bassett hound—Possibility of early diagnosis using muscle biopsy? Tierarztl. Prax. 1991, 19, 290–295. [Google Scholar]
- Davis, K.E.; Finnie, J.W.; Hooper, P.T. Lafora’s disease in a dog. Aust. Vet. J. 1990, 67, 192–193. [Google Scholar] [CrossRef]
- Hajek, I.; Kettner, F.; Simerdova, V.; Rusbridge, C.; Wang, P.; Minassian, B.A.; Palus, V. NHLRC1 repeat expansion in two beagles with Lafora disease. J. Small Anim. Pract. 2016, 57, 650–652. [Google Scholar] [CrossRef]
- Lohi, H.; Young, E.J.; Fitzmaurice, S.N.; Rusbridge, C.; Chan, E.M.; Vervoort, M.; Turnbull, J.; Zhao, X.C.; Ianzano, L.; Paterson, A.D.; et al. Expanded repeat in canine epilepsy. Science 2005, 307, 81. [Google Scholar] [CrossRef]
- Demeny, H.; Florea, B.; Tabaran, F.; Danciu, C.G.; Ognean, L. EEG Patterns Orienting to Lafora Disease Diagnosis-A Case Report in Two Beagles. Front. Vet. Sci. 2020, 7, 589430. [Google Scholar] [CrossRef] [PubMed]
- Mari, L.; Comero, G.; Mueller, E.; Kuehnlein, P.; Kehl, A. NHLRC1 homozygous dodecamer expansion in a Newfoundland dog with Lafora disease. J. Small Anim. Pract. 2021, 62, 1030–1032. [Google Scholar] [CrossRef]
- Schoeman, T.; Williams, J.; van Wilpe, E. Polyglucosan storage disease in a dog resembling Lafora’s disease. J. Vet. Intern. Med. 2002, 16, 201–207. [Google Scholar] [PubMed]
- Swain, L.; Key, G.; Tauro, A.; Ahonen, S.; Wang, P.; Ackerley, C.; Minassian, B.A.; Rusbridge, C. Lafora disease in miniature Wirehaired Dachshunds. PLoS ONE 2017, 12, e0182024. [Google Scholar] [CrossRef] [PubMed]
- Hegreberg, G.A.; Padgett, G.A. Inherited progressive epilepsy of the dog with comparisons to Lafora’s disease of man. Fed. Proc. 1976, 35, 1202–1205. [Google Scholar]
- Gredal, H.; Berendt, M.; Leifsson, P.S. Progressive myoclonus epilepsy in a beagle. J. Small Anim. Pract. 2003, 44, 511–514. [Google Scholar] [CrossRef]
- Ganesh, S.; Delgado-Escueta, A.V.; Sakamoto, T.; Avila, M.R.; Machado-Salas, J.; Hoshii, Y.; Akagi, T.; Gomi, H.; Suzuki, T.; Amano, K.; et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 2002, 11, 1251–1262. [Google Scholar] [CrossRef]
- Turnbull, J.; Wang, P.; Girard, J.M.; Ruggieri, A.; Wang, T.J.; Draginov, A.G.; Kameka, A.P.; Pencea, N.; Zhao, X.; Ackerley, C.A.; et al. Glycogen hyperphosphorylation underlies lafora body formation. Ann. Neurol. 2010, 68, 925–933. [Google Scholar] [CrossRef]
- Valles-Ortega, J.; Duran, J.; Garcia-Rocha, M.; Bosch, C.; Saez, I.; Pujadas, L.; Serafin, A.; Canas, X.; Soriano, E.; Delgado-Garcia, J.M.; et al. Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO Mol. Med. 2011, 3, 667–681. [Google Scholar] [CrossRef]
- Garcia-Cabrero, A.M.; Marinas, A.; Guerrero, R.; de Cordoba, S.R.; Serratosa, J.M.; Sanchez, M.P. Laforin and malin deletions in mice produce similar neurologic impairments. J. Neuropathol. Exp. Neurol. 2012, 71, 413–421. [Google Scholar] [CrossRef]
- Criado, O.; Aguado, C.; Gayarre, J.; Duran-Trio, L.; Garcia-Cabrero, A.M.; Vernia, S.; San Millan, B.; Heredia, M.; Roma-Mateo, C.; Mouron, S.; et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum. Mol. Genet. 2012, 21, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Austin, J.; Witmer, F.; Trueb, L. Studies in myoclonus epilepsy (Lafora body form). II. Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology 1970, 20, 160–176. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, F.; Santangelo, R.; Gasparo-Rippa, P.; Santoro, M. Biopsy findings (cerebral cortex, muscle, skin) in Lafora disease. Acta Neurol. 1987, 9, 81–94. [Google Scholar]
- Shirozu, M.; Hashimoto, M.; Tomimatsu, M.; Nakazawa, Y.; Anraku, S.; Nagata, M. Lafora disease diagnosed by skin biopsy. Kurume Med. J. 1985, 32, 311–313. [Google Scholar] [CrossRef]
- Carpenter, S.; Karpati, G. Ultrastructural findings in Lafora disease. Ann. Neurol. 1981, 10, 63–64. [Google Scholar] [CrossRef]
- Berard-Badier, M.; Pellissier, J.F.; Gambarelli, D.; de Barsy, T.; Roger, J.; Toga, M. The retina in Lafora disease: Light and electron microscopy. In Current Research in Ophthalmic Electron Microscopy; Springer: Berlin/Heidelberg, Germany, 1980; Volume 212, pp. 285–294. [Google Scholar]
- Andrade, D.M.; Ackerley, C.A.; Minett, T.S.; Teive, H.A.; Bohlega, S.; Scherer, S.W.; Minassian, B.A. Skin biopsy in Lafora disease: Genotype-phenotype correlations and diagnostic pitfalls. Neurology 2003, 61, 1611–1614. [Google Scholar] [CrossRef]
- von Klopmann, T.; Ahonen, S.; Espadas-Santiuste, I.; Matiasek, K.; Sanchez-Masian, D.; Rupp, S.; Vandenberghe, H.; Rose, J.; Wang, T.; Wang, P.; et al. Canine Lafora Disease: An Unstable Repeat Expansion Disorder. Life 2021, 11, 689. [Google Scholar] [CrossRef]
- Mitra, S.; Gumusgoz, E.; Minassian, B.A. Lafora disease: Current biology and therapeutic approaches. Rev. Neurol. 2022, 178, 315–325. [Google Scholar] [CrossRef]
- Drexler, W.; Fujimoto, J.G. State-of-the-art retinal optical coherence tomography. Prog. Retin. Eye Res. 2008, 27, 45–88. [Google Scholar] [CrossRef]
- Fischer, M.D.; Huber, G.; Beck, S.C.; Tanimoto, N.; Muehlfriedel, R.; Fahl, E.; Grimm, C.; Wenzel, A.; Reme, C.E.; van de Pavert, S.A.; et al. Noninvasive, in vivo assessment of mouse retinal structure using optical coherence tomography. PLoS ONE 2009, 4, e7507. [Google Scholar] [CrossRef]
- Grieve, K.; Thouvenin, O.; Sengupta, A.; Borderie, V.M.; Paques, M. Appearance of the Retina With Full-Field Optical Coherence Tomography. Investig. Ophthalmol. Vis. Sci. 2016, 57, OCT96–OCT104. [Google Scholar] [CrossRef] [PubMed]
- Jagodzinska, J.; Sarzi, E.; Cavalier, M.; Seveno, M.; Baecker, V.; Hamel, C.; Pequignot, M.; Delettre, C. Optical Coherence Tomography: Imaging Mouse Retinal Ganglion Cells In Vivo. J. Vis. Exp. 2017, 127, e55865. [Google Scholar]
- Fu, D.A.; Campbell-Thompson, M. Periodic Acid-Schiff Staining with Diastase. Methods Mol. Biol. 2017, 1639, 145–149. [Google Scholar] [PubMed]
- Ng, I.O.; Sturgess, R.P.; Williams, R.; Portmann, B. Ground-glass hepatocytes with Lafora body like inclusions—Histochemical, immunohistochemical and electronmicroscopic characterization. Histopathology 1990, 17, 109–115. [Google Scholar] [CrossRef]
- Ganesh, S.; Tsurutani, N.; Suzuki, T.; Hoshii, Y.; Ishihara, T.; Delgado-Escueta, A.V.; Yamakawa, K. The carbohydrate-binding domain of Lafora disease protein targets Lafora polyglucosan bodies. Biochem. Biophys. Res. Commun. 2004, 313, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.H.; Invergo, B.; Shimomura, K.; Takahashi, J.S.; Troy, J.B. Interpretation of the mouse electroretinogram. Doc. Ophthalmol. 2007, 115, 127–136. [Google Scholar] [CrossRef]
- Goto, Y.; Peachey, N.S.; Ziroli, N.E.; Seiple, W.H.; Gryczan, C.; Pepperberg, D.R.; Naash, M.I. Rod phototransduction in transgenic mice expressing a mutant opsin gene. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 1996, 13, 577–585. [Google Scholar] [PubMed]
- Robson, J.G.; Maeda, H.; Saszik, S.M.; Frishman, L.J. In vivo studies of signaling in rod pathways of the mouse using the electroretinogram. Vis. Res. 2004, 44, 3253–3268. [Google Scholar] [CrossRef] [PubMed]
- Green, D.G.; Guo, H.; Pillers, D.A. Normal photoresponses and altered b-wave responses to APB in the mdxCv3 mouse isolated retina ERG supports role for dystrophin in synaptic transmission. Vis. Neurosci. 2004, 21, 739–747. [Google Scholar] [CrossRef]
- Biel, M.; Seeliger, M.; Pfeifer, A.; Kohler, K.; Gerstner, A.; Ludwig, A.; Jaissle, G.; Fauser, S.; Zrenner, E.; Hofmann, F. Selective loss of cone function in mice lacking the cyclic nucleotide-gated channel CNG3. Proc. Natl. Acad. Sci. USA 1999, 96, 7553–7557. [Google Scholar] [CrossRef]
- Tanimoto, N.; Sothilingam, V.; Kondo, M.; Biel, M.; Humphries, P.; Seeliger, M.W. Electroretinographic assessment of rod- and cone-mediated bipolar cell pathways using flicker stimuli in mice. Sci. Rep. 2015, 5, 10731. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ball, S.L.; Peachey, N.S. Pharmacological studies of the mouse cone electroretinogram. Vis. Neurosci. 2005, 22, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Krishna, V.R.; Alexander, K.R.; Peachey, N.S. Temporal properties of the mouse cone electroretinogram. J. Neurophysiol. 2002, 87, 42–48. [Google Scholar] [CrossRef]
- Ekesten, B.; Gouras, P.; Moschos, M. Cone properties of the light-adapted murine ERG. Doc. Ophthalmol. 1998, 97, 23–31. [Google Scholar] [CrossRef]
- Garyali, P.; Siwach, P.; Singh, P.K.; Puri, R.; Mittal, S.; Sengupta, S.; Parihar, R.; Ganesh, S. The malin-laforin complex suppresses the cellular toxicity of misfolded proteins by promoting their degradation through the ubiquitin-proteasome system. Hum. Mol. Genet. 2009, 18, 688–700. [Google Scholar] [CrossRef]
- Aguado, C.; Sarkar, S.; Korolchuk, V.I.; Criado, O.; Vernia, S.; Boya, P.; Sanz, P.; de Cordoba, S.R.; Knecht, E.; Rubinsztein, D.C. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum. Mol. Genet. 2010, 19, 2867–2876. [Google Scholar] [CrossRef]
- Knecht, E.; Aguado, C.; Sarkar, S.; Korolchuk, V.I.; Criado-Garcia, O.; Vernia, S.; Boya, P.; Sanz, P.; Rodriguez de Cordoba, S.; Rubinsztein, D.C. Impaired autophagy in Lafora disease. Autophagy 2010, 6, 991–993. [Google Scholar] [CrossRef]
- Puri, R.; Ganesh, S. Laforin in autophagy: A possible link between carbohydrate and protein in Lafora disease? Autophagy 2010, 6, 1229–1231. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.N.; Maity, R.; Sharma, J.; Dey, P.; Shankar, S.K.; Satishchandra, P.; Jana, N.R. Sequestration of chaperones and proteasome into Lafora bodies and proteasomal dysfunction induced by Lafora disease-associated mutations of malin. Hum. Mol. Genet. 2010, 19, 4726–4734. [Google Scholar] [CrossRef]
- Puri, R.; Suzuki, T.; Yamakawa, K.; Ganesh, S. Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for Lafora disease. Hum. Mol. Genet. 2012, 21, 175–184. [Google Scholar] [CrossRef]
- Campbell, M.C.W.; Emptage, L.; Corapi, F. Effects of Lafora disease on the retina of a transgenic mouse model. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4319. [Google Scholar]
- Burns, S.A.; Elsner, A.E.; Sapoznik, K.A.; Warner, R.L.; Gast, T.J. Adaptive optics imaging of the human retina. Prog. Retin. Eye Res. 2019, 68, 1–30. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Dark Adapted ERG Parameters (10 Months) | ||||||
|---|---|---|---|---|---|---|
| Stimulus Flash in cd·s·m−2 | A Wave | B Wave | ||||
| Knockout | Wildtype | p-Value | Knockout | Wildtype | p-Value | |
| 0.0025 | - | - | 242 ± 38.76 | 258 ± 74.16 | 0.68 | |
| 0.006 | - | - | 340 ± 86.56 | 344 ± 102.19 | 0.95 | |
| 0.01 | 27 ± 9.91 | 32 ± 19.47 | 0.62 | 320 ± 47.77 | 348 ± 93.18 | 0.57 |
| 0.025 | 43 ± 20.98 | 55 ± 24.24 | 0.47 | 328 ± 47.30 | 357 ± 94.02 | 0.55 |
| 0.1 | 46 ± 15.13 | 58 ± 24.46 | 0.38 | 348 ± 59.59 | 372 ± 102.17 | 0.66 |
| 1 | 87 ± 34.40 | 108 ± 38.10 | 0.39 | 379 ± 100.68 | 409 ± 109.05 | 0.66 |
| 2 | 105 ± 36.65 | 105 ± 43.86 | 1.00 | 361 ± 106.10 | 378 ± 104.35 | 0.80 |
| 5 | 102 ± 34.97 | 103 ± 34.08 | 0.96 | 309 ± 130.79 | 345 ± 83.99 | 0.62 |
| 10 | 92 ± 30.89 | 96 ± 36.24 | 0.86 | 286 ± 131.38 | 304 ± 89.58 | 0.81 |
| Dark Adapted ERG Parameters (14 Months) | ||||||
| Stimulus Flash in cd·s·m−2 | A Wave | B Wave | ||||
| Knockout | Wildtype | p-Value | Knockout | Wildtype | p-Value | |
| 0.0025 | - | - | 232 ± 53.71 | 259 ± 64.01 | 0.49 | |
| 0.006 | - | - | 285 ± 85.24 | 282 ± 83.61 | 0.96 | |
| 0.01 | 30 ± 8.29 | 30 ± 13.33 | 1.00 | 296 ± 71.82 | 317 ± 99.56 | 0.71 |
| 0.025 | 63 ± 24.55 | 46 ± 27.68 | 0.33 | 328 ± 80.20 | 335 ± 109.26 | 0.91 |
| 0.1 | 63 ± 19.07 | 69 ± 30.05 | 0.72 | 334 ± 91.61 | 334 ± 125.15 | 1.00 |
| 1 | 112 ± 30.08 | 145 ± 26.07 | 0.10 | 438 ± 139.30 | 367 ± 127.21 | 0.42 |
| 2 | 129 ± 27.96 | 145 ± 35.95 | 0.45 | 437 ± 136.87 | 355 ± 123.90 | 0.35 |
| 5 | 111 ± 29.22 | 145 ± 29.12 | 0.10 | 369 ± 101.02 | 351 ± 117.61 | 0.80 |
| 10 | 110 ± 33.93 | 129 ± 24.05 | 0.34 | 313 ± 81.42 | 337 ± 109.48 | 0.70 |
| Light Adapted ERG Parameters (10 Months) | ||||||
| Stimulus Frequency in Hz | Trough to Peak Amplitude | |||||
| Knockout | Wildtype | p-Value | ||||
| 5 | 49.98 ± 25.47 | 47.93 ± 13.78 | 0.89 | |||
| 10 | 37.25 ± 20.57 | 36.72 ± 11.47 | 0.97 | |||
| 15 | 17.05 ± 4.79 | 18.27 ± 2.22 | 0.62 | |||
| 20 | 16.99 ± 6.21 | 12.9 ± 4.46 | 0.27 | |||
| Light Adapted ERG Parameters (14 Months) | ||||||
| Stimulus Frequency in Hz | Trough to Peak Amplitude | |||||
| Knockout | Wildtype | p-Value | ||||
| 5 | 48.74 ± 16.32 | 43.38 ± 13.65 | 0.59 | |||
| 10 | 30.7 ± 16.67 | 28.18 ± 11.45 | 0.79 | |||
| 15 | 15.5 ± 5.94 | 18.20 ± 6.22 | 0.50 | |||
| 20 | 14.1 ± 5.37 | 15.09 ± 2.96 | 0.73 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincent, A.; Ahmed, K.; Hussein, R.; Berberovic, Z.; Tumber, A.; Zhao, X.; Minassian, B.A. Retinal Phenotyping of a Murine Model of Lafora Disease. Genes 2023, 14, 854. https://doi.org/10.3390/genes14040854
Vincent A, Ahmed K, Hussein R, Berberovic Z, Tumber A, Zhao X, Minassian BA. Retinal Phenotyping of a Murine Model of Lafora Disease. Genes. 2023; 14(4):854. https://doi.org/10.3390/genes14040854
Chicago/Turabian StyleVincent, Ajoy, Kashif Ahmed, Rowaida Hussein, Zorana Berberovic, Anupreet Tumber, Xiaochu Zhao, and Berge A. Minassian. 2023. "Retinal Phenotyping of a Murine Model of Lafora Disease" Genes 14, no. 4: 854. https://doi.org/10.3390/genes14040854