
Role of Microbiota-Modified Bile Acids in the Regulation of Intracellular Organelles and Neurodegenerative Diseases

1
Kagawa School of Pharmaceutical Sciences, Tokushima Bunri University, Kagawa 769-2193, Japan
2
Institute of Neuroscience, Tokushima Bunri University, Kagawa 769-2193, Japan
*
Author to whom correspondence should be addressed.
Genes 2023, 14(4), 825; https://doi.org/10.3390/genes14040825
Submission received: 27 February 2023
/
Revised: 23 March 2023
/
Accepted: 28 March 2023
/
Published: 29 March 2023
(This article belongs to the Special Issue Human Microbiota: Current Updates on Pathogenetic Mechanisms and Methodological Advances)
{kind=link}
{kind=link}
Abstract
:Bile acids (BAs) are amphiphilic steroidal molecules generated from cholesterol in the liver and facilitate the digestion and absorption of fat-soluble substances in the gut. Some BAs in the intestine are modified by the gut microbiota. Because BAs are modified in a variety of ways by different types of bacteria present in the gut microbiota, changes in the gut microbiota can affect the metabolism of BAs in the host. Although most BAs absorbed from the gut are transferred to the liver, some are transferred to the systemic circulation. Furthermore, BAs have also been detected in the brain and are thought to migrate into the brain through the systemic circulation. Although BAs are known to affect a variety of physiological functions by acting as ligands for various nuclear and cell-surface receptors, BAs have also been found to act on mitochondria and autophagy in the cell. This review focuses on the BAs modified by the gut microbiota and their roles in intracellular organelles and neurodegenerative diseases.
1. Introduction
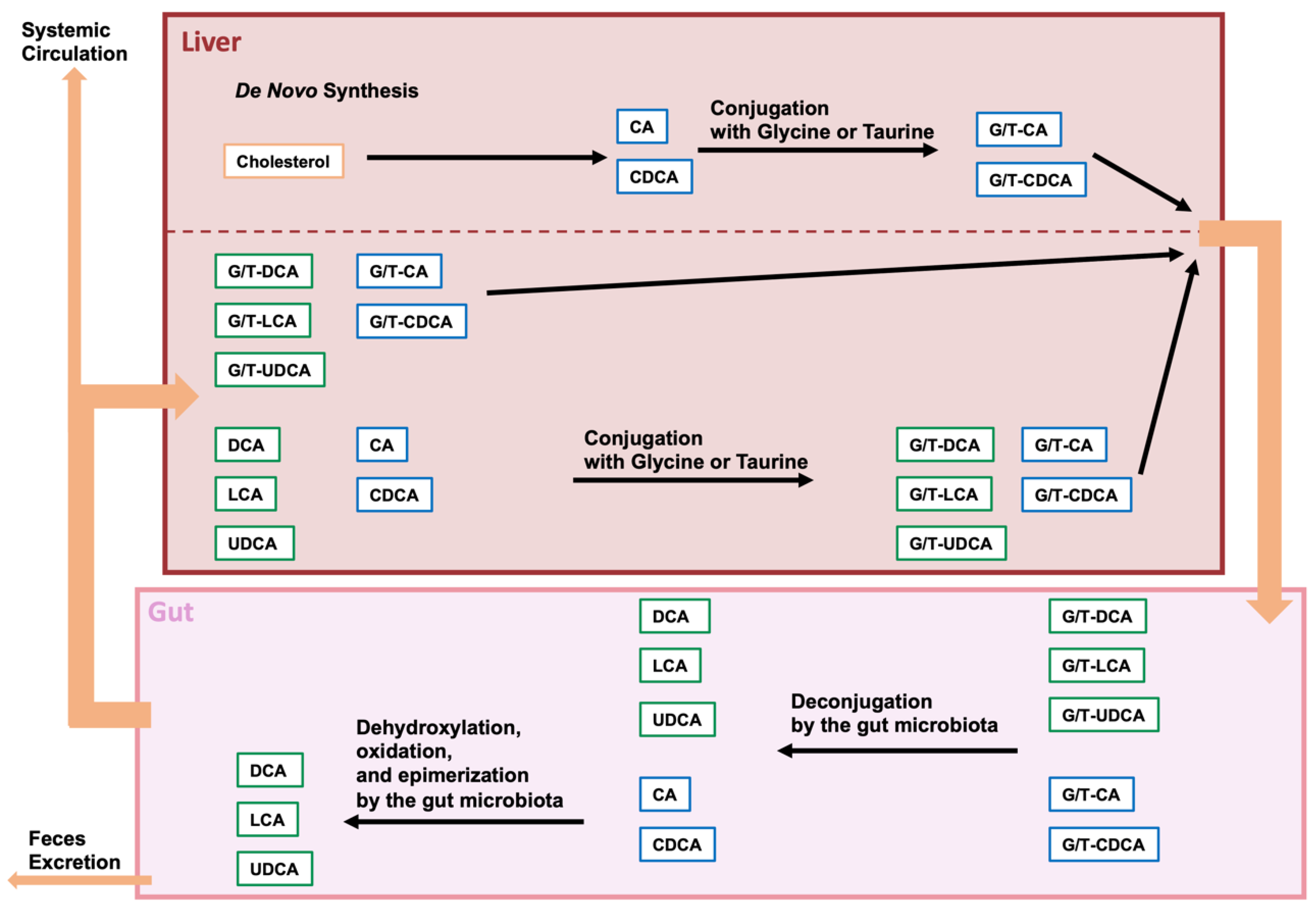
Bile acids (BAs) are important amphiphilic steroidal molecules generated from cholesterol in the liver and are important components of bile. BAs that are produced by de novo synthesis in the liver are called primary BAs. BAs move from the liver to the gallbladder and are secreted from the gallbladder into the small intestine in response to food intake. Because BAs are amphiphilic, they can function as surfactants to form micelles with cholesterol, lipids, and lipophilic vitamins in the intestine, facilitating the digestion and absorption of these fat-soluble substances [1,2]. Most of the BAs that are involved in facilitating fat digestion and absorption are absorbed before passing through the ileum [3]. However, some BAs that are not taken up by the intestine are transferred to the colon. During their transit to the colon, BAs undergo various modifications by the gut microbiota and microbiota-modified bile acids are called secondary BAs [4]. Because BAs are modified in a variety of ways by different types of bacteria present in the gut microbiota, changes in the gut microbiota can affect the metabolism of BAs in the host [5,6]. Some of the modified BAs are then absorbed. The absorbed BAs in the gut are then transported to the liver via the portal vein. This circulation of BAs between the liver and the intestine is called enterohepatic circulation. However, the BAs that are not absorbed in the gut are ultimately egested in the feces. Therefore, BAs play a role in the excretion of cholesterol. Furthermore, although most BAs absorbed from the gut are transferred to the liver, some are transferred to the systemic circulation [6,7] (Figure 1).
BAs have also been found in the brain and are thought to migrate into the brain through the systemic circulation. Thus, there is a possible link between BA and neurological function and neurological disease [8,9]. Not only do BAs affect a variety of physiological functions by acting as ligands for various nuclear and cell-surface receptors [6], but BAs also affect mitochondria [10,11,12] and autophagy [13,14,15]. This review focuses on the BAs modified by the gut microbiota and their functions in intracellular organelles and neurodegenerative diseases.
2. BAs and Gut Microbiota
2.1. Bile Acid Modification by the Gut Microbiota
BAs are biosynthesized from cholesterol in the liver, and the biosynthesis of BAs is conducted via using the classical (or neutral) or alternative (or acidic) pathway, using more than 16 enzymes [16,17,18,19]. In the classical pathway, cholesterol is converted to 7α-hydroxycholesterol with cytochrome P450 (CYP) 7A1 (CYP7A1), and 7α-hydroxycholestero is then converted to 7α-hydroxy-4-cholesten-3-one. Cholic acid (CA) is biosynthesized from 7α-hydroxy-4-cholesten-3-one, involving CYP8B1, and chenodeoxycholic acid (CDCA) is biosynthesized from 7α-hydroxy-4-cholesten-3-one, involving CYP27A1. In the alternative pathway, cholesterol is metabolized to (25R)-26-hydroxycholesterol by CYP27A1, and this is then converted to CDCA by CYP7B1. Bile acid-CoA: amino acid N-acyltransferase (BAAT) conjugates CA and CDCA with glycine or taurine [20]. After the conjugation of glycine or taurine to BAs, these BAs are sent to and stored in the gallbladder. BAs are discharged into the small intestine through the stimulation of food intake [1].
In the intestine, bile salt hydrolase (BSH) deconjugates conjugated BAs. Various gut bacteria have been shown to exhibit BSH activity [21,22]. Most of these bacteria, such as Lactobacillus, Bifidobacterium, Enterococcus, Clostridium, and Bacteroides spp., are found in the ileum and colon [23,24,25,26]. After deconjugation, the 7α-hydroxy group is removed from CA and CDCA, leading to a yield of deoxycholic acid (DCA) and lithocholic acid (LCA), respectively. A few bacteria among Clostridium spp. have been identified as capable of eliminating the 7α-hydroxy group. The α-dehydroxylation of the CA and CDCA is carried out by enzymes that are encoded in the bile acid-inducible (bai) operon. In addition, CDCA is converted to ursodeoxycholic acid (UDCA) by 7α-hydroxysteroid dehydrogenase (7α-HSDH) and 7β-HSDH. In addition, DCA and LCA are converted to iso-DCA and iso-LCA by HSDHs, respectively [27].
2.2. Analysis of the Composition of the Gut Microbiota and Bacterial Species with Enzymes That Modify BAs
The composition of gut microbiota and BAs is affected by diet, age, sex, antibiotics, and disease [7,28,29,30]. BAs and gut microbiota affect each other [5,6]. BAs are modified by gut bacteria through a variety of enzymatic reactions. Thus, the diversity of gut bacteria involved in the modification of bile acids has implications for host physiology and pathophysiology. Bacteria harboring BSH have been reported to be widely distributed across bacterial phyla and approximately 26% of bacteria strains in the Human Microbiome Project [31]. However, bacteria capable of conducting 7-α-dehydroxylation is limited to Clostridium spp. [27,32,33]. Furthermore, changes in the levels of microorganisms in gut microbiota are associated with neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease [34,35]. The relationship between gut microbiota and various diseases is currently being studied using multi-omics analysis, including metagenomics, metatranscriptomics, metaproteomics, and metabolomics [36]. There are two major types of metagenomic analyses that comprehensively analyze microbial communities using next-generation sequencers: 16S rRNA gene sequencing and shotgun metagenomic sequencing; these identify microorganisms and evaluate diversity and abundance [37]. 16S rRNA gene sequencing is the most widely used method of analyzing gut microbiota, and it is relatively inexpensive and simple to perform. In the 16S rRNA gene sequencing metagenomic analysis, after PCR amplification is conducted to target the 16S rRNA genes, the amplified PCR products are comprehensively sequenced using next-generation sequencing (NGS) to identify the diversity of gut microbiota and the types and composition ratios of its constituent bacteria. In shotgun metagenomic sequencing, DNA is randomly split into fragments, which are then sequenced by NGS. The sequenced DNA is linked using bioinformatics, resulting in the identification of species, strains, and functional genes [38,39]. These metagenomic analyses have revealed the association between changes in the composition of the gut microbiota and neurodegenerative diseases; furthermore, but their means, the association between changes in bacterial species with enzymes that modify BAs and neurodegenerative diseases are also being elucidated [34,35]. In patients with Alzheimer’s disease, Bacteroidetes is positively correlated with Alzheimer’s disease, while Firmicutes and Bifidobacterium are negatively correlated with Alzheimer’s disease [40,41,42]. In patients with Parkinson’s disease, Akkermansia is positively correlated with Parkinson’s disease, while Lactobacillus is negatively correlated with Parkinson’s disease [43,44,45]. In patients with Huntington’s disease, Intestinimonas, Bilophila, Lactobacillus, Oscillibacter, Gemmiger, and Dialister are positively correlated with Huntington’s disease [46] and Firmicutes, Lachnospiraceae, and Akkermansiaceae are negatively correlated with Huntington’s disease gene expansion carriers [47]. Furthermore, a recent study using shotgun metagenomic sequencing of Alzheimer’s disease patients has shown that Bacteroides spp., Alistipes spp., Odoribacter spp., and Barnesiella spp. increased and Lachnoclostridium spp. decreased in patients with Alzheimer’s disease [42]. Moreover, a recent study using shotgun metagenomic sequencing of Parkinson’s disease patients has shown that eighty-four species were associated with Parkinson’s disease, with 55 species more and 29 species less exist in Parkinson’s disease compared to healthy subjects. In addition, 34 genera more and 1 genus less present in Parkinson’s disease compared to healthy controls [48].
3. BAs as Regulatory Modulators
3.1. BAs as Regulatory Molecules of Nuclear and Cell-Surface Receptors
BAs are signaling molecules that bind to nuclear and cell-surface receptors, such as the farnesoid X receptor (FXR), a receptor for primary bile acids [49,50,51] and Takeda G protein receptor 5 (TGR5), also known as G protein-coupled bile acid receptor 1 and a receptor for secondary bile acids [50,51,52]. Among the nuclear receptors, BAs act as ligands for FXR, the constitutive androstane receptor, pregnane X receptor, vitamin D receptor, liver X receptor, glucocorticoid receptor, and retinoid-related orphan receptor γT. Moreover, BAs act as ligands for the cell-surface receptors TGR5, sphingosine-1-phosphate receptor 2, and the M2 and M3 muscarinic receptors. These receptors are expressed in various organs, including the brain [53,54,55,56,57,58]. Therefore, BAs play diverse physiological roles through the affect they have on these various receptors [7,59,60].
3.2. BAs as Regulatory Molecules of Intracellular Organelle
Mitochondrial and/or autophagic dysfunction affects neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease [61,62,63].
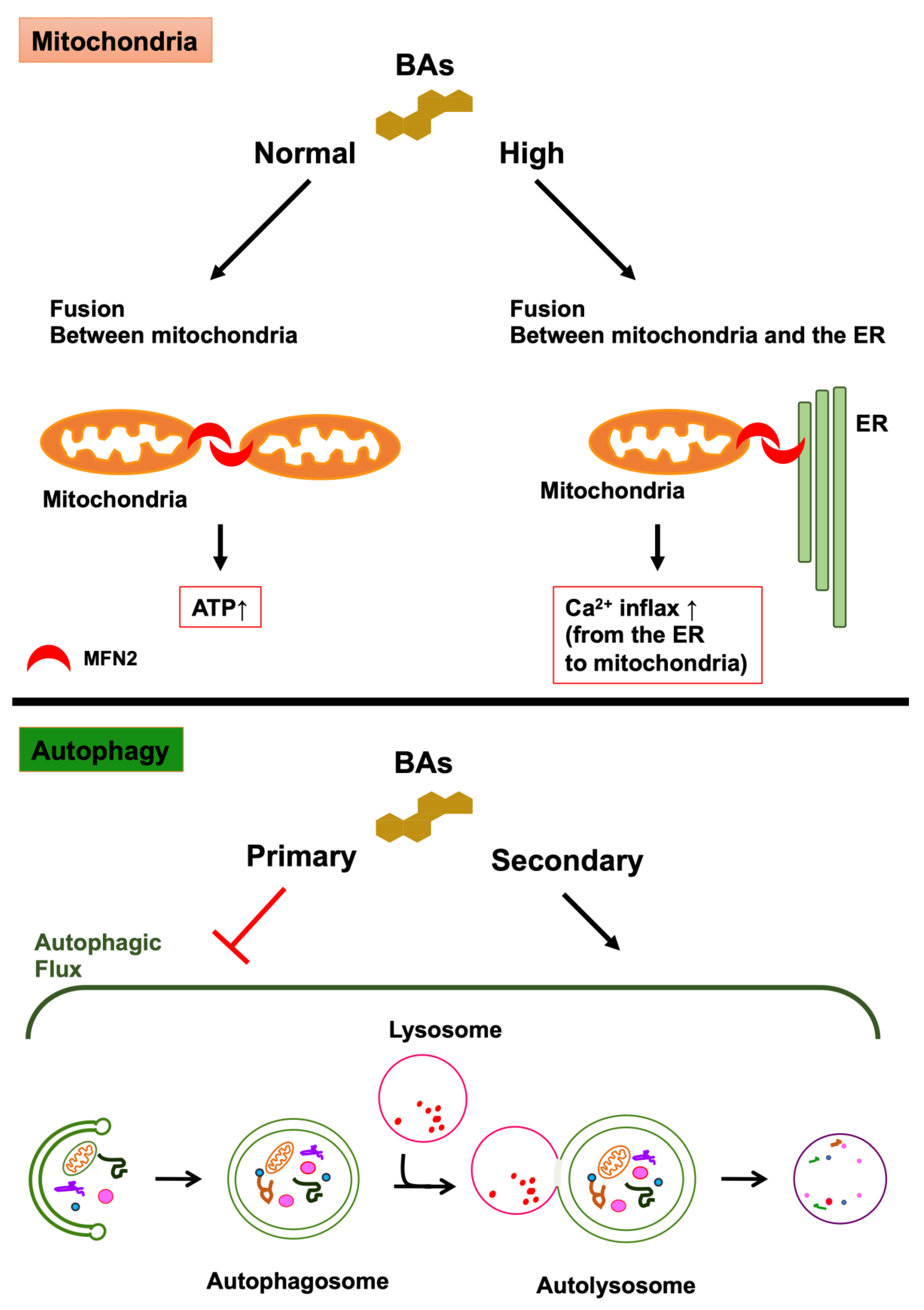
Mitochondria are intracellular organelles that have a double membrane consisting of an inner and outer membranes and function in the process of cellular ATP synthesis. They are involved in diverse and varied cellular functions, including apoptosis, regulation of intracellular calcium concentration, and metabolism of glucose, fatty acids, and amino acids [64]. Mitochondria maintain a functional population by repeating fusion and fission to eliminate dysfunctional mitochondria, and mitochondrial dysfunction is involved in various types of diseases [65,66]. The equilibrium between fission and fusion plays a crucial role in mitochondrial quality control. The fusion of mitochondria causes an exchange of mitochondrial DNA (mtDNA) and metabolites between damaged and healthy mitochondria to avoid the accumulation of damaged substances into one mitochondrion. Mitochondrial fusion undergoes when the outer mitochondrial membranes (OMM) of two mitochondria fuse with each other and with the inner mitochondrial membranes. Mitofusin (MFN) plays a crucial role in the fusion of the outer membrane. In addition, optic atrophy 1 (OPA1) plays a crucial role in the fusion of the inner membrane [67,68,69]. MFN is a dynamin-like GTPase and consists of two isoforms, namely, MFN1 and MFN2. Although both isoforms have important roles to play in the tethering and fusion of OMMs, the tethering activities of MFN1 are higher than those of MFN2. Therefore, MFN1 is the main tethering isoform for the fusion of OMMs [70,71]. MFN2 is located on the mitochondria-associated endoplasmic reticulum (ER) membrane (MAM), and it connects mitochondria to the ER, bringing Ca2+ influx from the ER to the mitochondria [72,73]. Mitochondrial fission tends to isolate damaged DNA and metabolites in mitochondria. Dynamin-related protein 1 (DRP1) is the major regulator of mitochondrial fission. DRP1 forms a ring-like structure composed of multimer at mitochondrial fission sites of the OMM and causes the constriction and scission of mitochondria. The activity of DRP1 is controlled by post-translational modifications, such as phosphorylation, ubiquitination, sumoylation, S-nitrosylation, and O-GlcNAcylation [74]. Glycocholic acid (GCA), taurocholic acid (TCA), glycochenodeoxycholic acid (GCDCA), and taurochenodeoxycholic acid (TCDCA) are abundant in the mitochondria of the liver of healthy subjects [75]. However, most BAs are toxic to mitochondria at high concentrations and can lead to a decrease in mitochondrial membrane potential [11,76,77]. By contrast, tauroursodeoxycholic acid (TUDCA) enhances mitochondrial biogenesis and protects against mitochondrial dysfunction [11,12]. Moreover, peroxisome proliferator-activated receptor γ coactivator 1α, nuclear respiratory factor 1, and mitochondrial transcription factor A are related to mitochondrial biogenesis [65,78]. TGR5 activation induces these molecules and promotes the functional gain and biogenesis of mitochondria [79]. Therefore, drugs that activate TGR5, including BA, may be potential therapeutic agents for neurodegenerative diseases. It has been recently reported that DCA, CDCA, and their taurine conjugates activate MFN2 by binding directly to MFN2, promoting mitochondria-to-mitochondria fusion and mitochondria-to-ER fusion in THP-1 cells that are differentiating into macrophages [10] (Figure 2). DCA and CDCA at physiological conditions (5 μM) promote the fusion between mitochondria, resulting in an increase in the generation of ATP. By contrast, DCA and CDCA with cholestatic conditions (100 μM) enhance the fusion between the mitochondria and the ER, leading to Ca2+ influx from the ER to the mitochondria, the activation of NLRP3 inflammasome and pyroptosis, and innate immunity. Furthermore, CA and UDCA antagonize the effects of DCA and CDCA on the GTPase activity of MFN2. The areas where mitochondria and ER contact are called mitochondria-associated membranes or mitochondria-associated ER membranes (MAMs) and an association between MAMs and neurodegenerative diseases has been indicated [80,81]. MAMs are initiation sites of autophagosome formation [82,83]. In addition, knockdown of MFN2 evokes impaired autophagy [84,85]. By contrast, overexpression of MFN2 leads to the induction of autophagy [85]. In addition, it is critical to maintain a balance between mitochondrial fusion and fission to support both mitochondrial and cellular function, and mitochondrial structural changes are associated with neurodegenerative diseases. Because MFN2 plays a crucial role in controlling mitochondrial structure, MFN2 is related to neurodegenerative diseases [86].
Autophagosomes have an important role in autophagy. Autophagy is a major intracellular degradation process in which autophagosomes sequester cytoplasmic components such as damaged proteins and intracellular organelles and then fuse with lysosomes to degrade those cytoplasmic components. The intracellular organelles in which autophagosomes and lysosomes are fused together are called autolysosomes [87]. The mechanistic (or mammalian) target of rapamycin complex 1 (mTORC1) is the key regulator of the initiation phase of autophagy, although numerous other molecules, such as microtubule-associated protein 1 light chain 3 (LC3), are also involved in autophagy. The inhibition of mTORC1 leads to the activation of the Unc-51-like kinase 1/2 (ULK1/2), resulting in the conjugation of phosphatidylethanolamine to LC3 and the formation of autophagosomes [61,88]. The formation of an autolysosome in autophagosome-lysosome fusion is conducted by soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) [89,90], Rab7 [91,92], UV radiation resistance-associated gene protein (UVRAG) [93], the homotypic fusion and protein sorting (HOPS) complex [94], and LC3s [95,96]. DCA induces autophagy in the human esophageal epithelial cell line [14], and UDCA also induces autophagy in the human liver [15]. By contrast, GCDCA inhibits autophagy by suppressing transcription factor E3 (TFE3) in the human liver cell line [97]. Moreover, CA, CDCA, and TCA interfere with the formation of autolysosomes in hepatocytes and the liver [13,15]. The activation of FXR, a receptor for primary bile acids, leads to the inhibition of autophagy [98,99]. By contrast, the activation of TGR5, a receptor for secondary BAs, leads to the activation of autophagy [100] (Figure 2). The activation of TGR5 leads to the activation of cAMP-response element binding protein (CREB). CREB promotes autophagy and upregulates autophagy-related genes such as Atg7, Ulk1, and Tfeb, while FXR represses autophagy and the expression of these genes by binding to CREB [98]. Furthermore, CREB-regulated transcription coactivator 1 induces autophagy-related genes by binding to CREB in neurons in competition with FXR [101].
4. Microbiota-Modified BAs in Neurodegenerative Diseases
The source of the BAs found in the brain has not been identified, but conjugated and unconjugated BAs have been found there [102,103,104]. BAs present in the brain may be produced there or migrate through the circulatory system. Primary BAs (CA and CDCA) are also produced by de novo synthesis in the brain. These primary BAs are responsible for the majority of cholesterol metabolism in the brain. This third pathway in the brain to produce primary BAs by de novo synthesis is called neural pathway [105]. Because secondary BAs are generated by the gut microbiota, the secondary BAs detected in the brain are thought to be transferred from the circulation. Furthermore, because a correlation has been identified between brain bile acid concentrations and serum bile acid concentrations, it is now believed that the majority of BAs in the brain migrate through circulation [106,107]. However, because there is neural pathway to BA synthesis, primary BAs produced by de novo synthesis in the brain may also play a role in the physiological and pathophysiological condition [105]. BAs are transported from the circulation, either through the blood–brain barrier (BBB) or through BA transporters [16,108]. Lipophilic BAs can pass through the BBB through passive diffusion. By contrast, hydrophilic BAs can pass through the BBB through transporters [109,110]. Therefore, the brain can be influenced by the gut microbiota via BAs.
4.1. Alzheimer’s Disease
Alzheimer’s disease is a progressive and irreversible neurodegenerative disease characterized by dementia, memory loss, and morphological changes in multiple regions of the brain. The pathological features of patients with Alzheimer’s disease are the accumulation of amyloid β peptide and tau protein entanglement in the brain [111]. Amyloid β peptide is produced from amyloid precursor protein (APP) with β- and γ-secretase [112,113]. The γ-secretase complex contains presenilin 1 (PS1). Autophagy refers to the process of removing the accumulation of misfolded proteins, and the suppression of autophagy is connected with Alzheimer’s disease [114]. Therefore, BAs can affect Alzheimer’s disease by influencing autophagy. LCA levels in plasma are higher in patients with Alzheimer’s disease. In addition, LCA levels in plasma increase by approximately 3 fold within 8–9 years from when healthy subjects develop Alzheimer’s disease [115]. By contrast, the levels of CA in plasma and the TCA levels in the brain are significantly lower in patients with Alzheimer’s disease [104]. However, the neuroprotective effect of TUDCA, which is a taurine-conjugated secondary BA, has been demonstrated in neurodegenerative disease [116]. TUDCA lowers amyloid β peptides and relieves memory deterioration in APP/PS1 double-knockout mice used as a model for Alzheimer’s disease [117,118]. The ratios for both unconjugated and conjugated secondary BAs, including LCA, GCDCA, taurodeoxycholic acid (TDCA), glycodeoxycholic acid (GDCA), and UDCA, to CA were higher in the brain of patients with Alzheimer’s disease [102]. Furthermore, the ratios of DCA, GDCA, TDCA, and glycolithocholic acid. GLCA) were significantly higher in the serum of patients with Alzheimer’s disease [119]. Therefore, increased secondary BAs may be associated with Alzheimer’s disease, and the status of the gut microbiome may affect the progression and suppression of Alzheimer’s disease. In addition, the increase in Lactobacillus, which has BSH that deconjugates conjugated-BAs, in feces is related to Alzheimer’s disease [120,121].
4.2. Parkinson’s Disease
Parkinson’s disease is a progressive neurodegenerative disease that manifests itself in resting tremor, muscle rigidity, bradykinesia, akinesia, and postural reflex disorder [122]. It results in the loss of dopaminergic neurons in the substantia nigra [123]. The dysfunction of phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) and Parkin is considered a major cause of parkisonisms [124], and this dysfunction results in the impairment of mitophagy, suggesting that the quality control of mitochondria plays an crucial role in the suppression of Parkinson’s disease [61,125]. BAs are associated with autophagy and the quality control of mitochondria. Furthermore, plasma levels of CA, DCA, TDCA, and GDCA in patients with Parkinson’s disease are significantly higher when compared with healthy subjects [126,127]. However, the plasma levels of GUDCA in patients with Parkinson’s disease are decreased [128]. Moreover, levels of DCA and LCA are higher in the appendix and LCA is higher in the ileum of patients with Parkinson’s disease [129]. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and rotenone treatment to rodents are commonly used to model Parkinson’s disease. These chemicals prevent the function of complex I in mitochondria, causing oxidative stress and mitochondrial damage in neurons, leading to neurotoxicity and parkinsonism [130,131]. TUDCA protects dopaminergic neurons from MPTP-induced neurotoxicity [132]. It also suppresses MPTP-induced loss of dopaminergic neurons and mitochondrial membrane potential to improve the motor symptoms induced by MPTP [133]. Furthermore, rat models of Parkinson’s disease induced by rotenone show mitochondrial swelling, and the loss of mitochondrial cristae and UDCA improves these mitochondrial abnormalities [134]. In addition, several reports have indicated that BSH-containing bacteria, such as Lactobacillus, Bifidobacterium, Enterococcus, and Bacteroides spp., are increased in patients with Parkinson’s disease [34,35]. This indicates a relationship between unconjugated BAs and Parkinson’s disease.
4.3. Huntington’s Disease
Huntington’s disease is an autosomal-dominant neurodegenerative disease that manifests in cognitive disability, and psychiatric disturbance, and motor dysfunctions [135]. Huntington’s disease is caused by cytosine-adenine-guanine (CAG) expansion which encodes a polyglutamine at the N-terminus of huntingtin (HTT) [136]. HTT has a similar structure to the three autophagy proteins of yeast, Atg11, Atg23, and Vac8 [137,138] and acts as an autophagy initiator and enhancer [139]. The mutation of HTT leads to a reduction in mitophagy [140]. BAs are associated with mitophagy. In addition, 3-nitropropionic acid (3-NP) selectively damages neurons in the striatum and is involved in the development of Huntington’s disease. 3-NP inhibits succinate dehydrogenase in mitochondria and leads to the degeneration of the caudate-putamen [141,142]. TUDCA improves 3-NP-induced neural mitochondrial damage, neural cell death, and sensorimotor deficits [143]. Moreover, TUDCA ameliorates striatal apoptosis, cerebral and striatal atrophy, and sensorimotor deficits in R6/2 transgenic mice that carry the human huntingtin gene which has 144 CAG repeats and are commonly used as a model for Huntington’s disease [144].
5. Concluding Remarks
Primary BAs are synthesized in the liver and these BAs are modified by the involvement of various microorganisms present in the gut microbiota. Therefore, a variety of bile acids exist in the human body. Bile acids have been shown to exert diverse physiological effects by binding to various nuclear and cell-surface receptors. Therefore, bile acids have come to be recognized as signaling molecules. In addition, it has recently been reported that BAs are present in mitochondria and affect mitochondrial ATP synthesis. Furthermore, some BAs have been shown to act on mitochondrial fusion as ligands for MFN2, which is involved in the fusion between mitochondria and between mitochondria and ER. BAs have also been shown to regulate the progression of autophagy. Although the causes of neurodegenerative diseases are still incompletely elucidated, mitochondrial and mitophagy dysfunction in neurons is considered one of the causes of neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. Therefore, further elucidation of the function of BAs and the gut microbiota which modifies BAs may lead to new treatments for neurodegenerative diseases.
Author Contributions
Y.K. conceptualized and wrote this manuscript. H.N. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Tokushima Bunri University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Di Ciaula, A.; Garruti, G.; Baccetto, R.L.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, S4–S14. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y.L. Bile Acid Signaling in Metabolic Disease and Drug Therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef] [Green Version]
- Vallim, T.Q.D.A.; Tarling, E.J.; Edwards, P.A. Pleiotropic Roles of Bile Acids in Metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larabi, A.B.; Masson, H.L.P.; Bäumler, A.J. Bile acids as modulators of gut microbiota composition and function. Gut Microbes 2023, 15, 2172671. [Google Scholar] [CrossRef]
- Perino, A.; Demagny, H.; Velazquez-Villegas, L.A.; Schoonjans, K. Molecular physiology of bile acid signaling in health, disease, and aging. Physiol. Rev. 2021, 101, 683–731. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. Physiological Role of Bile Acids Modified by the Gut Microbiome. Microorganisms 2021, 10, 68. [Google Scholar] [CrossRef]
- Grant, S.; DeMorrow, S. Bile Acid Signaling in Neurodegenerative and Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 5982. [Google Scholar] [CrossRef]
- McMillin, M.; DeMorrow, S. Effects of bile acids on neurological function and disease. FASEB J. 2016, 30, 3658–3668. [Google Scholar] [CrossRef] [Green Version]
- Che, Y.; Xu, W.; Ding, C.; He, T.; Xu, X.; Shuai, Y.; Huang, H.; Wu, J.; Wang, Y.; Wang, C.; et al. Bile acids target mitofusin 2 to differentially regulate innate immunity in physiological versus cholestatic conditions. Cell Rep. 2023, 42, 112011. [Google Scholar] [CrossRef]
- Rolo, A.P.; Palmeira, C.M.; Holy, J.M.; Wallace, K.B. Role of Mitochondrial Dysfunction in Combined Bile Acid-Induced Cytotoxicity: The Switch Between Apoptosis and Necrosis. Toxicol. Sci. 2004, 79, 196–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, I.; Gordino, G.; Moreira, S.; Nunes, M.J.; Azevedo, C.; Gama, M.J.; Rodrigues, E.; Rodrigues, C.M.P.; Castro-Caldas, M. Tauroursodeoxycholic Acid Protects Against Mitochondrial Dysfunction and Cell Death via Mitophagy in Human Neuroblastoma Cells. Mol. Neurobiol. 2016, 54, 6107–6119. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Ni, H.-M.; Kong, B.; Apte, U.; Guo, G.; Ding, W.-X. Suppression of Autophagic Flux by Bile Acids in Hepatocytes. Toxicol. Sci. 2013, 137, 478–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roesly, H.B.; Khan, M.R.; Chen, H.D.R.; Hill, K.A.; Narendran, N.; Watts, G.S.; Chen, X.; Dvorak, K. The decreased expression of Beclin-1 correlates with progression to esophageal adenocarcinoma: The role of deoxycholic acid. Am. J. Physiol. Liver Physiol. 2012, 302, G864–G872. [Google Scholar] [CrossRef] [PubMed]
- Panzitt, K.; Jungwirth, E.; Krones, E.; Lee, J.M.; Pollheimer, M.; Thallinger, G.G.; Kolb-Lenz, D.; Xiao, R.; Thorell, A.; Trauner, M.; et al. FXR-dependent Rubicon induction impairs autophagy in models of human cholestasis. J. Hepatol. 2020, 72, 1122–1131. [Google Scholar] [CrossRef] [Green Version]
- Kiriyama, Y.; Nochi, H. The Biosynthesis, Signaling, and Neurological Functions of Bile Acids. Biomolecules 2019, 9, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.W. Fifty years of advances in bile acid synthesis and metabolism. J. Lipid Res. 2009, 50, S120–S125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katafuchi, T.; Makishima, M. Molecular Basis of Bile Acid-FXR-FGF15/19 Signaling Axis. Int. J. Mol. Sci. 2022, 23, 6046. [Google Scholar] [CrossRef]
- Pandak, W.M.; Kakiyama, G. The acidic pathway of bile acid synthesis: Not just an alternative pathway. Liver Res. 2019, 3, 88–98. [Google Scholar] [CrossRef]
- Styles, N.A.; Shonsey, E.M.; Falany, J.L.; Guidry, A.L.; Barnes, S.; Falany, C.N. Carboxy-terminal mutations of bile acid CoA:N-acyltransferase alter activity and substrate specificity. J. Lipid Res. 2016, 57, 1133–1143. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.M.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef] [Green Version]
- O’Flaherty, S.; Crawley, A.B.; Theriot, C.M.; Barrangou, R. The Lactobacillus Bile Salt Hydrolase Repertoire Reveals Niche-Specific Adaptation. Msphere 2018, 3, e00140-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, G.; Sandhu, K.V.; Griffin, B.T.; Dinan, T.G.; Cryan, J.F.; Hyland, N.P. Gut Reactions: Breaking Down Xenobiotic–Microbiome Interactions. Pharmacol. Rev. 2019, 71, 198–224. [Google Scholar] [CrossRef] [Green Version]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Okpara, E.S.; Hu, W.; Yan, C.; Wang, Y.; Liang, Q.; Chiang, J.Y.L.; Han, S. Interactive Relationships between Intestinal Flora and Bile Acids. Int. J. Mol. Sci. 2022, 23, 8343. [Google Scholar] [CrossRef]
- Begley, M.; Hill, C.; Gahan, C.G.M. Bile Salt Hydrolase Activity in Probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doden, H.; Ridlon, J. Microbial Hydroxysteroid Dehydrogenases: From Alpha to Omega. Microorganisms 2021, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Pérez, O.; Cruz-Ramón, V.; Chinchilla-López, P.; Méndez-Sánchez, N. The Role of the Gut Microbiota in Bile Acid Metabolism. Ann. Hepatol. 2017, 16, s15–s20. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, S.; Wang, M.; Hu, H.; Yin, J.; Liu, C.; Huang, Y. Gut Microbiota Dysbiosis Associated with Bile Acid Metabolism in Neonatal Cholestasis Disease. Sci. Rep. 2020, 10, 7686. [Google Scholar] [CrossRef]
- Majait, S.; Nieuwdorp, M.; Kemper, M.; Soeters, M. The Black Box Orchestra of Gut Bacteria and Bile Acids: Who Is the Conductor? Int. J. Mol. Sci. 2023, 24, 1816. [Google Scholar] [CrossRef]
- Song, Z.; Cai, Y.; Lao, X.; Wang, X.; Lin, X.; Cui, Y.; Kalavagunta, P.K.; Liao, J.; Jin, L.; Shang, J.; et al. Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome 2019, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, J.L.; Cummings, B.P. The 7-α-dehydroxylation pathway: An integral component of gut bacterial bile acid metabolism and potential therapeutic target. Front. Microbiol. 2023, 13, 1093420. [Google Scholar] [CrossRef] [PubMed]
- Guzior, D.V.; Quinn, R.A. Review: Microbial transformations of human bile acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef]
- Spichak, S.; Bastiaanssen, T.F.; Berding, K.; Vlckova, K.; Clarke, G.; Dinan, T.G.; Cryan, J.F. Mining microbes for mental health: Determining the role of microbial metabolic pathways in human brain health and disease. Neurosci. Biobehav. Rev. 2021, 125, 698–761. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, L.; Cantoni, C.; Rotondo, E.; Galimberti, D. The Gut Microbiome–Brain Crosstalk in Neurodegenerative Diseases. Biomedicines 2022, 10, 1486. [Google Scholar] [CrossRef]
- Jansson, J.K.; Baker, E.S. A multi-omic future for microbiome studies. Nat. Microbiol. 2016, 1, 16049. [Google Scholar] [CrossRef] [PubMed]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [Green Version]
- Laudadio, I.; Fulci, V.; Palone, F.; Stronati, L.; Cucchiara, S.; Carissimi, C. Quantitative Assessment of Shotgun Metagenomics and 16S rDNA Amplicon Sequencing in the Study of Human Gut Microbiome. OMICS J. Integr. Biol. 2018, 22, 248–254. [Google Scholar] [CrossRef]
- D’Argenio, V. Human Microbiome Acquisition and Bioinformatic Challenges in Metagenomic Studies. Int. J. Mol. Sci. 2018, 19, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Wu, L.; Peng, G.; Han, Y.; Tang, R.; Ge, J.; Zhang, L.; Jia, L.; Yue, S.; Zhou, K.; et al. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain Behav. Immun. 2019, 80, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Haran, J.P.; Bhattarai, S.K.; Foley, S.E.; Dutta, P.; Ward, D.V.; Bucci, V.; McCormick, B.A. Alzheimer’s Disease Microbiome Is Associated with Dysregulation of the Anti-Inflammatory P-Glycoprotein Pathway. Mbio 2019, 10, e00632-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedarf, J.R.; Hildebrand, F.; Coelho, L.P.; Sunagawa, S.; Bahram, M.; Goeser, F.; Bork, P.; Wüllner, U. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Cui, L.; Yang, Y.; Miao, J.; Zhao, X.; Zhang, J.; Cui, G.; Zhang, Y. Gut Microbiota Differs Between Parkinson’s Disease Patients and Healthy Controls in Northeast China. Front. Mol. Neurosci. 2019, 12, 171. [Google Scholar] [CrossRef] [Green Version]
- Barichella, M.; Severgnini, M.; Cilia, R.; Cassani, E.; Bolliri, C.; Caronni, S.; Ferri, V.; Cancello, R.; Ceccarani, C.; Faierman, S.; et al. Unraveling gut microbiota in Parkinson’s disease and atypical parkinsonism. Mov. Disord. 2018, 34, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Dong, W.; Yang, Q.; Yu, X.; Ma, J.; Gu, W.; Huang, Y. Altered Gut Microbiota Related to Inflammatory Responses in Patients With Huntington’s Disease. Front. Immunol. 2021, 11, 603594. [Google Scholar] [CrossRef] [PubMed]
- Wasser, C.I.; Mercieca, E.-C.; Kong, G.; Hannan, A.J.; McKeown, S.J.; Glikmann-Johnston, Y.; Stout, J.C. Gut dysbiosis in Huntington’s disease: Associations among gut microbiota, cognitive performance and clinical outcomes. Brain Commun. 2020, 2, 110. [Google Scholar] [CrossRef] [PubMed]
- Wallen, Z.D.; Demirkan, A.; Twa, G.; Cohen, G.; Dean, M.N.; Standaert, D.G.; Sampson, T.R.; Payami, H. Metagenomics of Parkinson’s disease implicates the gut microbiome in multiple disease mechanisms. Nat. Commun. 2022, 13, 6958. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, D.; Li, X.; Cao, Y.; Yi, C.; Ocansey, D.K.W.; Zhou, Y.; Mao, F. Farnesoid-X receptor as a therapeutic target for inflammatory bowel disease and colorectal cancer. Front. Pharmacol. 2022, 13, 1016836. [Google Scholar] [CrossRef]
- Biagioli, M.; Fiorucci, S. Bile acid activated receptors: Integrating immune and metabolic regulation in non-alcoholic fatty liver disease. Liver Res. 2021, 5, 119–141. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E. Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol. Med. 2015, 21, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Di Ciaula, A.; Garruti, G.; Vacca, M.; De Angelis, M.; Wang, D.Q.-H. Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome. Nutrients 2020, 12, 3709. [Google Scholar] [CrossRef]
- Duboc, H.; Taché, Y.; Hofmann, A.F. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig. Liver Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Thathiah, A. Regulation of neuronal communication by G protein-coupled receptors. FEBS Lett. 2015, 589, 1607–1619. [Google Scholar] [CrossRef] [Green Version]
- Tayebati, S.K. Phospholipid and Lipid Derivatives as Potential Neuroprotective Compounds. Molecules 2018, 23, 2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, L.; Antel, J. Sphingosine-1-Phosphate Receptors in the Central Nervous and Immune Systems. Curr. Drug Targets 2016, 17, 1841–1850. [Google Scholar] [CrossRef]
- Maria, A.A.; Andréas, M.-R.O.; Haider, N.B. Role of Nuclear Receptors in Central Nervous System Development and Associated Diseases. J. Exp. Neurosci. 2015, 9 (Suppl. S2), 93–121. [Google Scholar] [CrossRef]
- Frye, C.A.; Paris, J.J.; A Walf, A.; Rusconi, J.C. Effects and Mechanisms of 3α,5α,-THP on Emotion, Motivation, and Reward Functions Involving Pregnane Xenobiotic Receptor. Front. Neurosci. 2012, 5, 136. [Google Scholar] [CrossRef] [Green Version]
- Thibaut, M.M.; Bindels, L.B. Crosstalk between bile acid-activated receptors and microbiome in entero-hepatic inflammation. Trends Mol. Med. 2022, 28, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wang, S.; Wang, P.; Tang, C.; Wang, Z.; Chen, L.; Luo, G.; Chen, H.; Liu, Y.; Feng, B.; et al. Bile acids and their receptors in regulation of gut health and diseases. Prog. Lipid Res. 2023, 89, 101210. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. The Function of Autophagy in Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 16, 26797–26812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. Intra- and Intercellular Quality Control Mechanisms of Mitochondria. Cells 2017, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial Diseases: Hope for the Future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 Mediate Sequential Steps in Mitochondrial Membrane Fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2020, 31, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhou, X.; Hu, X.; Joshi, A.S.; Guo, X.; Zhu, Y.; Chen, Q.; Prinz, W.A.; Hu, J. Sequences flanking the transmembrane segments facilitate mitochondrial localization and membrane fusion by mitofusin. Proc. Natl. Acad. Sci. USA 2017, 114, E9863–E9872. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Zhao, F.; Hsia, J.; Ma, X.; Liu, Y.; Torres, S.; Fujioka, H.; Zhu, X. The role of Mfn2 in the structure and function of endoplasmic reticulum-mitochondrial tethering in vivo. J. Cell Sci. 2021, 134, jcs253443. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Shutt, T.E. The Role of Impaired Mitochondrial Dynamics in MFN2-Mediated Pathology. Front. Cell Dev. Biol. 2022, 10, 858286. [Google Scholar] [CrossRef] [PubMed]
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature 2021, 590, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhou, Y.; Lu, Y.; Zhou, K.; Cai, W. PHB2 interacts with LC3 and SQSTM1 is required for bile acids-induced mitophagy in cholestatic liver. Cell Death Dis. 2018, 9, 160. [Google Scholar] [CrossRef] [Green Version]
- Palmeira, C.M.; Rolo, A.P. Mitochondrially-mediated toxicity of bile acids. Toxicology 2004, 203, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Perez, M.-J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Popov, L. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.-J.; Zhang, S.-F. Activation of TGR5 promotes mitochondrial biogenesis in human aortic endothelial cells. Biochem. Biophys. Res. Commun. 2018, 500, 952–957. [Google Scholar] [CrossRef]
- Liu, J.; Yang, J. Mitochondria-associated membranes: A hub for neurodegenerative diseases. Biomed. Pharmacother. 2022, 149, 112890. [Google Scholar] [CrossRef]
- Markovinovic, A.; Greig, J.; Martín-Guerrero, S.M.; Salam, S.; Paillusson, S. Endoplasmic reticulum–mitochondria signaling in neurons and neurodegenerative diseases. J. Cell Sci. 2022, 135, jcs248534. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Paillusson, S.; Miller, C.C.J. ER-mitochondria signaling regulates autophagy. Autophagy 2017, 13, 1250–1251. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Gao, H.; Zhao, L.; Wang, X.; Zheng, M. Mitofusin 2-Deficiency Suppresses Cell Proliferation through Disturbance of Autophagy. PLoS ONE 2015, 10, e0121328. [Google Scholar] [CrossRef]
- Ashraf, R.; Kumar, S. Mfn2-mediated mitochondrial fusion promotes autophagy and suppresses ovarian cancer progression by reducing ROS through AMPK/mTOR/ERK signaling. Cell. Mol. Life Sci. 2022, 79, 573. [Google Scholar] [CrossRef]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 330. [Google Scholar] [CrossRef] [Green Version]
- Patra, S.; Patil, S.; Klionsky, D.J.; Bhutia, S.K. Lysosome signaling in cell survival and programmed cell death for cellular homeostasis. J. Cell. Physiol. 2022, 238, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The Hairpin-type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atlashkin, V.; Kreykenbohm, V.; Eskelinen, E.-L.; Wenzel, D.; Fayyazi, A.; von Mollard, G.F. Deletion of the SNARE vti1b in Mice Results in the Loss of a Single SNARE Partner, Syntaxin 8. Mol. Cell. Biol. 2003, 23, 5198–5207. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, M.G.; Munafó, D.B.; Berón, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.-L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Lee, J.-S.; Inn, K.-S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nature 2008, 10, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome–lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 2014, 25, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.S.; Jariwala, J.S.; Anderson, E.; Mitra, K.; Meisenhelder, J.; Chang, J.T.; Ideker, T.; Hunter, T.; Nizet, V.; Dillin, A.; et al. Phosphorylation of LC3 by the Hippo Kinases STK3/STK4 Is Essential for Autophagy. Mol. Cell 2014, 57, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, H.-Q.; Zhu, X.; Zhang, L.; Albanesi, J.; Levine, B.; Yin, H. GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion. Proc. Natl. Acad. Sci. USA 2015, 112, 7015–7020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, W.; Chen, Z.; Chen, Y.; Tan, M.; Chen, Y.; Chen, J.; Chi, X.; Chen, Y. Glycochenodeoxycholic acid impairs transcription factor E3 -dependent autophagy-lysosome machinery by disrupting reactive oxygen species homeostasis in L02 cells. Toxicol. Lett. 2020, 331, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Seok, S.; Fu, T.; Choi, S.-E.; Li, Y.; Zhu, R.; Kumar, S.; Sun, X.; Yoon, G.; Kang, Y.; Zhong, W.; et al. Transcriptional regulation of autophagy by an FXR–CREB axis. Nature 2014, 516, 108–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef] [Green Version]
- Carino, A.; Marchianò, S.; Biagioli, M.; Scarpelli, P.; Bordoni, M.; Di Giorgio, C.; Roselli, R.; Fiorucci, C.; Monti, M.C.; Distrutti, E.; et al. The bile acid activated receptors GPBAR1 and FXR exert antagonistic effects on autophagy. FASEB J. 2020, 35, e21271. [Google Scholar] [CrossRef]
- Pan, Y.; He, X.; Li, C.; Li, Y.; Li, W.; Zhang, H.; Wang, Y.; Zhou, G.; Yang, J.; Li, J.; et al. Neuronal activity recruits the CRTC1/CREB axis to drive transcription-dependent autophagy for maintaining late-phase LTD. Cell Rep. 2021, 36, 109398. [Google Scholar] [CrossRef] [PubMed]
- Baloni, P.; Funk, C.C.; Yan, J.; Yurkovich, J.T.; Kueider-Paisley, A.; Nho, K.; Heinken, A.; Jia, W.; Mahmoudiandehkordi, S.; Louie, G.; et al. Metabolic Network Analysis Reveals Altered Bile Acid Synthesis and Metabolism in Alzheimer’s Disease. Cell Rep. Med. 2020, 1, 100138. [Google Scholar] [CrossRef]
- Mano, N.; Goto, T.; Uchida, M.; Nishimura, K.; Ando, M.; Kobayashi, N.; Goto, J. Presence of protein-bound unconjugated bile acids in the cytoplasmic fraction of rat brain. J. Lipid Res. 2004, 45, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Elliott, C.T.; McGuinness, B.; Passmore, P.; Kehoe, P.G.; Hölscher, C.; McClean, P.L.; Graham, S.F.; Green, B.D. Metabolomic Profiling of Bile Acids in Clinical and Experimental Samples of Alzheimer’s Disease. Metabolites 2017, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Mertens, K.L.; Kalsbeek, A.; Soeters, M.R.; Eggink, H.M. Bile Acid Signaling Pathways from the Enterohepatic Circulation to the Central Nervous System. Front. Neurosci. 2017, 11, 617. [Google Scholar] [CrossRef] [Green Version]
- Higashi, T.; Watanabe, S.; Tomaru, K.; Yamazaki, W.; Yoshizawa, K.; Ogawa, S.; Nagao, H.; Minato, K.; Maekawa, M.; Mano, N. Unconjugated bile acids in rat brain: Analytical method based on LC/ESI-MS/MS with chemical derivatization and estimation of their origin by comparison to serum levels. Steroids 2017, 125, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Kamp, F.; Hamilton, J.A. Movement of fatty acids, fatty acid analogs, and bile acids across phospholipid bilayers. Biochemistry 1993, 32, 11074–11085. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Cardoso, V.F.; Corlianò, M.; Singaraja, R.R. Bile Acids: A Communication Channel in the Gut-Brain Axis. NeuroMol. Med. 2020, 23, 99–117. [Google Scholar] [CrossRef]
- Benedetti, A.; Di Sario, A.; Marucci, L.; Svegliati-Baroni, G.; Schteingart, C.D.; Ton-Nu, H.T.; Hofmann, A.F. Carrier-mediated transport of conjugated bile acids across the basolateral membrane of biliary epithelial cells. Am. J. Physiol. Liver Physiol. 1997, 272, G1416–G1424. [Google Scholar] [CrossRef]
- St-Pierre, M.V.; Kullak-Ublick, G.A.; Hagenbuch, B.; Meier, P.J. Transport of Bile Acids in Hepatic and Non-Hepatic Tissues. J. Exp. Biol. 2001, 204, 1673–1686. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s Disease: The Challenge of the Second Century. Sci. Transl. Med. 2011, 3, 77sr1. [Google Scholar] [CrossRef] [Green Version]
- Vassar, R.; Cole, S. The Basic Biology of BACE1: A Key Therapeutic Target for Alzheimers Disease. Curr. Genom. 2007, 8, 509–530. [Google Scholar] [CrossRef] [Green Version]
- Tomita, T. Molecular mechanism of intramembrane proteolysis by γ-secretase. J. Biochem. 2014, 156, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yang, X.; Song, Y.-Q.; Tu, J. Autophagy in Alzheimer’s disease pathogenesis: Therapeutic potential and future perspectives. Ageing Res. Rev. 2021, 72, 101464. [Google Scholar] [CrossRef] [PubMed]
- Marksteiner, J.; Blasko, I.; Kemmler, G.; Koal, T.; Humpel, C. Bile acid quantification of 20 plasma metabolites identifies lithocholic acid as a putative biomarker in Alzheimer’s disease. Metabolomics 2017, 14, 1. [Google Scholar] [CrossRef] [Green Version]
- Mulak, A. Bile Acids as Key Modulators of the Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 84, 461–477. [Google Scholar] [CrossRef]
- Nunes, A.F.; Amaral, J.D.; Lo, A.C.; Fonseca, M.B.; Viana, R.J.; Callaerts-Vegh, Z.; D’Hooge, R.; Rodrigues, C.M. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol. Neurobiol. 2012, 45, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.C.; Callaerts-Vegh, Z.; Nunes, A.F.; Rodrigues, C.M.; D’Hooge, R. Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol. Dis. 2013, 50, 21–29. [Google Scholar] [CrossRef] [PubMed]
- MahmoudianDehkordi, S.; Arnold, M.; Nho, K.; Ahmad, S.; Jia, W.; Xie, G.; Louie, G.; Kueider-Paisley, A.; Moseley, M.A.; Thompson, J.W.; et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease—An emerging role for gut microbiome. Alzheimers Dement. 2019, 15, 76–92. [Google Scholar] [CrossRef]
- Wu, S.; Liu, X.; Jiang, R.; Yan, X.; Ling, Z. Roles and Mechanisms of Gut Microbiota in Patients With Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 650047. [Google Scholar] [CrossRef]
- D’Argenio, V.; Veneruso, I.; Gong, C.; Cecarini, V.; Bonfili, L.; Eleuteri, A.M. Gut Microbiome and Mycobiome Alterations in an In Vivo Model of Alzheimer’s Disease. Genes 2022, 13, 1564. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brichta, L.; Greengard, P.; Flajolet, M. Advances in the pharmacological treatment of Parkinson’s disease: Targeting neurotransmitter systems. Trends Neurosci. 2013, 36, 543–554. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldeeb, M.A.; Thomas, R.A.; Ragheb, M.A.; Fallahi, A.; Fon, E.A. Mitochondrial quality control in health and in Parkinson’s disease. Physiol. Rev. 2022, 102, 1721–1755. [Google Scholar] [CrossRef]
- Yakhine-Diop, S.M.; Morales-García, J.A.; Niso-Santano, M.; González-Polo, R.A.; Uribe-Carretero, E.; Martinez-Chacon, G.; Durand, S.; Maiuri, M.C.; Aiastui, A.; Zulaica, M.; et al. Metabolic alterations in plasma from patients with familial and idiopathic Parkinson’s disease. Aging 2020, 12, 16690–16708. [Google Scholar] [CrossRef]
- Shao, Y.; Li, T.; Liu, Z.; Wang, X.; Xu, X.; Li, S.; Xu, G.; Le, W. Comprehensive metabolic profiling of Parkinson’s disease by liquid chromatography-mass spectrometry. Mol. Neurodegener. 2021, 16, 4. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, C.; Zhao, N.; Li, W.; Yang, Z.; Liu, X.; Le, W.; Zhang, X. Potential biomarkers of Parkinson’s disease revealed by plasma metabolic profiling. J. Chromatogr. B 2018, 1081–1082, 101–108. [Google Scholar] [CrossRef]
- Li, P.; Killinger, B.; Ensink, E.; Beddows, I.; Yilmaz, A.; Lubben, N.; Lamp, J.; Schilthuis, M.; Vega, I.; Woltjer, R.; et al. Gut Microbiota Dysbiosis Is Associated with Elevated Bile Acids in Parkinson’s Disease. Metabolites 2021, 11, 29. [Google Scholar] [CrossRef]
- Martinez, T.N.; Greenamyre, J.T.; Ritz, B.R.; Paul, K.C.; Bronstein, J.M.; Lee, J.; Fifel, K.; Piggins, H.; Deboer, T.; Martella, G.; et al. Toxin Models of Mitochondrial Dysfunction in Parkinson’s Disease. Antioxid. Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolaños, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 2017, 25, 542–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Caldas, M.; Carvalho, A.N.; Rodrigues, E.; Henderson, C.; Wolf, C.R.; Rodrigues, C.; Gama, M.J. Tauroursodeoxycholic Acid Prevents MPTP-Induced Dopaminergic Cell Death in a Mouse Model of Parkinson’s Disease. Mol. Neurobiol. 2012, 46, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.I.; Duarte-Silva, S.; Silva-Fernandes, A.; Nunes, M.J.; Carvalho, A.N.; Rodrigues, E.; Gama, M.J.; Rodrigues, C.M.P.; Maciel, P.; Castro-Caldas, M. Tauroursodeoxycholic Acid Improves Motor Symptoms in a Mouse Model of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 9139–9155. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, N.F.; Safar, M.M.; Salem, H.A. Ursodeoxycholic Acid Ameliorates Apoptotic Cascade in the Rotenone Model of Parkinson’s Disease: Modulation of Mitochondrial Perturbations. Mol. Neurobiol. 2014, 53, 810–817. [Google Scholar] [CrossRef]
- Dayalu, P.; Albin, R.L. Huntington Disease. Neurol. Clin. 2015, 33, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nature 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffan, J.S. Does Huntingtin play a role in selective macroautophagy? Cell Cycle 2010, 9, 3401–3413. [Google Scholar] [CrossRef] [PubMed]
- Wanker, E.E.; Ast, A.; Schindler, F.; Trepte, P.; Schnoegl, S. The pathobiology of perturbed mutant huntingtin protein–protein interactions in Huntington’s disease. J. Neurochem. 2019, 151, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2020, 17, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Túnez, I.; Tasset, I.; La Cruz, V.P.-D.; Santamaría, A. 3-Nitropropionic Acid as a Tool to Study the Mechanisms Involved in Huntington’s Disease: Past, Present and Future. Molecules 2010, 15, 878–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouillet, E.; Jacquard, C.; Bizat, N.; Blum, D. 3-Nitropropionic acid: A mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J. Neurochem. 2005, 95, 1521–1540. [Google Scholar] [CrossRef] [PubMed]
- Keene, C.; Rodrigues, C.M.; Eich, T.; Linehan-Stieers, C.; Abt, A.; Kren, B.T.; Steer, C.J.; Low, W.C. A Bile Acid Protects against Motor and Cognitive Deficits and Reduces Striatal Degeneration in the 3-Nitropropionic Acid Model of Huntington’s Disease. Exp. Neurol. 2001, 171, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Keene, C.D.; Rodrigues, C.M.P.; Eich, T.; Chhabra, M.S.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10671–10676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Modification of bile acids by the gut microbiome. Cholesterol is converted in the liver to cholic acid (CA) or chenodeoxycholic acid (CDCA), and these bile acids are then con-jugated with glycine or taurine (G/T). Conjugated CA and CDCA are transferred to the gallbladder and secreted from the gallbladder into the intestine upon food intake. Conjugated BA is deconjugated by intestinal bacterial bile salt hydrolases (BSHs), and CA and CDCA are further dehydrogenated to deoxycholic acid (DCA) and lithocholic acid (LCA), respectively. CDCA is also dehydrogenated and epimerized by intestinal bacteria and converted to ursodeoxycholic acid (UDCA). Approximately 95% of the BAs in the gut are absorbed and transferred to the liver, while the remaining BAs are excreted in the feces. Some of the BAs reabsorbed from the gut are effluxed into the systemic circulation.
Figure 1.
Modification of bile acids by the gut microbiome. Cholesterol is converted in the liver to cholic acid (CA) or chenodeoxycholic acid (CDCA), and these bile acids are then con-jugated with glycine or taurine (G/T). Conjugated CA and CDCA are transferred to the gallbladder and secreted from the gallbladder into the intestine upon food intake. Conjugated BA is deconjugated by intestinal bacterial bile salt hydrolases (BSHs), and CA and CDCA are further dehydrogenated to deoxycholic acid (DCA) and lithocholic acid (LCA), respectively. CDCA is also dehydrogenated and epimerized by intestinal bacteria and converted to ursodeoxycholic acid (UDCA). Approximately 95% of the BAs in the gut are absorbed and transferred to the liver, while the remaining BAs are excreted in the feces. Some of the BAs reabsorbed from the gut are effluxed into the systemic circulation.

Figure 2.
The effects of bile acids (BAs) on mitochondria and autophagy. The effects of BAs on mitochondria: DCA, CDCA, and their taurine conjugates activate MFN2 by binding directly to MFN2, promoting mitochondria-to-mitochondria fusion and mitochondria-to-ER fusion. These BAs, at normal concentration, promote the fusion between mitochondria, leading to the generation of ATP. By contrast, these BAs, at high concentration, promote the fusion between the mitochondria and the ER, leading to Ca2+ influx from the ER to the mitochondria. The effects of BAs on autophagy: secondary BAs (DCA and UDCA) induce autophagy and primary bile acids (CA, CDCA, and TCA) inhibit autophagy by preventing the formation of autolysosomes. In addition, the activation of TGR5, a receptor for secondary bile acids, leads to the activation of autophagy. By contrast, the activation of FXR, a receptor for primary bile acids, leads to the inhibition of autophagy.
Figure 2.
The effects of bile acids (BAs) on mitochondria and autophagy. The effects of BAs on mitochondria: DCA, CDCA, and their taurine conjugates activate MFN2 by binding directly to MFN2, promoting mitochondria-to-mitochondria fusion and mitochondria-to-ER fusion. These BAs, at normal concentration, promote the fusion between mitochondria, leading to the generation of ATP. By contrast, these BAs, at high concentration, promote the fusion between the mitochondria and the ER, leading to Ca2+ influx from the ER to the mitochondria. The effects of BAs on autophagy: secondary BAs (DCA and UDCA) induce autophagy and primary bile acids (CA, CDCA, and TCA) inhibit autophagy by preventing the formation of autolysosomes. In addition, the activation of TGR5, a receptor for secondary bile acids, leads to the activation of autophagy. By contrast, the activation of FXR, a receptor for primary bile acids, leads to the inhibition of autophagy.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kiriyama, Y.; Nochi, H. Role of Microbiota-Modified Bile Acids in the Regulation of Intracellular Organelles and Neurodegenerative Diseases. Genes 2023, 14, 825. https://doi.org/10.3390/genes14040825
AMA Style
Kiriyama Y, Nochi H. Role of Microbiota-Modified Bile Acids in the Regulation of Intracellular Organelles and Neurodegenerative Diseases. Genes. 2023; 14(4):825. https://doi.org/10.3390/genes14040825
Chicago/Turabian StyleKiriyama, Yoshimitsu, and Hiromi Nochi. 2023. "Role of Microbiota-Modified Bile Acids in the Regulation of Intracellular Organelles and Neurodegenerative Diseases" Genes 14, no. 4: 825. https://doi.org/10.3390/genes14040825
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.