
Genomic Characterization and Genetic Profiles of Salmonella Gallinarum Strains Isolated from Layers with Fowl Typhoid in Colombia

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Isolation
2.2. DNA Extraction and PCR Identification
2.3. Whole-Genome Sequencing, Assembly, and Annotation
2.4. Bioinformatics Analysis: Phylogenetics. Molecular Typing. Virulome, Resistome, and Mobile Genetic Element Characterization; and Comparative Genome Analyses
3. Results
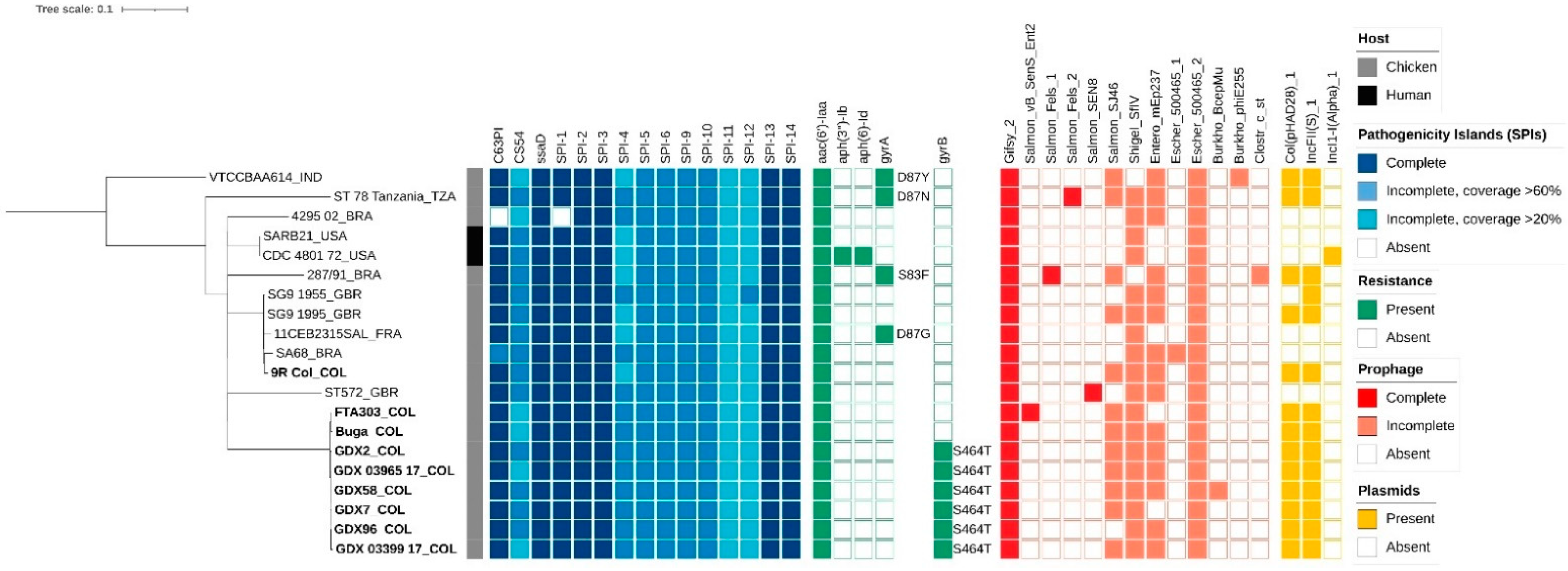
3.1. Genome Assembly, Annotation, and Molecular Typing
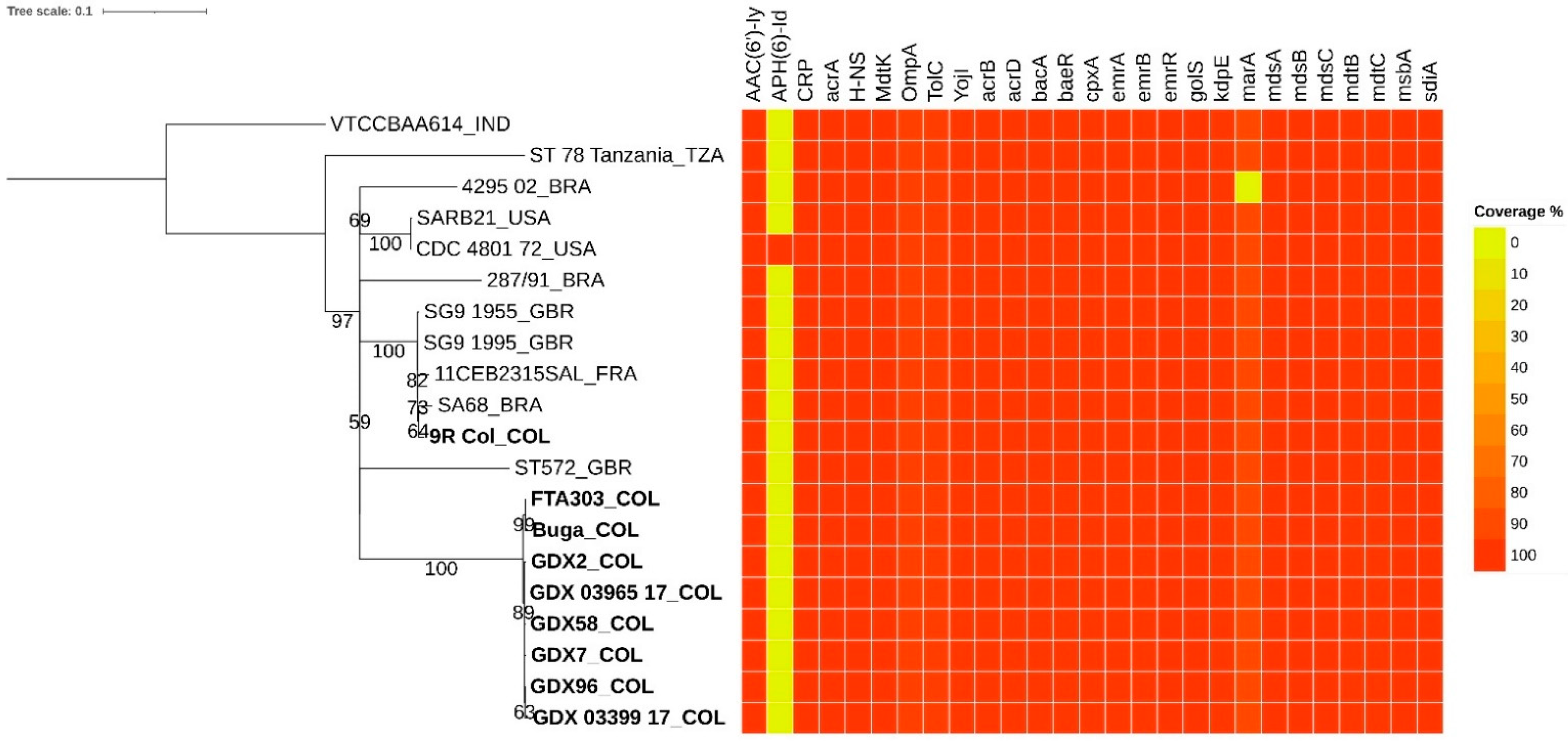
3.2. Antimicrobial Resistance Genes
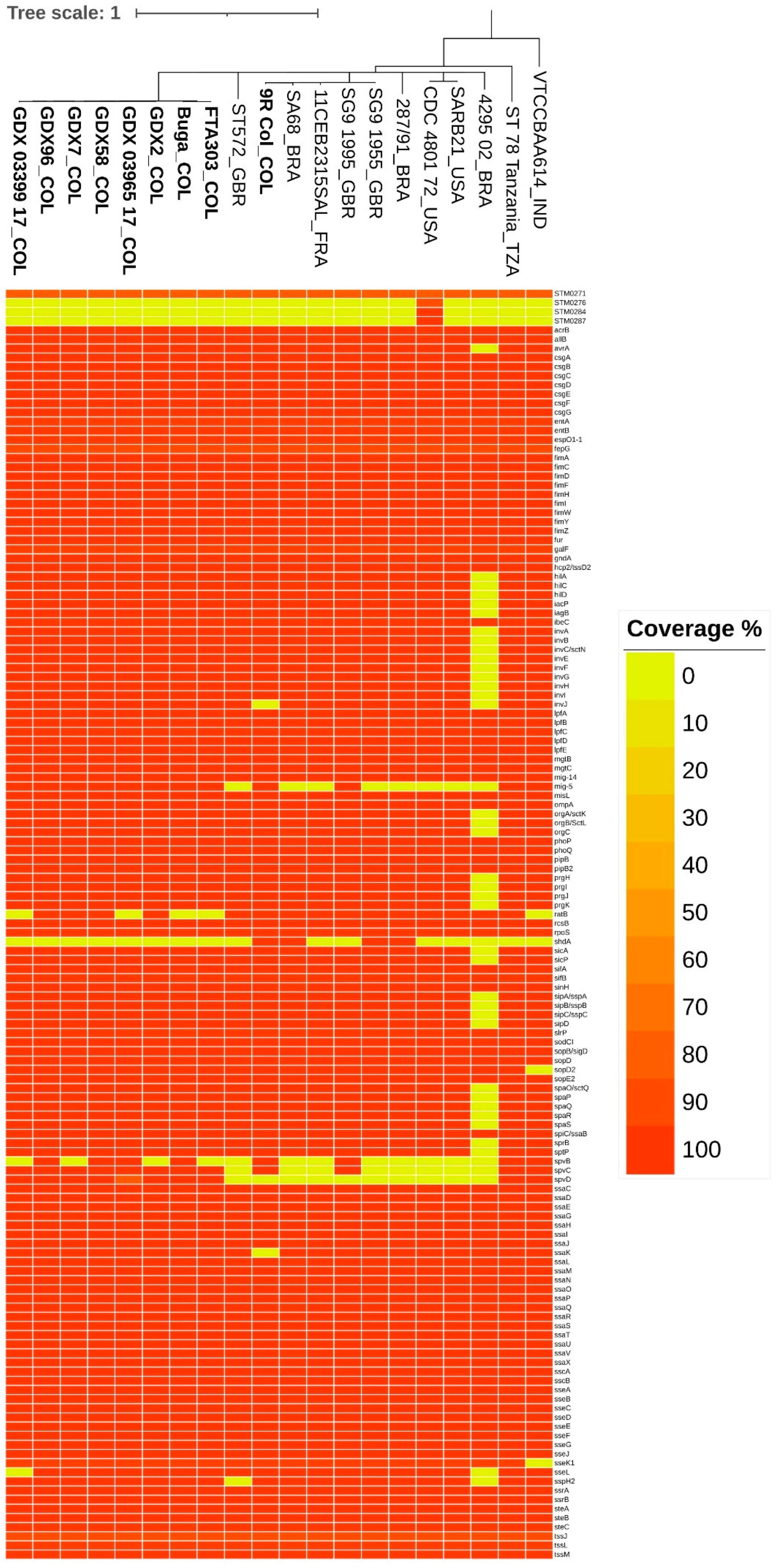
3.3. Virulence Genes
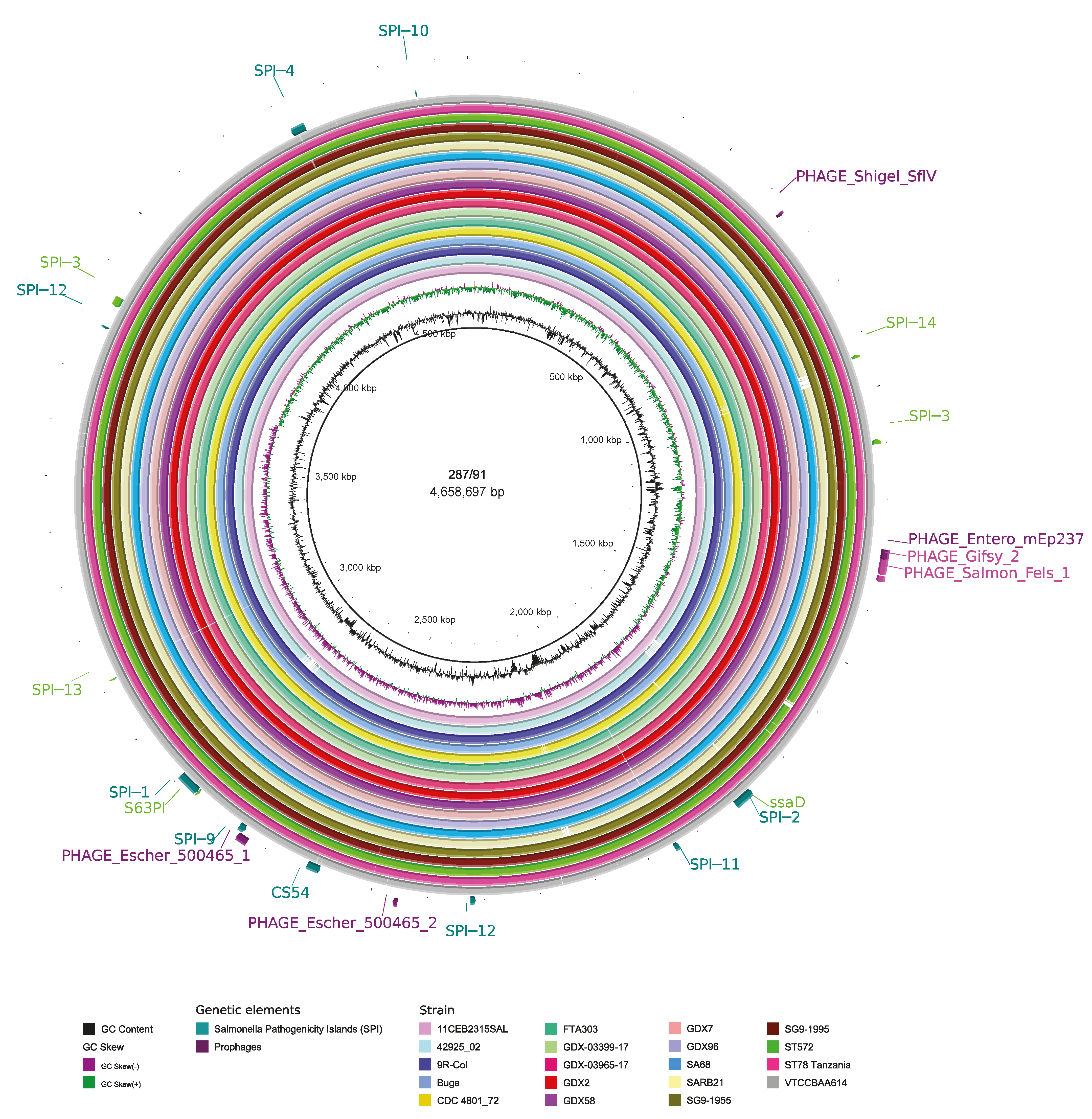
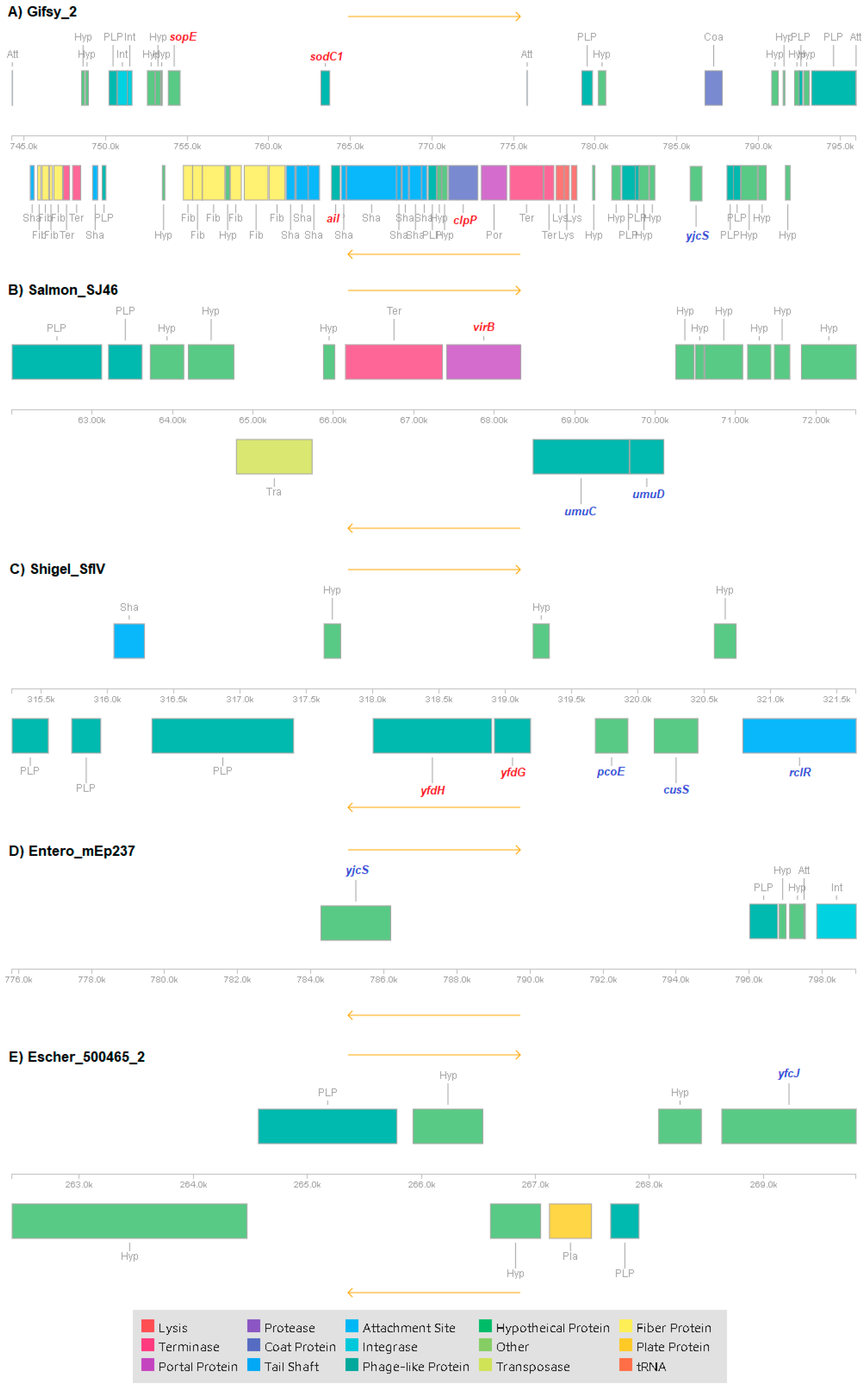
3.4. Mobile Genetic Elements
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guibourdenche, M.; Roggentin, P.; Mikoleit, M.; Fields, P.I.; Bockemühl, J.; Grimont, P.A.D.; Weill, F.-X. Supplement 2003–2007 (No. 47) to the White-Kauffmann-Le Minor Scheme. Res. Microbiol. 2010, 161, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Thomson, N.R.; Clayton, D.J.; Windhorst, D.; Vernikos, G.; Davidson, S.; Churcher, C.; Quail, M.A.; Stevens, M.; Jones, M.A.; Watson, M.; et al. Comparative Genome Analysis of Salmonella Enteritidis PT4 and Salmonella Gallinarum 287/91 Provides Insights into Evolutionary and Host Adaptation Pathways. Genome. Res. 2008, 18, 1624–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivaprasad, H.L.; Barrow, P.A. Pullorum Disease and Fowl Typhoid. In Diseases of Poultry, 13th ed.; Swayne, D.E., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 678–693. [Google Scholar]
- Barrow, P.A.; Freitas Neto, O.C. Pullorum Disease and Fowl Typhoid—New Thoughts on Old Diseases: A Review. Avian Pathol. 2011, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chappell, L.; Kaiser, P.; Barrow, P.; Jones, M.A.; Johnston, C.; Wigley, P. The Immunobiology of Avian Systemic Salmonellosis. Vet. Immunol. Immunopathol. 2009, 128, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, S.C.; Forest, C.G.; Lepage, C.; Leclerc, J.-M.; Daigle, F. So Similar, yet so Different: Uncovering Distinctive Features in the Genomes of Salmonella Enterica Serovars Typhimurium and Typhi. FEMS Microbiol. Lett. 2010, 305, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Marcus, S.L.; Brumell, J.H.; Pfeifer, C.G.; Finlay, B.B. Salmonella Pathogenicity Islands: Big Virulence in Small Packages. Microbes Infect. 2000, 2, 145–156. [Google Scholar] [CrossRef]
- Cheng, R.A.; Eade, C.R.; Wiedmann, M. Embracing Diversity: Differences in Virulence Mechanisms, Disease Severity, and Host Adaptations Contribute to the Success of Nontyphoidal Salmonella as a Foodborne Pathogen. Front. Microbiol. 2019, 10, 1368. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.H.; Lee, M.-J.; Park, J.-H.; Lee, J.-H.; Eo, S.-K.; Kwon, J.-T.; Chae, J.-S. Identification of Salmonella Gallinarum Virulence Genes in a Chicken Infection Model Using PCR-Based Signature-Tagged Mutagenesis. Microbiology 2005, 151, 3957–3968. [Google Scholar] [CrossRef] [Green Version]
- Rychlik, I.; Gregorova, D.; Hradecka, H. Distribution and Function of Plasmids in Salmonella Enterica. Vet. Microbiol. 2006, 112, 1–10. [Google Scholar] [CrossRef]
- Figueroa-Bossi, N.; Uzzau, S.; Maloriol, D.; Bossi, L. Variable Assortment of Prophages Provides a Transferable Repertoire of Pathogenic Determinants in Salmonella. Mol. Microbiol. 2001, 39, 260–271. [Google Scholar] [CrossRef]
- Ehrbar, K.; Hardt, W.-D. Bacteriophage-Encoded Type III Effectors in Salmonella Enterica Subspecies 1 Serovar Typhimurium. Infect. Genet. Evol. 2005, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Jain, S.K. Role of Antigens and Virulence Factors of Salmonella Enterica Serovar Typhi in Its Pathogenesis. Microbiol. Res. 2012, 167, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Revolledo, L.; Ferreira, A.J.P.; Mead, G.C. Prospects in Salmonella Control: Competitive Exclusion, Probiotics, and Enhancement of Avian Intestinal Immunity. J. Appl. Poult. Res. 2006, 15, 341–351. [Google Scholar] [CrossRef]
- Smith, H.W. The Use of Live Vaccines in Experimental Salmonella Gallinarum Infection in Chickens with Observations on Their Interference Effect. J. Hyg. 1956, 54, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Barrow, P.A.; Lovell, M.A.; Stocker, B.A. Protection against Experimental Fowl Typhoid by Parenteral Administration of Live SL5828, an AroA-SerC (Aromatic Dependent) Mutant of a Wild-Type Salmonella Gallinarum Strain Made Lysogenic for P22 Sie. Avian. Pathol. 2000, 29, 423–431. [Google Scholar] [CrossRef]
- Rosu, V.; Chadfield, M.S.; Santona, A.; Christensen, J.P.; Thomsen, L.E.; Rubino, S.; Olsen, J.E. Effects of Crp Deletion in Salmonella Enterica Serotype Gallinarum. Acta Vet. Scand. 2007, 49, 14. [Google Scholar] [CrossRef] [Green Version]
- Penha Filho, R.A.C.; de Paiva, J.B.; da Silva, M.D.; de Almeida, A.M.; Berchieri, A. Control of Salmonella Enteritidis and Salmonella Gallinarum in Birds by Using Live Vaccine Candidate Containing Attenuated Salmonella Gallinarum Mutant Strain. Vaccine 2010, 28, 2853–2859. [Google Scholar] [CrossRef] [PubMed]
- Le Bouquin, S.; Bonifait, L.; Thépault, A.; Ledein, T.; Guillon, F.; Rouxel, S.; Souillard, R.; Chemaly, M. Epidemiological and Bacteriological Investigations Using Whole-Genome Sequencing in a Recurrent Outbreak of Pullorum Disease on a Quail Farm in France. Animals 2020, 11, 29. [Google Scholar] [CrossRef]
- Li, Y.; Kang, X.; Ed-Dra, A.; Zhou, X.; Jia, C.; Müller, A.; Liu, Y.; Kehrenberg, C.; Yue, M. Genome-Based Assessment of Antimicrobial Resistance and Virulence Potential of Isolates of Non-Pullorum/Gallinarum Salmonella Serovars Recovered from Dead Poultry in China. Microbiol. Spectr. 2022, 10, e0096522. [Google Scholar] [CrossRef]
- Matthews, T.D.; Schmieder, R.; Silva, G.G.Z.; Busch, J.; Cassman, N.; Dutilh, B.E.; Green, D.; Matlock, B.; Heffernan, B.; Olsen, G.J.; et al. Genomic Comparison of the Closely-Related Salmonella Enterica Serovars Enteritidis, Dublin and Gallinarum. PLoS ONE 2015, 10, e0126883. [Google Scholar] [CrossRef]
- Batista, D.F.A.; Freitas Neto, O.C.; Barrow, P.A.; de Oliveira, M.T.; Almeida, A.M.; Ferraudo, A.S.; Berchieri, A. Identification and Characterization of Regions of Difference between the Salmonella Gallinarum Biovar Gallinarum and the Salmonella Gallinarum Biovar Pullorum Genomes. Infect. Genet. Evol. 2015, 30, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Vaid, R.K.; Thakur, Z.; Anand, T.; Kumar, S.; Tripathi, B.N. Comparative Genome Analysis of Salmonella Enterica Serovar Gallinarum Biovars Pullorum and Gallinarum Decodes Strain Specific Genes. PLoS ONE 2021, 16, e0255612. [Google Scholar] [CrossRef]
- Van Immerseel, F.; Studholme, D.J.; Eeckhaut, V.; Heyndrickx, M.; Dewulf, J.; Dewaele, I.; Van Hoorebeke, S.; Haesebrouck, F.; Van Meirhaeghe, H.; Ducatelle, R.; et al. Salmonella Gallinarum Field Isolates from Laying Hens Are Related to the Vaccine Strain SG9R. Vaccine 2013, 31, 4940–4945. [Google Scholar] [CrossRef]
- De Carli, S.; Gräf, T.; Kipper, D.; Lehmann, F.K.M.; Zanetti, N.; Siqueira, F.M.; Cibulski, S.; Fonseca, A.S.K.; Ikuta, N.; Lunge, V.R. Molecular and Phylogenetic Analyses of Salmonella Gallinarum Trace the Origin and Diversification of Recent Outbreaks of Fowl Typhoid in Poultry Farms. Vet. Microbiol. 2017, 212, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.-H.; Ha, E.-J.; Ko, D.-S.; Lee, C.-Y.; Kim, J.-H.; Kwon, H.-J. Molecular Evolution of Salmonella Enterica Subsp. Enterica Serovar Gallinarum Biovar Gallinarum in the Field. Vet. Microbiol. 2019, 235, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Chacón, R.D.; Moura, Q.; Astolfi-Ferreira, C.S.; De la Torre, D.I.; Guerrero, L.M.C.; Martínez, L.A.R.; Corzo, A.N.; Morales López, N.H.; Ramírez, M.; Lincopan, N.; et al. Draft Genome Sequences of Four Salmonella Enterica Subsp. Enterica Serovar Gallinarum Strains Isolated from Layer Breeder Flocks in an Outbreak of Fowl Typhoid in Colombia. Microbiol. Resour. Announc. 2019, 8, e00122-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.-S.; Kwon, Y.-K.; Kim, H.-R.; Oh, J.-Y.; Kim, M.-J.; An, B.-K.; Shin, E.-G.; Kwon, J.-H.; Park, C.-K. Differential Identification of Salmonella Enterica Serovar Gallinarum Biovars Gallinarum and Pullorum and the Biovar Gallinarum Live Vaccine Strain 9R. Vet. Microbiol. 2012, 160, 491–495. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 30 October 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI Prokaryotic Genome Annotation Pipeline. Nucleic. Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic. Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Yoshida, C.E.; Kruczkiewicz, P.; Laing, C.R.; Lingohr, E.J.; Gannon, V.P.J.; Nash, J.H.E.; Taboada, E.N. The Salmonella In Silico Typing Resource (SISTR): An Open Web-Accessible Tool for Rapidly Typing and Subtyping Draft Salmonella Genome Assemblies. PLoS ONE 2016, 11, e0147101. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [Green Version]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic. Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic. Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids Using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Roer, L.; Hendriksen, R.S.; Leekitcharoenphon, P.; Lukjancenko, O.; Kaas, R.S.; Hasman, H.; Aarestrup, F.M. Is the Evolution of Salmonella Enterica Subsp. Enterica Linked to Restriction-Modification Systems? mSystems 2016, 1, e00009-16. [Google Scholar] [CrossRef] [Green Version]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic. Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple Prokaryote Genome Comparisons. BMC Genomics 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- USDA. Livestock and Poultry: World Markets and Trade. United States Department of Agriculture 2022. Available online: https://www.fas.usda.gov/data/livestock-and-poultry-world-markets-and-trade (accessed on 30 November 2022).
- Pulido-Landínez, M.; Sánchez-Ingunza, R.; Guard, J.; do Nascimento, V.P. Presence of Salmonella Enteritidis and Salmonella Gallinarum in Commercial Laying Hens Diagnosed with Fowl Typhoid Disease in Colombia. Avian. Dis. 2014, 58, 165–170. [Google Scholar] [CrossRef]
- Davin-Regli, A.; Bolla, J.-M.; James, C.E.; Lavigne, J.-P.; Chevalier, J.; Garnotel, E.; Molitor, A.; Pagès, J.-M. Membrane Permeability and Regulation of Drug “Influx and Efflux” in Enterobacterial Pathogens. Curr. Drug. Targets 2008, 9, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Vilela, F.P.; Rodrigues, D.D.P.; Allard, M.W.; Falcão, J.P. Prevalence of Efflux Pump and Heavy Metal Tolerance Encoding Genes among Salmonella Enterica Serovar Infantis Strains from Diverse Sources in Brazil. PLoS ONE 2022, 17, e0277979. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, R.; Barh, D.; Weimer, B.C.; Viana, M.V.C.; Profeta, R.; Sousa, T.J.; Aburjaile, F.F.; Quino, W.; Souza, R.P.; Mestanza, O.; et al. WGS-Based Lineage and Antimicrobial Resistance Pattern of Salmonella Typhimurium Isolated during 2000–2017 in Peru. Antibiotics 2022, 11, 1170. [Google Scholar] [CrossRef]
- Chacón, R.D.; Chacón, J.L.; Ramírez, M.; Cueva, C.L.R.; Quispe-Rojas, W.U.; Reyes-Moreno, C.B.; Astolfi-Ferreira, C.S.; Ferreira, A.J.P. Complete Genome Sequence Data of Two Salmonella Enterica Subsp. Enterica Serovar Gallinarum: A 9R Vaccine Strain and a Virulent Brazilian Field Strain. Data Brief 2023, 47, 108959. [Google Scholar] [CrossRef] [PubMed]
- Randall, L.P.; Woodward, M.J. The Multiple Antibiotic Resistance (Mar) Locus and Its Significance. Res. Vet. Sci. 2002, 72, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kern, W.V.; Oethinger, M.; Jellen-Ritter, A.S.; Levy, S.B. Non-Target Gene Mutations in the Development of Fluoroquinolone Resistance in Escherichia Coli. Antimicrob. Agents Chemother. 2000, 44, 814–820. [Google Scholar] [CrossRef] [Green Version]
- Cloeckaert, A.; Chaslus-Dancla, E. Mechanisms of Quinolone Resistance in Salmonella. Vet. Res. 2001, 32, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.S.; Kim, A.; Jung, B.Y.; Her, M.; Jeong, W.; Cho, Y.M.; Oh, J.Y.; Lee, Y.J.; Kwon, J.H.; Kwon, Y.K. Characterization of Antimicrobial Resistance of Recent Salmonella Enterica Serovar Gallinarum Isolates from Chickens in South Korea. Avian Pathol. 2010, 39, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Seo, K.W.; Mo, I.P.; Lee, Y.J. Genetic Characterization of Fluoroquinolone Resistance in Salmonella Enterica Serovar Gallinarum Isolates from Chicken in Korea. Avian Dis. 2019, 63, 584–590. [Google Scholar] [CrossRef]
- Gensberg, K.; Jin, Y.F.; Piddock, L.J. A Novel GyrB Mutation in a Fluoroquinolone-Resistant Clinical Isolate of Salmonella Typhimurium. FEMS Microbiol. Lett. 1995, 132, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.L.; Panzenhagen, P.; Ferrari, R.G.; Dos Santos, A.; Paschoalin, V.M.F.; Conte-Junior, C.A. Frequency of Antimicrobial Resistance Genes in Salmonella From Brazil by in Silico Whole-Genome Sequencing Analysis: An Overview of the Last Four Decades. Front. Microbiol. 2020, 11, 1864. [Google Scholar] [CrossRef] [PubMed]
- Magnet, S.; Courvalin, P.; Lambert, T. Activation of the Cryptic Aac(6′)-Iy Aminoglycoside Resistance Gene of Salmonella by a Chromosomal Deletion Generating a Transcriptional Fusion. J. Bacteriol. 1999, 181, 6650–6655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nhung, N.T.; Chansiripornchai, N.; Carrique-Mas, J.J. Antimicrobial Resistance in Bacterial Poultry Pathogens: A Review. Front. Vet. Sci. 2017, 4, 126. [Google Scholar] [CrossRef] [Green Version]
- van Duijkeren, E.; Schwarz, C.; Bouchard, D.; Catry, B.; Pomba, C.; Baptiste, K.E.; Moreno, M.A.; Rantala, M.; Ružauskas, M.; Sanders, P.; et al. The Use of Aminoglycosides in Animals within the EU: Development of Resistance in Animals and Possible Impact on Human and Animal Health: A Review. J. Antimicrob. Chemother. 2019, 74, 2480–2496. [Google Scholar] [CrossRef]
- Jones, M.A.; Wigley, P.; Page, K.L.; Hulme, S.D.; Barrow, P.A. Salmonella Enterica Serovar Gallinarum Requires the Salmonella Pathogenicity Island 2 Type III Secretion System but Not the Salmonella Pathogenicity Island 1 Type III Secretion System for Virulence in Chickens. Infect. Immun. 2001, 69, 5471–5476. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Hardt, W.D.; Galán, J.E. Salmonella Typhimurium Encodes a Putative Iron Transport System within the Centisome 63 Pathogenicity Island. Infect. Immun. 1999, 67, 1974–1981. [Google Scholar] [CrossRef]
- Juhas, M.; van der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic Islands: Tools of Bacterial Horizontal Gene Transfer and Evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amavisit, P.; Lightfoot, D.; Browning, G.F.; Markham, P.F. Variation between Pathogenic Serovars within Salmonella Pathogenicity Islands. J. Bacteriol. 2003, 185, 3624–3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, S.L.; Johnson, T.J.; Ricke, S.C.; Nayak, R.; Danzeisen, J. Salmonella Pathogenicity and Host Adaptation in Chicken-Associated Serovars. Microbiol. Mol. Biol. Rev. 2013, 77, 582–607. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.-J.; Cho, S.-H. Pathogenicity of SG 9R, a Rough Vaccine Strain against Fowl Typhoid. Vaccine 2011, 29, 1311–1318. [Google Scholar] [CrossRef]
- Grabe, G.J.; Zhang, Y.; Przydacz, M.; Rolhion, N.; Yang, Y.; Pruneda, J.N.; Komander, D.; Holden, D.W.; Hare, S.A. The Salmonella Effector SpvD Is a Cysteine Hydrolase with a Serovar-Specific Polymorphism Influencing Catalytic Activity, Suppression of Immune Responses, and Bacterial Virulence. J. Biol. Chem. 2016, 291, 25853–25863. [Google Scholar] [CrossRef] [Green Version]
- Rüssmann, H.; Kubori, T.; Sauer, J.; Galán, J.E. Molecular and Functional Analysis of the Type III Secretion Signal of the Salmonella Enterica InvJ Protein. Mol. Microbiol. 2002, 46, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Wee, D.H.; Hughes, K.T. Molecular Ruler Determines Needle Length for the Salmonella Spi-1 Injectisome. Proc. Natl. Acad. Sci. USA 2015, 112, 4098–4103. [Google Scholar] [CrossRef] [Green Version]
- Hensel, M.; Shea, J.E.; Raupach, B.; Monack, D.; Falkow, S.; Gleeson, C.; Kubo, T.; Holden, D.W. Functional Analysis of SsaJ and the SsaK/U Operon, 13 Genes Encoding Components of the Type III Secretion Apparatus of Salmonella Pathogenicity Island 2. Mol. Microbiol. 1997, 24, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.-S.; Kwon, Y.-K.; Kim, H.-R.; Oh, J.-Y.; Kim, M.-J.; An, B.-K.; Shin, E.-G.; Kwon, J.-H.; Park, C.-K. Comparative Proteome and Transcriptome Analyses of Wild-Type and Live Vaccine Strains of Salmonella Enterica Serovar Gallinarum. Vaccine 2012, 30, 6368–6375. [Google Scholar] [CrossRef]
- Fiegen, U.; Klein, G.; de Jong, A.; Kehrenberg, C. Detection of a Novel QnrB19-Carrying Plasmid Variant Mediating Decreased Fluoroquinolone Susceptibility in Salmonella Enterica Serovar Hadar. Microb. Drug. Resist. 2017, 23, 280–284. [Google Scholar] [CrossRef]
- Astolfi-Ferreira, C.S.; Pequini, M.R.S.; Nuñez, L.F.N.; Parra, S.H.S.; Chacon, R.; de la Torre, D.I.D.; Pedroso, A.C.; Ferreira, A.J.P. A Comparative Survey between Non-Systemic Salmonella Spp. (Paratyphoid Group) and Systemic Salmonella Pullorum and S. Gallinarum with a Focus on Virulence Genes. Pesq. Vet. Bras. 2017, 37, 1064–1068. [Google Scholar] [CrossRef]
- Mottawea, W.; Duceppe, M.-O.; Dupras, A.A.; Usongo, V.; Jeukens, J.; Freschi, L.; Emond-Rheault, J.-G.; Hamel, J.; Kukavica-Ibrulj, I.; Boyle, B.; et al. Salmonella Enterica Prophage Sequence Profiles Reflect Genome Diversity and Can Be Used for High Discrimination Subtyping. Front. Microbiol. 2018, 9, 836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingsley, R.A.; Humphries, A.D.; Weening, E.H.; De Zoete, M.R.; Winter, S.; Papaconstantinopoulou, A.; Dougan, G.; Bäumler, A.J. Molecular and Phenotypic Analysis of the CS54 Island of Salmonella Enterica Serotype Typhimurium: Identification of Intestinal Colonization and Persistence Determinants. Infect. Immun. 2003, 71, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rytkönen, A.; Poh, J.; Garmendia, J.; Boyle, C.; Thompson, A.; Liu, M.; Freemont, P.; Hinton, J.C.D.; Holden, D.W. SseL, a Salmonella Deubiquitinase Required for Macrophage Killing and Virulence. Proc. Natl. Acad. Sci. USA 2007, 104, 3502–3507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäumler, A.J.; Heffron, F. Identification and Sequence Analysis of LpfABCDE, a Putative Fimbrial Operon of Salmonella Typhimurium. J. Bacteriol. 1995, 177, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suez, J.; Porwollik, S.; Dagan, A.; Marzel, A.; Schorr, Y.I.; Desai, P.T.; Agmon, V.; McClelland, M.; Rahav, G.; Gal-Mor, O. Virulence Gene Profiling and Pathogenicity Characterization of Non-Typhoidal Salmonella Accounted for Invasive Disease in Humans. PLoS ONE 2013, 8, e58449. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Farm | Isolated Strain | Source | Type of Bird | Age (Weeks) |
|---|---|---|---|---|
| 1 | Buga | Liver | Layer breeder | 1 |
| 2 | GDX7 | Liver | Layer breeder | 13 |
| 3 | FTA303 | Liver | Layer breeder | 15 |
| 4 | GDX58 | Liver | Layer breeder | 25 |
| 5 | GDX2 | Liver | Layer hen | 1 |
| 6 | GDX96 | Liver | Layer hen | 11 |
| 7 | GDX-03965-17 | Brain | Layer hen | 23 |
| 8 | GDX-03399-17 | Coelomic cavity | Layer hen | 63 |
| Strain | Country | Year Isolated | SRA Accession No. |
|---|---|---|---|
| Buga | Colombia | 2017 | SRR8521219 |
| GDX7 | Colombia | 2017 | SRR8521149 |
| FTA303 | Colombia | 2017 | SRR8521220 |
| GDX58 | Colombia | 2017 | SRR8521221 |
| GDX2 | Colombia | 2017 | SRR22245387 |
| GDX96 | Colombia | 2017 | SRR22245388 |
| GDX-03965-17 | Colombia | 2017 | SRR22245385 |
| GDX-03399-17 | Colombia | 2017 | SRR22245386 |
| 9R-Col | Colombia | 2017 | SRR22245384 |
| SA68 | Brazil | 1990 | CP110192 B |
| 4295/02 | Brazil | 2002 | SRR17885111 |
| 287/91 | Brazil | 2010 | SRR21618241 |
| SG9-1955 | UK | 1955 | CM001153-CM001154 B |
| SG9-1995 | UK | 1995 | SRR1045136 |
| ST572 | UK | 2009 | SRR2121409 |
| 11CEB2315SAL | France | 2011 | ERR9714929 |
| SARB21 A | USA | 1972 | ERR424914 |
| CDC 4801/72 A | USA | 1972 | SRR1122702 |
| ST 78 Tanzania | Tanzania | 2017 | SRR11005774 |
| VTCCBAA614 | India | 2012 | JSWQ00000000 B |
| Strain | Genome Status | No. of Bases | No. of Contigs | N50 | Depth | % GC | No. of CDS | MLST |
|---|---|---|---|---|---|---|---|---|
| Buga | Draft | 4,703,728 | 43 | 290,630 | 58× | 53.1 | 4509 | 78 |
| GDX7 | Draft | 4,703,746 | 41 | 243,065 | 61× | 51.5 | 4506 | 78 |
| FTA303 | Draft | 4,769,563 | 140 | 163,753 | 77× | 50.1 | 4589 | 78 |
| GDX58 | Draft | 4,705,375 | 49 | 401,050 | 70× | 51.1 | 4593 | 78 |
| GDX2 | Draft | 4,704,264 | 34 | 400,863 | 70× | 51.9 | 4505 | 78 |
| GDX96 | Draft | 4,704,639 | 39 | 400,863 | 74× | 51.2 | 4506 | 78 |
| GDX-03965-17 | Draft | 4,702,114 | 56 | 181,715 | 60× | 52.3 | 4506 | 78 |
| GDX-03399-17 | Draft | 4,701,504 | 81 | 116,199 | 65× | 51.7 | 4509 | 78 |
| 9R-Col | Draft | 4,738,050 | 175 | 206,437 | 49× | 51.9 | 4534 | 78 |
| SA68 | Complete | 4,657,435 | NA * | NA * | NA * | 52.2 | 4412 | 78 |
| 4295/02 | Draft | 4,477,714 | 37 | 283,382 | 36× | 52.3 | 4372 | 78 |
| 287/91 | Complete | 4,658,697 | NA * | NA * | NA * | 52.2 | 4510 | 331 |
| SG9-1955 | Complete | 4,658,698 | NA * | NA * | NA * | 52.2 | 4392 | 78 |
| SG9-1995 | Draft | 4,701,135 | 62 | 195,494 | 70× | 50.1 | 4502 | 78 |
| ST572 | Draft | 4,641,016 | 31 | 495,945 | 67× | 51.7 | 4418 | 78 |
| 11CEB2315SAL | Draft | 4,614,697 | 41 | 284,067 | 148× | 51.7 | 4400 | 78 |
| SARB21 | Draft | 4,560,140 | 47 | 235,893 | 32× | 51.8 | 4336 | 78 |
| CDC 4801/72 | Draft | 4,757,193 | 264 | 185,274 | 184× | 50.9 | 4334 | 78 |
| ST 78 Tanzania | Draft | 4,737,077 | 35 | 403,192 | 113× | 51.5 | 4549 | 78 |
| VTCCBAA614 | Draft | 4,701,135 | 62 | 195,494 | 70× | 50.1 | 4504 | 78 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chacón, R.D.; Ramírez, M.; Rodríguez-Cueva, C.L.; Sánchez, C.; Quispe-Rojas, W.U.; Astolfi-Ferreira, C.S.; Piantino Ferreira, A.J. Genomic Characterization and Genetic Profiles of Salmonella Gallinarum Strains Isolated from Layers with Fowl Typhoid in Colombia. Genes 2023, 14, 823. https://doi.org/10.3390/genes14040823
Chacón RD, Ramírez M, Rodríguez-Cueva CL, Sánchez C, Quispe-Rojas WU, Astolfi-Ferreira CS, Piantino Ferreira AJ. Genomic Characterization and Genetic Profiles of Salmonella Gallinarum Strains Isolated from Layers with Fowl Typhoid in Colombia. Genes. 2023; 14(4):823. https://doi.org/10.3390/genes14040823
Chicago/Turabian StyleChacón, Ruy D., Manuel Ramírez, Carmen L. Rodríguez-Cueva, Christian Sánchez, Wilma Ursula Quispe-Rojas, Claudete S. Astolfi-Ferreira, and Antonio J. Piantino Ferreira. 2023. "Genomic Characterization and Genetic Profiles of Salmonella Gallinarum Strains Isolated from Layers with Fowl Typhoid in Colombia" Genes 14, no. 4: 823. https://doi.org/10.3390/genes14040823