
Chromosomal Microarray in Patients with Non-Syndromic Autism Spectrum Disorders in the Clinical Routine of a Tertiary Hospital
 and
and
Abstract
:1. Introduction
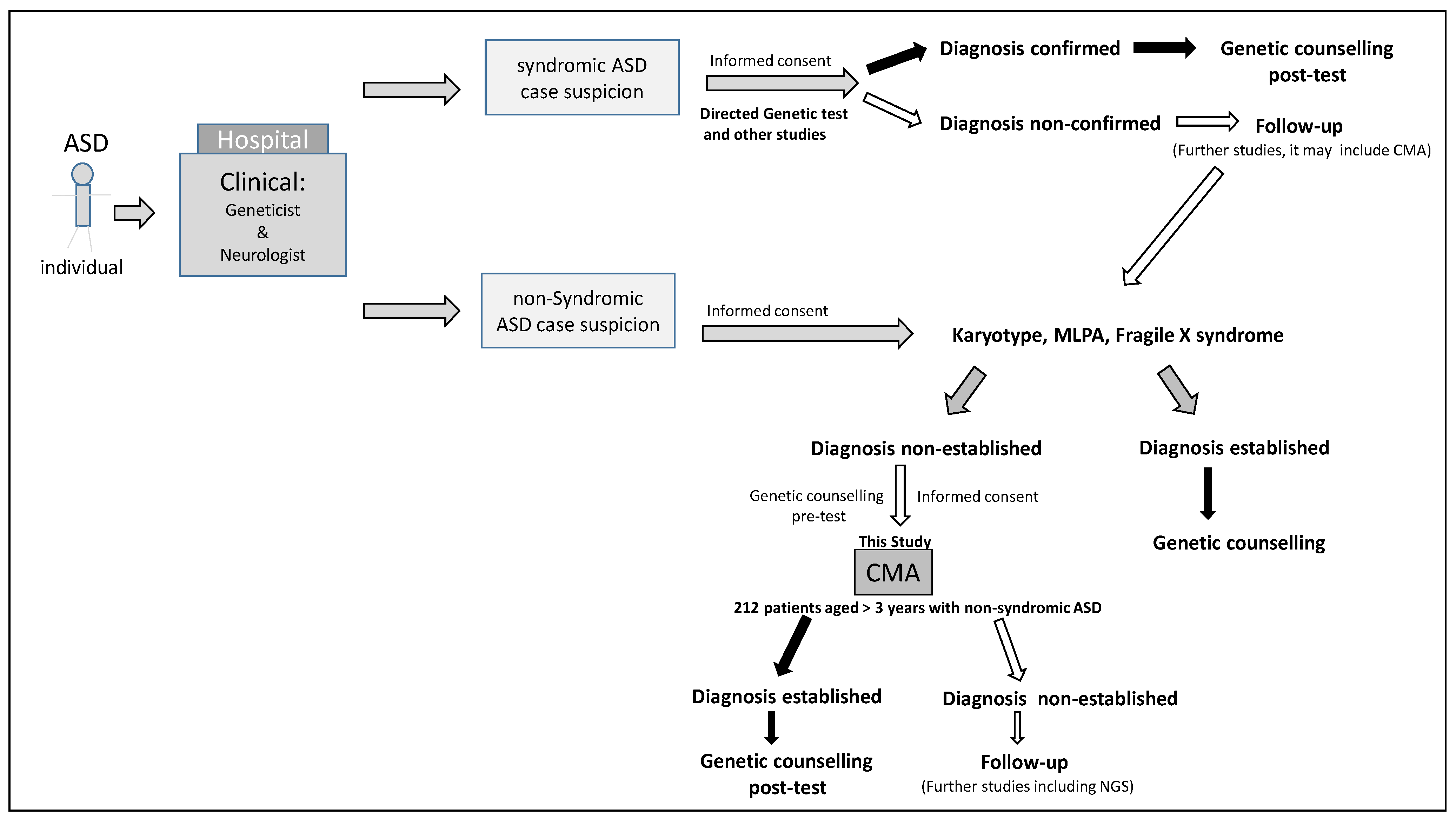
2. Materials and Methods
Subjects
3. Genetic Studies
3.1. Karyotyping and FISH Analyses
3.2. Chromosomal Microarray Analysis (CMA)
3.3. MLPA (Multiplex Ligation-Dependent Probe Amplification)
3.4. Fragile-X Syndrome Analysis
3.5. Statistical Analysis
3.6. Limitation of the Study
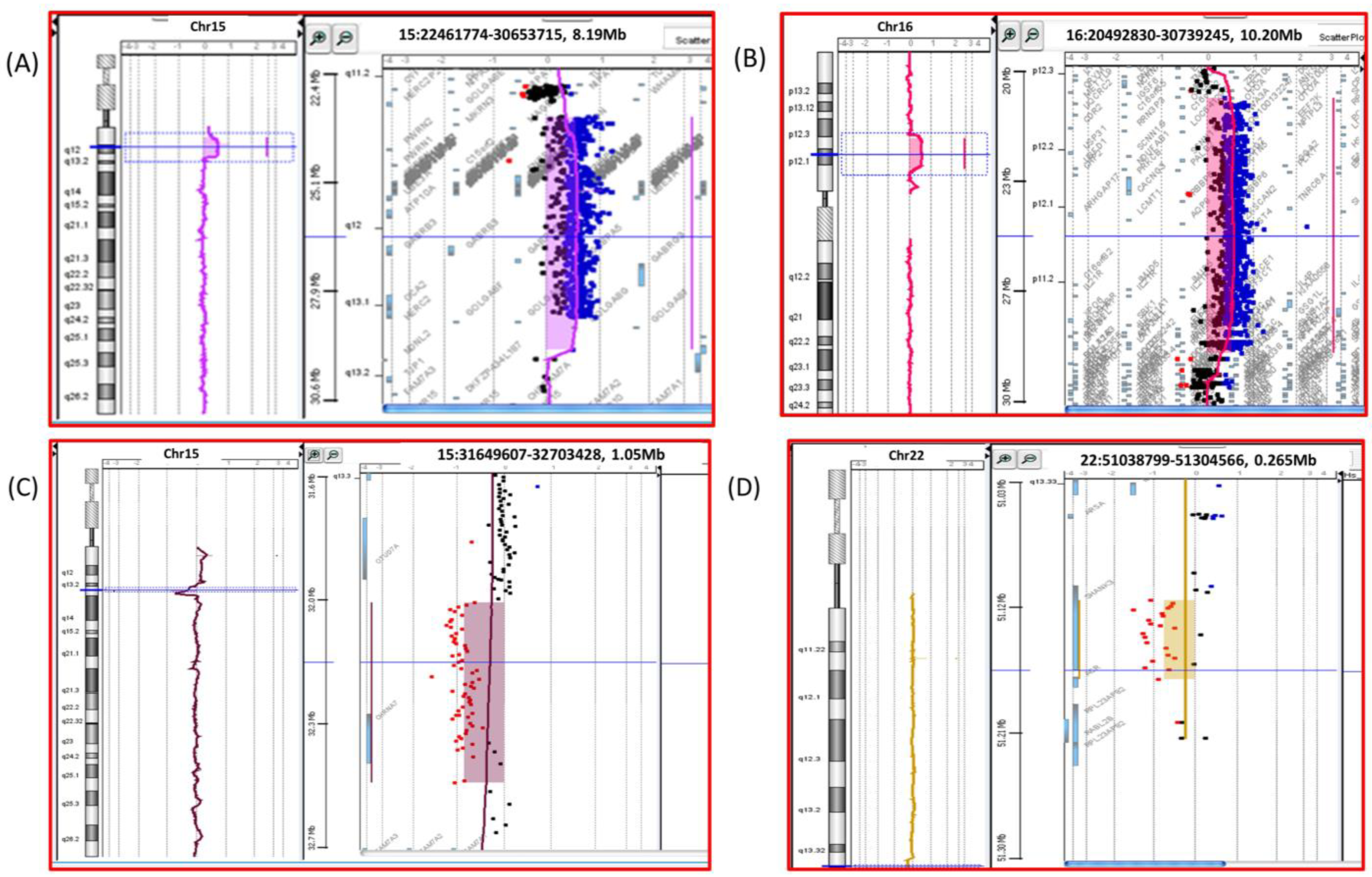
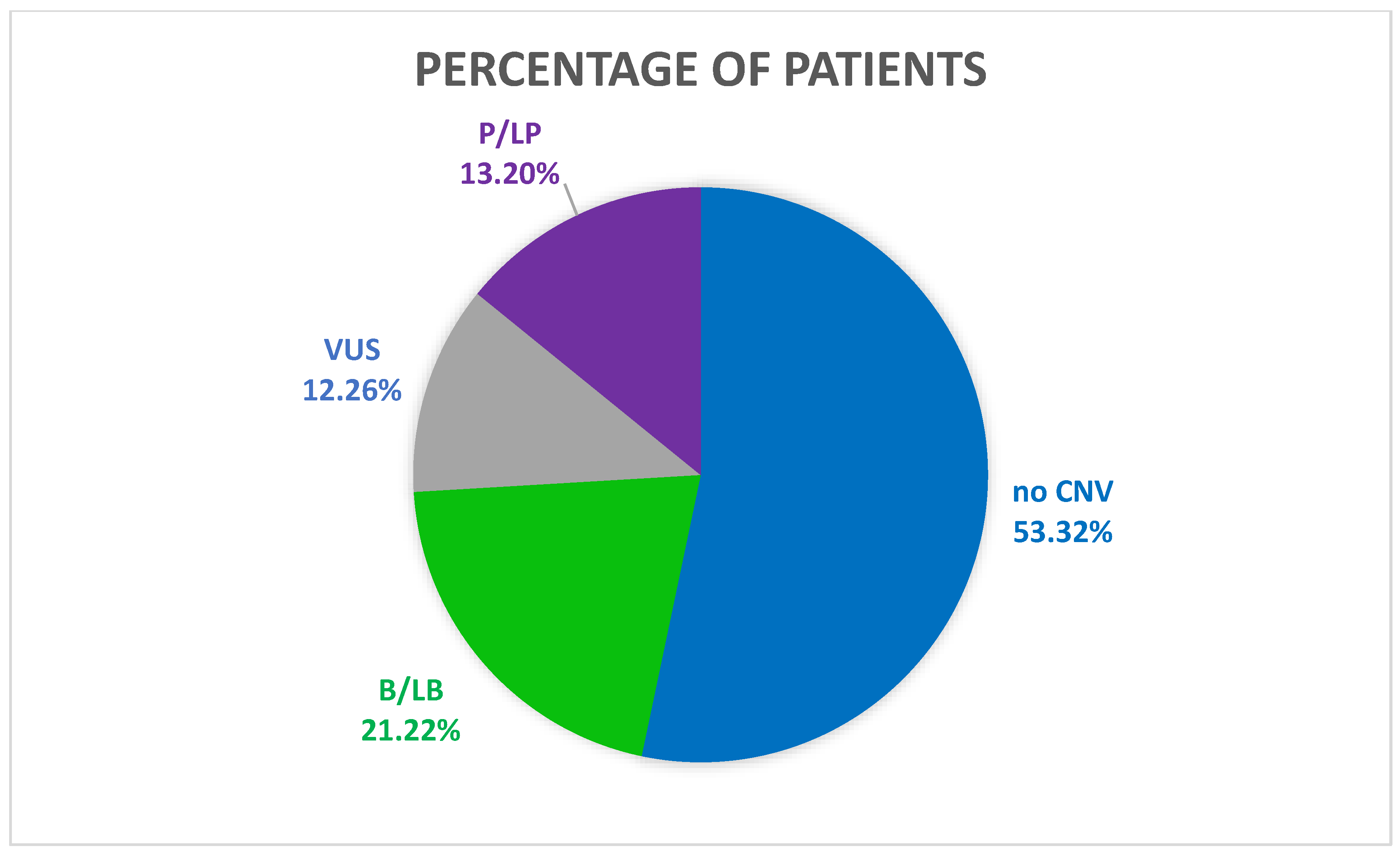
4. Results
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Afif, I.Y.; Farkhan, M.; Kurdi, O.; Maula, M.I.; Ammarullah, M.I.; Setiyana, B.; Jamari, J.; Winarni, T.I. Effect of Short-Term Deep-Pressure Portable Seat on Behavioral and Biological Stress in Children with Autism Spectrum Disorders: A Pilot Study. Bioengineering 2022, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Márquez, C.M.; Albores, G.L. Autistic spectrum disorders: Diagnostic and therapeutic challenges in Mexico. Salud Ment. 2011, 34, 435–441. [Google Scholar]
- Gazzellone, M.J.; Zhou, X.; Lionel, A.C.; Uddin, M.; Thiruvahindrapuram, B.; Liang, S. Copy number variation in Han Chinese individuals with autism spectrum disorder. J. Neurodev. Disord. 2014, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, J.H. Autism spectrum disorders—A genetics review. Genet. Med. 2011, 13, 278–294. [Google Scholar] [CrossRef] [Green Version]
- Fortea Sevilla, M.S.; Escandell Bermúdez, M.O.; Castro Sánchez, J.J. Estimated prevalence of autism spectrum disorders in the Canary Islands. An. Pediatr. 2013, 79, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Carballal Mariño, M.; Gago Ageitos, A.; Ares Alvarez, J.; Del Rio Garma, M.; García Cendón, C.; Goicoechea Castaño, A.; Pena Nieto, J.; en representación del Grupo de Trabajo de Psiquiatría Infantil de la Asociación Galega de Pediatría de Atención Primaria (AGAPap). Prevalence of neurodevelopmental, behavioural and learning disorders in Pediatric Primary Care. An. Pediatr. 2018, 89, 153–161. [Google Scholar] [CrossRef]
- Canal-Bedia, R.; García-Primo, P.; Martín-Cilleros, M.V.; Santos-Borbujo, J.; Guisuraga-Fernández, Z.; Herráez-García, L.; Herráez-García, M.D.M.; Boada-Muñoz, L.; Fuentes-Biggi, J.; La Paz, M.P.-D. Modified Checklist for Autism in Toddlers: Cross-Cultural Adaptation and Validation in Spain. J. Autism Dev. Disord. 2010, 41, 1342–1351. [Google Scholar] [CrossRef]
- Morales, P.; Domènech-Llaberia, E.; Jané, M.C.; Canals, J. Mild autism spectrum disorders in preschool children: Prevalence, co-occurrent symptoms and psychosocial development. Rev. Psicopatol. Psicol. Clin. 2014, 18, 217–231. [Google Scholar]
- Kawa, R.; Saemundsen, E.; Jónsdóttir, S.L.; Hellendoorn, A.; Lemcke, S.; Canal-Bedia, R.; García-Primo, P.; Moilanen, I. European studies on prevalence and risk of autism spectrum disorders according to immigrant status-a review. Eur. J. Public Health 2017, 27, 101–110. [Google Scholar]
- AlAyadhi, L.Y.; Hashmi, J.A.; Iqbal, M.; Albalawi, A.M.; Samman, M.I.; Elamin, N.E.; Bashir, S.; Basit, S. High-resolution SNP genotyping platform identified recurrent and novel CNVs in autism multiplex families. Neuroscience 2016, 339, 561–570. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Cigudosa, J.C.; Lapunzina, P. ; IAGEC. Consenso para la Implementación de los Arrays; Instituto Roche: Madrid, Spain, 2012; Volume 1, p. 178. Available online: https://www.institutoroche.es/ (accessed on 14 March 2023).
- Devlin, B.; Scherer, S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012, 22, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auranen, M.; Nieminen, T.; Majuri, S.; Vanhala, R.; Peltonen, L.; Järvelä, I. Analysis of autism susceptibility gene loci on chromosomes 1p, 4p, 6q, 7q, 13q, 15q, 16p, 17q, 19q and 22q in Finnish multiplex families. Mol. Psychiatry 2000, 5, 320–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, A.; Nagai, M.; Mizuno, S.; Ogishima, S.; Tamiya, G.; Ueki, M.; Sakurai, R.; Makino, S.; Obara, T.; Ishikuro, M.; et al. Clustering by phenotype and genome-wide association study in autism. Transl. Psychiatry 2020, 10, 290. [Google Scholar] [CrossRef]
- Álvarez-Mora, M.I.; Escalona, R.C.; Navarro, O.P.; Madrigal, I.; Quintela, I.; Amigo, J.; Martinez-Elurbe, D.; Linder-Lucht, M.; Aznar Lain, G.; Carracedo, A. Comprehensive molecular testing in patients with high functioning autism spectrum disorder. Mutat. Res. 2016, 784, 46–52. [Google Scholar] [CrossRef]
- Cuscó, I.; Medrano, A.; Gener, B.; Vilardell, M.; Gallastegui, F.; Villa, O.; González, E.; Rodríguez-Santiago, B.; Vilella, E.; Del Campo, M.; et al. Autism-specific copy number variants further implicate the phosphatidylinositol signaling pathway and the glutamatergic synapse in the etiology of the disorder. Hum. Mol. Genet. 2009, 18, 1795–1804. [Google Scholar] [CrossRef] [Green Version]
- Toma, C.; Torrico, B.; Hervás, A.; Valdés-Mas, R.; Tristán-Noguero, A.; Padillo, V.; Maristany, M.; Salgado, M.; Arenas, C.; Puente, X.S.; et al. Exome sequencing in multiplex autism families suggests a major role for heterozygous truncating mutations. Mol. Psychiatry 2013, 19, 784–790. [Google Scholar] [CrossRef] [Green Version]
- Amado-Puentes, A.; Reparaz-Andrade, A.; Del Campo-García, A.; Blanco-Barca, M.Ó.; Salgado-Barreira, Á.; Del Campo-Pérez, V.; Fernández-Lorenzo, J.R. Neurodevelopmental Disorders and Array-Based Comparative Genomic Hybridization: Sensitivity and Specificity using a Criteria Checklist for Genetic Test Performance. Neuropediatrics 2019, 50, 164–169. [Google Scholar] [CrossRef]
- Arteche-López, A.; Gómez Rodríguez, M.J.; Sánchez Calvin, M.T.; Quesada-Espinosa, J.F.; Lezana Rosales, J.M.; Palma Milla, C.; Gómez-Manjón, I.; Hidalgo Mayoral, I.; de la Fuente, R.P.; de Bustamante, A.D.; et al. Towards a Change in the Diagnostic Algorithm of Autism Spectrum Disorders: Evidence Supporting Whole Exome Sequencing as a First-Tier Test. Genes 2021, 12, 560. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Granero, F.; Blanco-Kelly, F.; Sanchez-Jimeno, C.; Avila-Fernandez, A.; Arteche, A.; Bustamante-Aragones, A.; Rodilla, C.; Rodríguez-Pinilla, E.; Riveiro-Alvarez, R.; Tahsin-Swafiri, S.; et al. Comparison of the diagnostic yield of aCGH and genome-wide sequencing across different neurodevelopmental disorders. NPJ Genom. Med. 2021, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Quintela, I.; Eirís, J.; Gómez-Lado, C.; Pérez-Gay, L.; Dacruz, D.; Cruz, R.; Castro-Gago, M.; Míguez, L.; Carracedo, Á.; Barros, F. Copy number variation analysis of patients with intellectual disability from North-West Spain. Gene 2017, 626, 189–199. [Google Scholar] [CrossRef]
- Marshall, C.; Noor, A.; Vicent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural Variation of Chromosomes in Autism Spectrum Disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Griswold, A.J.; Ma, D.; Cukier, H.N.; Nations, L.D.; Schmidt, M.A.; Chung, R.-H.; Jaworski, J.M.; Salyakina, D.; Konidari, I.; Whitehead, P.L.; et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum. Mol. Genet. 2012, 21, 3513–3523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanduri, C.; Kantojärvi, K.; Salo, P.M.; Vanhala, R.; Buck, G.; Blancher, C.; Lähdesmäki, H.; Järvelä, I. The Landscape of Copy Number Variations in Finnish Families with Autism Spectrum Disorders. Autism Res. 2015, 9, 9–16. [Google Scholar] [CrossRef]
- Annunziata, S.; Bulgheroni, S.; ·D’Arrigo, S.; Esposito, S.; Taddei, M.; Saletti, V.; Alfei, E.; Sciacca, F.L.; Rizzo, A.; Pantaleoni, C.; et al. CGH Findings in Children with Complex and Essential Autistic Spectrum Disorder. J. Autism Dev. Disord. 2023, 53, 615–623. [Google Scholar] [CrossRef]
- Monteiro, S.; Pinto, J.; Mira Coelho, A.; Leão, M.; Dória, S. Identification of Copy Number Variation by Array-CGHin Portuguese Children and Adolescents Diagnosed with Autism Spectrum Disorders. Neuropediatrics 2019, 50, 367–377. [Google Scholar]
- Lee, C.L.; Chuang, C.K.; Tu, R.Y.; Chiu, H.C.; Lo, Y.T.; Chang, Y.H.; Chen, Y.J.; Chou, C.L.; Wu, P.S.; Chen, C.P.; et al. Increased Diagnostic Yield of Array Comparative Genomic Hybridization for Autism Spectrum Disorder in One Institution in Taiwan. Medicina 2022, 58, 15. [Google Scholar] [CrossRef]
- Napoli, E.; Russo, S.; Casula, L.; Alesi, V.; Amendola, F.A.; Angioni, A.; Novelli, A.; Valeri, G.; Menghini, D.; Vicari, S. Array-CGH Analysis in a Cohort of Phenotypically Well- Characterized Individuals with “Essential” Autism Spectrum Disorders. J. Autism Dev. Disord 2018, 48, 442–449. [Google Scholar] [CrossRef]
- Vicari, S.; Napoli, E.; Cordeddu, C.; Menghini, D.; Alesi, V.; Loddo, S.; Novelli, A.; Tartaglia, M. Copy number variants in autism spectrum disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 92, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Chehbani, F.; Tomaiuolo, P.; Picinelli, C.; Baccarin, M.; Castronovo, P.; Scattoni, M.L.; Gaddour, N.; Persico, A.M. Yield of array- CGH analysis in Tunisian children with autism spectrum disorder. Mol. Genet. Genom. Med. 2022, 10, e1939. [Google Scholar] [CrossRef] [PubMed]
- Vallespín, E.; Palomares Bralo, M.; Mori, M.Á.; Martín, R.; García-Miñaúr, S.; Fernández, L.; de Torres, M.L.; García-Santiago, F.; Mansilla, E.; Santos, F.; et al. Customized high resolution CGH-array for clinical diagnosis reveals additional genomic imbalances in previous well-defined pathological samples. Am. J. Med. Genet A 2013, 161, 1950–1960. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Revenga, L.; Vallespín, E.; Madrigal, I.; Palomares, M.; Mur, A.; García-Miñaur, S.; Santos, F.; Mori, M.Á.; Lapunzina, P.; Mila, M.; et al. A parallel study of different array-CGH platforms in a set of Spanish patients with developmental delay and intellectual disability. Gene 2013, 521, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Posar, A.; Visconti, P. Early Motor Signs in Autism Spectrum Disorder. Children 2022, 9, 294. [Google Scholar] [CrossRef]
- Prasad, A.; Merico, D.; Thiruvahindrapuram, B.; Wei, J.; Lionel, A.C.; Sato, D.; Rickaby, J.; Lu, C.; Szatmari, P.; Roberts, W.; et al. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 2012, 2, 1665–1685. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Li, J.; Wu, Q.; Yang, X.; Huang, A.Y.; Zhang, J.; Ye, A.Y.; Dou, Y.; Yan, L.; Zhou, W.-Z.; et al. AutismKB 2.0: A knowledgebase for the genetic evidence of autism spectrum disorder. Database 2018, 2018, bay106. [Google Scholar] [CrossRef]
- Roohi, J.; Montagna, C.; Tegay, D.; Palmer, L.E.; DeVincent, C.; Pomeroy, J.C.; Christian, S.L.; Nowak, N.; Hatchwell, E. Disruption of contactin 4 in three subjects with autism spectrum disorder. J. Med. Genet. 2008, 46, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, C.; Zhou, B.; Hu, C.; Xu, X. Novel microduplication of CHL1 gene in a patient with autism spectrum disorder: A case report and a brief literature review. Mol. Cytogenet. 2016, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremer, A.; Giacobini, M.; Eriksson, M.; Gustavsson, P.; Nordin, V.; Fernell, E.; Gillberg, C.; Nordgren, A.; Uppströmer, Å.; Anderlid, B.-M.; et al. Copy number variation characteristics in subpopulations of patients with autism spectrum disorders. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 156, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Gillentine, M.A.; Berry, L.N.; Berry, L.N.; Goin-Kochel, R.P.; Ali, M.A.; Ge, J.; Guffey, D.; Rosenfeld, J.A.; Hannig, V.; Bader, P.; et al. The Cognitive and Behavioral Phenotypes of Individuals with CHRNA7 Duplications. J. Autism Dev. Disord. 2017, 47, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, J.C.K.; Hall, V.; Maloney, V.K.; Huang, S.; Roberts, A.; Brady, A.F.; Foulds, N.; Bewes, B.; Volleth, M.; Liehr, T.; et al. 16p11.2–p12.2 duplication syndrome; a genomic condition differentiated from euchromatic variation of 16p11.2. Eur. J. Hum. Genet. 2012, 21, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, A.; Novelli, A.; Bernardini, L.; Igliozzi, R.; Parrini, B. Further characterization of the new microdeletion syndrome of 16p11.2-p12.2. Am. J. Med. Genet. Part A 2009, 149A, 1200–1204. [Google Scholar] [CrossRef]
- Shinawi, M.; Liu, P.; Kang, S.-H.L.; Shen, J.; Belmont, J.W.; A Scott, D.; Probst, F.J.; Craigen, W.J.; Graham, B.H.; Pursley, A.; et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet. 2009, 47, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Baker, P.; Piven, J.; Schwartz, S.; Patil, S. Brief report: Duplication of chromosome 15q11-13 in two individuals with autistic disorder. J. Autism Dev. Disord. 1994, 24, 529–553. [Google Scholar] [CrossRef]
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 2013, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Riley, K.N.; Catalano, L.M.; Bernat, J.A.; Adams, S.D.; Martin, D.M.; Lalani, S.R.; Patel, A.; Burnside, R.D.; Innis, J.W.; Rudd, M.K. Recurrent deletions and duplications of chromosome 2q11.2 and 2q13 are associated with variable outcomes. Am. J. Med. Genet. Part A 2015, 167, 2664–2673. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.A.; Ballif, B.C.; Torchia, B.S.; Sahoo, T.; Ravnan, J.B.; Schultz, R.; Lamb, A.; Bejjani, B.A.; Shaffer, L.G. Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genet. Med. 2010, 12, 694–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogart, A.; Nagarajan, R.P.; Patzel, K.A.; Yasui, D.H.; LaSalle, J.M. 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum. Mol. Genet. 2007, 16, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Drayer, M.; Kamps, A.; ten Kate, P.; Kuipers, G.; Gouw, L. Microcephaly, ocular hypertelorism, low-set ears, hand and feet anomalies, short stature and mental retardation. Syndr. Identif. 1977, 5, 9–11. [Google Scholar]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [Green Version]
- Krumm, N.; Turner, T.N.; Baker, C.; Vives, L.; Mohajeri, K.; Witherspoon, K.; Raja, A.; Coe, B.P.; Stessman, H.A.; He, Z.X.; et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet. 2015, 47, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Motavalli, M.N.; Herguner, S. Autistic disorder and 22q11.2 duplication. World J. Biol. Psychiatry 2007, 8, 127–130. [Google Scholar] [CrossRef]
- Paylor, R.; Glaser, B.; Mupo, A.; Ataliotis, P.; Spencer, C.; Sobotka, A.; Sparks, C.; Choi, C.-H.; Oghalai, J.; Curran, S.; et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: Implications for 22q11 deletion syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 7729–7734. [Google Scholar] [CrossRef] [Green Version]
- Nevado, J.; Bel-Fenellós, C.; Sandoval-Talamantes, A.K.; Hernández, A.; Biencinto-López, C.; Martínez-Fernández, M.L.; Barrúz, P.; Santos-Simarro, F.; Mori-Álvarez, M.Á.; Mansilla, E.; et al. Deep Phenotyping and Genetic Characterization of a Cohort of 70 Individuals With 5p Minus Syndrome. Front. Genet. 2021, 12, 645595. [Google Scholar] [CrossRef]
- Krgovic, D.; Kokalj Vokac, N.; Zagorac, A.; Gregoric Kumperscak, H. Rare structural variants in the DOCK8 gene identified in a cohort of 439 patients with neurodevelopmental disorders. Sci. Rep. 2018, 8, 9449. [Google Scholar] [CrossRef] [Green Version]
- Glessner, J.T.; Li, J.; Wang, D.; March, M.; Lima, L.; Desai, A.; Hadley, D.; Kao, C.; Gur, R.E.; Cohen, N.; et al. Copy number variation meta-analysis reveals a novel duplication at 9p24 associated with multiple neurodevelopmental disorders. Genome Med. 2017, 9, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.N.; Kollu, R.; Banerjee-Basu, S. AutDB: A gene reference resource for autism research. Nucleic Acids Res. 2008, 37, D832–D836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Córdoba, M.; Rodriguez, S.; Morón, D.G.; Medina, N.; Kauffman, M. Expanding the spectrum of Grik2 mutations: Intellectual disability, behavioural disorder, epilepsy and dystonia. Clin. Genet. 2014, 87, 293–295. [Google Scholar] [CrossRef]
- Nord, A.S.; Network, S.P.; Roeb, W.; Dickel, D.; Walsh, T.; Kusenda, M.; O’Connor, K.L.; Malhotra, D.; E McCarthy, S.; Stray, S.M.; et al. Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur. J. Hum. Genet. 2011, 19, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.H.; Bird, L.M.; Thibert, R.L.; Williams, C.A. If not Angelman, what is it? A review of Angelman-like syndromes. Am. J. Med. Genet. Part A 2014, 164, 975–992. [Google Scholar] [CrossRef]
- Ma, W.; Mao, J.; Wang, X.; Duan, L.; Song, Y.; Lian, X.; Zheng, J.; Liu, Z.; Nie, M.; Wu, X. Novel Microdeletion in the X Chromosome Leads to Kallmann Syndrome, Ichthyosis, Obesity, and Strabismus. Mol. Syndr. 2016, 6, 236–241. [Google Scholar] [CrossRef]
- Xu, S. Association of copy number variations in autism spectrum disorders: A Systematic Review. Chin. J. Biol. 2014, 1, 9. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Patient | Clinical Features | Results | Size (Kb) | Genes. OMIM Genes Underlined | Classification |
|---|---|---|---|---|---|
| AUT24 | ASD, seizures, and plagiocephaly | arr[hg19] 16p12.2p11.2(21475060_29182196)x3 | 8000 | OTOA, UQCRC2, SCNN1G, SCNN1B, COG7, EARS2, PALB2, TNRC6A, IL4R, IL21R, KIAA0556, CLN3, TUFM, ATP2A1, CD19, LAT | P |
| AUT28 | ASD, developmental delay and joint laxity | arr[hg19] chrXp22.33(194896_2658336)x3 | 2400 | PLCXD1, GTPBP6, NCRNA00107, PPP2R3B, SHOX,CRLF2, CSF2RA, IL3RA, SLC25A6, NCRNA00105, ASMTL, P2RY8, SFRS17A, ASMT, DHRSX, ZBED1, CD99, XG, GYG2, ARSD, ARSE | LP |
| AUT33 | ASD, developmental delay, seizures, and joint laxity | arr[hg19] chr3p26.3(297696_927607)x3 | 629.9 | CHL1 | LP |
| AUT35 | ASD and hyperactivity | arr[hg19] 6q16.3(101869595_102582366)x3 | 712.7 | GRIK2 | LP |
| AUT36 | ASD, microcephaly, developmental delay, hypotonia, and seizures | arr[hg19] 18q21.2(51091726_51331417)x1 | 240 | TCF4 | P |
| AUT45 | ASD Asperger | arr[hg19] 3p26.3-p26.2(2163261_2876491)x3, mat | 713 | CNTN4 | LP |
| arr [hg19] 19p13.2(12388254_12403612)x3 | 15.3 | ZNF44 | LB | ||
| AUT46 | ASD | arr[hg19] 9p24.3(300306_371857)x1 | 72 | DOCK8 | LP |
| AUT49 | ASD, developmental delay, ectopic kidney, hyperactivity, dolichocephaly, prominent forehead, hypoplasia of the middle third of the face, and short palpebral fissures | arr[hg19] 2q37.2q37.3(236959860_242852625)x1 | 5892 | AGAP1, GBX2, ASB18, IQCA1, CXCR7, COPS8, COL6A3, MLPH, PRLH, RAB17, LRRFIP1, RBM44, RAMP1, UBE2F, UBE2F, SCLY, ESPNL, KLHL30, ILKAP, LOC151174, LOC643387, HES6, PER2, TRAF3IP1, ASB1, LOC151171, TWIST2, FLJ43879, HDAC4, MGC16025, MIR4269, LOC150935, NDUFA10, OR6B2, PRR21, OR6B3, MYEOV2, OTOS, GPC1, PP14571, MIR149, ANKMY1, DUSP28, RNPEPL1, CAPN10, GPR35, AQP12B, AQP12A, KIF1A, AGXT, C2orf54, LOC200772, SNED1, MTERFD2, PASK, PPP1R7, ANO7, HDLBP, SEPT2, FARP2, STK25, BOKAS1, BOK, THAP4, ATG4B, DTYMK, ING5, D2HGDH, GAL3ST2, NEU4, PDCD1, C2orf85 | P |
| AUT52 | ASD, developmental delay, hypotonia, and hearing loss | arr[hg19] 2p25.3(39193_4234087)x3 | 4190 | FAM110C, SH3YL1, ACP1, FAM150B, TMEM18, C2orf90, SNTG2, TPO, PXDN, MYT1L, LOC730811, TSSC1, TTC15, ADI1, RNASEH1, RPS7, COLEC11, ALLC | P |
| arr[hg19] 5p15.33(204849_3876457)x1 | 3670 | CCDC127, SDHA, PDCD6, AHRR, LOC100310782, C5orf55, EXOC3, LOC25845, SLC9A3, CEP72, TPPP, ZDHHC11, BRD9, TRIP13, NKD2, SLC12A7, SLC6A19, SLC6A18, TERT, CLPTM1L, SLC6A3, LPCAT1, SDHAP3, LOC728613, MRPL36, NDUFS6, IRX4, IRX2, C5orf38, IRX1 | P | ||
| arr[hg19] 15q11.2(25420800_25491412)x3 | 70.61 | SNORD115-4, SNORD115-5, SNORD115-10, SNORD115-9, SNORD115-12, SNORD115-6, SNORD115-7, SNORD115-8, SNORD115-36, SNORD115-43, SNORD115-29, SNORD115-11, SNORD115-13, SNORD115-14, SNORD115-16, SNORD115-18, SNORD115-19, SNORD115-17, SNORD115-21, SNORD115-20, SNORD115-15, SNORD115-22, PAR4, SNORD115-23, HBII-52-24, SNORD115-25, SNORD115-26, HBII-52-27, HBII-52-28, SNORD115-30, SNORD115-31, SNORD115-32, SNORD115-33, SNORD115-34, SNORD115-35, SNORD115-37, SNORD115-38, SNORD115-39, SNORD115-40, SNORD115-41. | B | ||
| AUT59 | ASD, seizures, absence of speech, psychomotor delay | arr [hg19] 16p11.2(28846555_31713018)x3 | 2800 | ATXN2L, TUFM,SH2B, ATP2A1, CD19, NFATC2IP, SPNS1, LAT, RRN3P2, RUNDC2C, LOC606724, BOLA2, BOLA2B, GIYD2, GIYD1, SULT1A4, SULT1A3, LOC388242, LOC613038, LOC440354, SLC7A5P1, SPN, QPRT, C16orf54, ZG16, KIF22, MAZ, PRRT2, C16orf53, MVP, CDIPT, LOC440356, SEZ6L2, ASPHD1, KCTD13, TMEM219, TAOK2, HIRIP3, INO80E, DOC2A, C16orf92, FAM57B, ALDOA, PPP4C, TBX6, YPEL3, GDPD3, MAPK3, LOC100271831, CORO1A,LOC613037, LOC595101, CD2BP2, TBC1D10B, MYLPF, SEPT1, ZNF48, ZNF771, DCTPP1, SEPHS2, ITGAL, ZNF768, ZNF747, ZNF764, ZNF688, ZNF785, ZNF689, PRR14, FBRS, SRCAP, SNORA30, PHKG2, C16orf93, RNF40, ZNF629, BCL7C, MIR762, CTF1, NCRNA00095, FBXL19, ORAI3, SETD1A, HSD3B7, STX1B, STX4, ZNF668, ZNF646, POL3S, VKORC1, BCKDK, MYST1, PRSS8, PRSS36, FUS, PYCARD, TRIM72, PYDC1, ITGAM, ITGAX, ITGAD, COX6A2, ZNF843, ARMC5, TGFB1I1, SLC5A2, C16orf58, AHSP, CSDAP1, C16orf67 | P |
| AUT71 | Asperger, left sensorineural hearing loss, astigmatism, obesity, and speech abnormalities | arr[hg19] 15q11.2(22880274_23648846)x3 | 768.5 | CYFIP1, NIPA2, NIPA1, WHAMML1, GOLGA8IP, HERC2P2, HERC2P7, GOLGA8E | VUS-LP |
| AUT81 | ASD, psychomotor delay, and microcephaly | arr[hg19] 15q26.1(90719940_93509788)x1 | 2780 | SEMA4A, CIB1, GDPGP1, NGR1, IQGAP1, CRTC3, FURIN, RECQL1, FES, MAN2A2, RCCD1,PEC1, BLM, UNC45A, VPS33B, CHD2, SV2B, SLCO3A1, STX, RGMA | P |
| AUT90 | ASD, Mild ID, expressive dysphasia | arr[hg19] 22q13.33(51123491_51178264)x1 | 54.77 | SHANK3 | P |
| AUT95 | ASD, ID | arr[hg19] Xp11.22(52841226_5308135)x4 | 240.1 | XAGE5, XAGE3, FAM156A, FAM156B, GPR173. | P |
| arr[hg19] 9q34.13(134378819_134399206)x3 | 20.3 | POMT1, UCK1 | LP | ||
| arr[hg19] 22q11.23(24276174_24332956)x3 | 56.7 | GSTT2, GSTT2B, DDTL, DDT. | B | ||
| AUT97 | ASD | arr[hg19] 15q11.2-q13.1(23641200_29331964)x3 | 5690 | MKRN3, MAGEL2, NDN, SNRPN, UBE3A, GABRB3, GABRA5, OCA2, HERC2, APBA2, NSMCE3 | P |
| AUT116 | ASD | arr[hg19] 2q11.1-q13(95635960_110301275)x2-3 mosaic 75% | 14,660 | ASTL, STARD7, TMEM127, SNRNP200, NCAPH, LMAN2L, CNNM4, ZAP70, VWA3B, CNGA3, C2orf64, TSGA10, LIPT1, AFF3, POU3F3, SLC5A7, RANBP2, EDAR, NPHP1 | P |
| AUT136 | ASD (one brother with ASD, another with ID, mother with schizophrenia) | arr[hg19] 15q11.2-q13.1(23641200_29331964)x3 | 5690 | MKRN3, MAGEL2, NDN, SNRPN, UBE3A, GABRB3, GABRA5, OCA2, HERC2, APBA2, NSMCE3 | P |
| AUT167 | ASD, short stature, ID, and macrocephaly | arr[hg19] Xp22.33-p22.2(60679_9711656)x0 | 9600 | PLCXD1, GTPBP6, NCRNA00107, PPP2R3B, SHOX, CRLF2, CSF2RA, IL3RA, SLC25A6, NCRNA00105, ASMTL, P2RY8, SFRS17A, ASMT, DHRSX, ZBED1, CD99, XG, XGPY2, XG, GYG2, ARSD, ARSE, ARSH, ARSF, MXRA5, PRKX, NLGN4X, VCX3A, HDHD1A, STS, VCX, PNPLA4, MIR651, VCX2, VCX3B, KAL1, FAM9A, FAM9B, TBL1X, GPR143, SHROOM2, CLCN4. | P |
| AUT169 | ASD, severe ID, absence of speech, macrocephaly, and joint laxity | arr[hg19] 3p25.3-p25.2(10975732_11885558)x1 | 910 | SLC6A11, SLC6A1, HRH1, ATG7, VGLL4, C3orf31. | LP |
| AUT177 | ASD, ID, and psychomotor delay | arr[hg19] 16p11.2(29350787_30198123)x3 90% mosaic dn | 850 | BOLA2, SLX1B, SLX1A, SPN, QPR1, ZG16, KIF22, MAZ, PRRT2, PAGR1, MVP, CDIPT, SEZ6L2, KCTD13, TAOK2, HRIP3, DOC2A, C16orf92, TLCD38, ALDOA, PPP4C, TBX6,YPEL3, GDPD3, MAPK3, CORO1A, SULT1A4, SULT1A3 | P |
| AUT185 | ASD | arr [hg19] 7q21.13(89865734_90897065)x3 | 1030 | STEAP2, C7orf63, GTPBP10, CLDN12, CDK14, FZD1. | VUS |
| arr[hg19] 22q11.21 (19710115_19755528)x3 | 45 | SEPT5, GP1BB, TBX1 | LP | ||
| AUT194 | ASD and absence of speech | arr[hg19] 9p24.3(220253_315028)x3 | 94.7 | DOCK8 | LP |
| AUT218 | ASD, psychomotor delay | arr[hg19] 2q23.1(148790680_148879681)x1 | 90 | MBD5 | LP |
| AUT219 | ASD | arr[hg19] 8q21(82572090_82618539)x1 | 46.45 | IMPA1, SLC10A5, ZFAND1 | LP |
| arr[hg19] 10q21.3(68516519_68613775)x1 | 97.26 | CTNNA3 | LP | ||
| AUT223 | ASD and mild ID | arr[hg19] 22q13.33(51116128_51219009)x1 | 102.88 | SHANK3 | P |
| AUT228 | ASD, hypotonia, and psychomotor delay | arr[hg19] 15q13.3(32098670_32423858)x1 | 325.18 | CHRNA7 | LP |
| AUT230 | ASD and psychomotor delay | arr[hg19]15q11.2(22,880,274_23,648,846)x3 | 768.5 | CYFIP1, NIPA2, NIPA1 | VUS-LP |
| arr[hg19] 15q13.3(32011762_32514890)x3 | 503 | CHRNA7 | VUS | ||
| arr[hg19] Xq22.2(103220412_103269195)x2 | 49 | MIR1256, TMSB15B, H2BFXP, H2BFWT | LB | ||
| AUT232 | ASD, hypotonia, psychomotor delay, and myoclonus | arr[hg19] 3q29(195697011_197335597)x1 | 1610 | TNK2, TPRC, ZDHH19, SLC51A, TM45F19, UBXN7, SMCO1, WDR53, FBX045, CEP19, PIGX, PAK2, NCBP2, PIGZ, MELTF, DLG1, MIR4797, RUBCN, BDH1, KIAA8226, FYTTD1, BDH1 | LP |
| Patient | Results | Size (Kb) | Genes. OMIM Genes Underlined |
|---|---|---|---|
| AUT7 | arr[hg19] 22q11.21(18889039_19010508)x3 | 121 | DGCR6, PRODH, DGCR5, DGCR9, DGCR10 |
| AUT29 | arr[hg19] 6q11.1 (61982931_62917272)x3 | 934.3 | KHDRBS2 |
| AUT43 | arr[hg19] 5q21.2-q21.3(103185551_107582929)x1 mat | 4390 | RAB9BP9, EFNA5, FBXL17 |
| arr[hg19] Xq22(107976048_107979726)x2 dn | 3.68 | IRS4 (benign) | |
| AUT55 | arr [hg19] 14q31.1 (80521415_81855721)x3 | 1334 | DIO2, C14orf145, TSHR, GTF2A1, SNORA79, STON2 |
| AUT56 | arr [hg19] 4p16.3 273646_1033610)x3 | 759.96 | ZNF732, ZNF141, ABCA11P, ZNF721, PIGG, PDE6B, ATP5I, MYL5, MFSD7, PCGF3, CPLX1, GAK, TMEM175, DGKQ, SLC26A1, IDUA, FGFRL1 |
| arr[hg19] 1p36.33(1627899_1663760)x1 | 35.8 | CDK11B, MMP23A, CDK11A and SLC35E2 (benign) | |
| AUT62 | arr[hg19] 14q13.2 (35404289_35780723)x3pat | 376.4 | C14orf19, SRP54, FAM177A1, PPP2R3C, KIAA0391, PSMA6 |
| AUT75 | arr[hg19] 1q44(249,104,658_249,212,668)x4 | 108 | SH3BP5L, MIR3124, ZNF672, ZNF692, PGBD2 |
| arr[hg19] 5p14.3(21,269,398_21,691,673)x3 mat | 422.77 | GUSBP1 (benign) | |
| arr[hg19] 17p12(15,183,668_15,185,073)x1, mat | 1.40 | (benign) | |
| arr[hg19] Xq22.2(103,220,412_103,269,195)x3, mat | 48.78 | MIR1256, TMSB15B, H2BFXP, H2BFWT (benign) | |
| AUT83 | arr [hg19] 2q11.2 (97728447_98021593)x1, | 295 | FAHD2B, ANKRD36 (benign) |
| arr [hg19] 4q25 (110170903_110390360)x3 | 219 | COL25A1, SEC24B, (benign) | |
| arr [hg19] 10p12.1 (27613431_27694710)x1 | 81 | PTCHD3 (benign) | |
| arr[hg19] 17p13.3(1693_296541)x3 | 293 | DOC2B, RPH3AL, C17orf97, FAM101B | |
| AUT98 | arr[hg19] 20p13(4666856_4674849)x1 | 7.99 | PRNP |
| AUT100 | arr[hg19] 1p32.3(54231745_54325010)x3 | 93.26 | TMEM48, YIPF1 |
| AUT103 | arr[hg19] 8p11.21.1(429374004_43055737)x1 | 118.33 | FNTA, SGK196 (POMK), HGSNAT |
| AUT104 | arr[hg19] 8p11.21.1(429374004_43055737)x1 | 118.33 | FNTA, SGK196 (POMK), HGSNAT |
| AUT129 | arr[hg19] 10q24.32(103366911_103453040)x3 | 86.13 | DPCD, FBXW4. |
| AUT146 | arr[hg19] 15q12(27311913_27340486)x3 | 28.6 | GABRG3 |
| AUT148 | arr[hg19] 14q24.3(74036805_74550384)x3 | 513.58 | ACOT2, ACOT4, ACOT6, DNAL1, PNMA1, C14orf43, PTGR2, ZNF410, FAM161B, COQ6, ENTPD5, C14orf45, ALDH6A1 |
| AUT152 | arr[hg19] 12p13.33(323258_658266)x2-3 mat | 335 | SLC6A12, SLC6A13, KDM5A, CCDC77, B4GALNT3 |
| AUT154 | arr[hg19] 21q11.2(15588480_15624577)x3 | 36.1 | RBM11 |
| AUT175 | arr[hg19] 17p11.2-p11.1(21193939_22205821)x3, pat | 1010 | MAP2K3, KCNJ12, KCNJ18, C17orf51, FAM27L, FLJ36000 |
| AUT179 | arr[hg19] 12q24.33(132321161_132811002)x3 | 489.8 | MMP17, ULK1, PUS1, EP400, SNORA49, EP400NL, DDX51, NOC4L, GALNT9 |
| AUT180 | arr[hg19] 12q24.33(132321161_132811002)x3 | 489.8 | MMP17, ULK1, PUS1, EP400, SNORA49, EP400NL, DDX51, NOC4L, GALNT9 |
| AUT195 | arr[hg19] Xp22.2(13567942_13767555)x3 | 200 | EGFL6, RAB9A, TRAPPC2, OFD1 |
| arr[hg19] Xq21.33(93842754_94388922)x1 | 546.17 | - | |
| AUT224 | arr[hg19] 15q13.3(32019696_32575866)x3 | 556.17 | CHRNA7 |
| AUT225 | arr[hg19] 15q13.3(32011762_32575866)x3 | 564.10 | CHRNA7 |
| arr[hg19] 16p12.2(22629047_22709775)x3 | 80.73 | - | |
| arr[hg19] 15q13.2(30938215_30971589)x3 | 33.37 | - | |
| AUT227 | arr[hg19] 15q13.3(32011762_32514890)x3 | 503.00 | CHRNA7 |
| AUT229 | arr[hg19] 15q13.3(32059475_3253966)x3 | 480.20 | CHRNA7 |
| AUT231 | arr[hg19] 15q13.3(32011762_32514890)x3 | 503.22 | OTUD7A, CHRNA7 |
| CNV Type | DELETIONS | #CNV ˂ 100 Kb/Median SIZE (Kb) | #CNV 100–1000 Kb/Median SIZE (Kb) | #CNV ˃ 1000 Kb/Median SIZE (Kb) |
| B/LB | 32 | 22/28.74 | 8/159.62 | 2/1290 |
| VUS | 2 | 1/8.00 | 0/- | 1/4390 |
| P/LP | 16 | 4/58.42 * | 7/400.61 * | 5/4643.80 * |
| Total | 50 | 27 | 15 | 8 |
| CNV Type | DUPLICATIONS | #CNV˂100 Kb/Median SIZE (Kb) | #CNV 100–1000 Kb/Median SIZE (Kb) | #CNV ˃ 1000 Kb/Median SIZE (Kb) |
| B/LB | 37 | 21/48.90 | 15/224.64 | 1/1024 |
| VUS | 25 | 6/50.0 | 16/460.93 | 3/1124.67 |
| P/LP | 18 | 3/53.33 | 7/669.10 * | 8/5630.1 * |
| Total | 80 | 30 | 38 | 12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandoval-Talamantes, A.K.; Mori, M.Á.; Santos-Simarro, F.; García-Miñaur, S.; Mansilla, E.; Tenorio, J.A.; Peña, C.; Adan, C.; Fernández-Elvira, M.; Rueda, I.; et al. Chromosomal Microarray in Patients with Non-Syndromic Autism Spectrum Disorders in the Clinical Routine of a Tertiary Hospital. Genes 2023, 14, 820. https://doi.org/10.3390/genes14040820
Sandoval-Talamantes AK, Mori MÁ, Santos-Simarro F, García-Miñaur S, Mansilla E, Tenorio JA, Peña C, Adan C, Fernández-Elvira M, Rueda I, et al. Chromosomal Microarray in Patients with Non-Syndromic Autism Spectrum Disorders in the Clinical Routine of a Tertiary Hospital. Genes. 2023; 14(4):820. https://doi.org/10.3390/genes14040820
Chicago/Turabian StyleSandoval-Talamantes, Ana Karen, María Ángeles Mori, Fernando Santos-Simarro, Sixto García-Miñaur, Elena Mansilla, Jair Antonio Tenorio, Carolina Peña, Carmen Adan, María Fernández-Elvira, Inmaculada Rueda, and et al. 2023. "Chromosomal Microarray in Patients with Non-Syndromic Autism Spectrum Disorders in the Clinical Routine of a Tertiary Hospital" Genes 14, no. 4: 820. https://doi.org/10.3390/genes14040820