
Transcriptomic and Chromatin Landscape Analysis Reveals That Involvement of Pituitary Level Transcription Factors Modulate Incubation Behaviors of Magang Geese
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
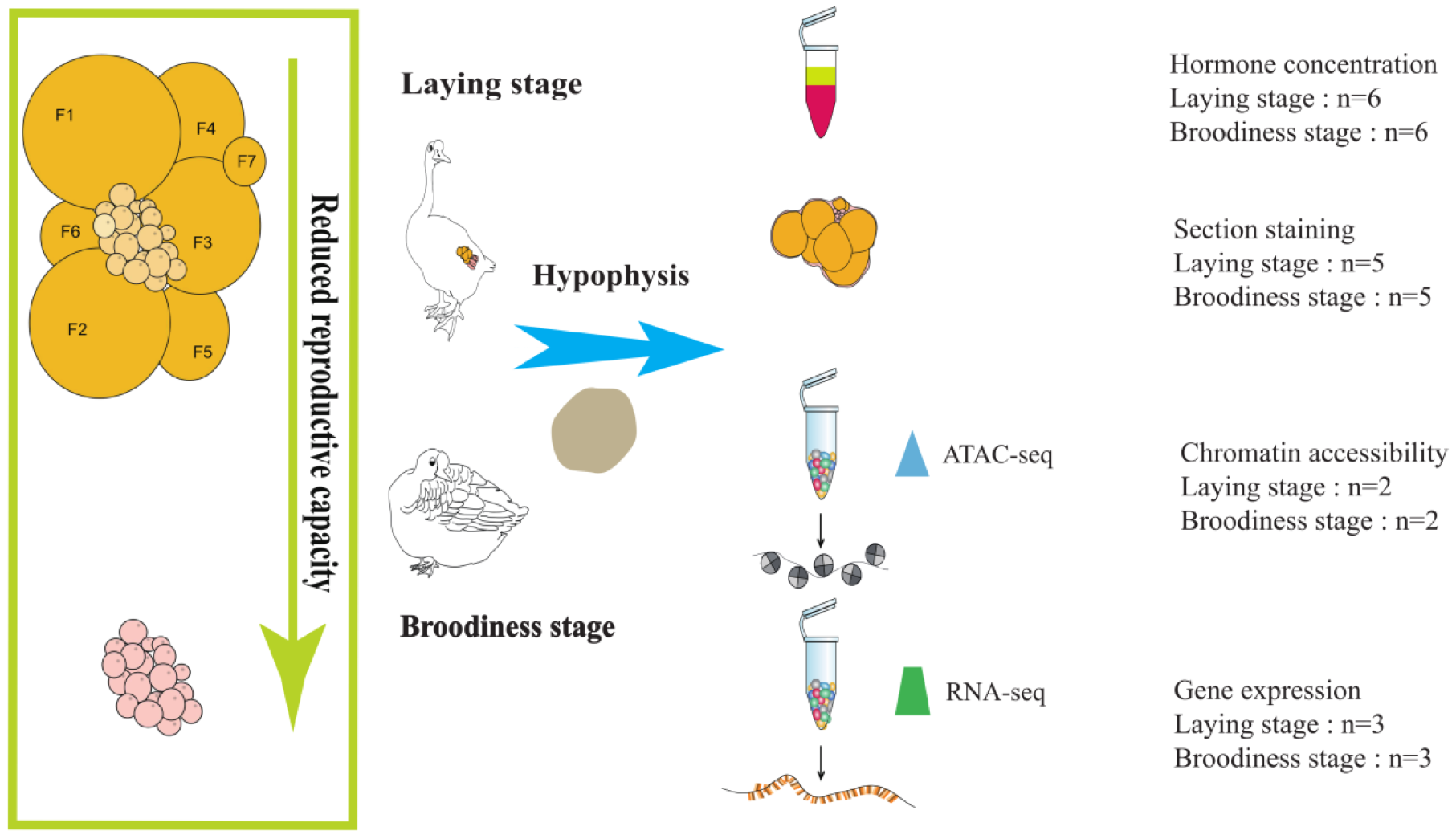
2.2. Animals and Sample Collection
2.3. ATAC-seq Library for Frozen Pituitary Tissues
2.4. ATAC-seq Data Analysis
2.5. The Identification of DARs and Peak Annotation
2.6. Computational Footprint Analysi
2.7. RNA-seq and Differential Gene Expression Analysis
2.8. GO and KEGG Pathway Enrichment Analyses
2.9. Ovarian Histology and Serum Hormone
3. Results
3.1. Ovarian Histology Structure and Hormone Concentration
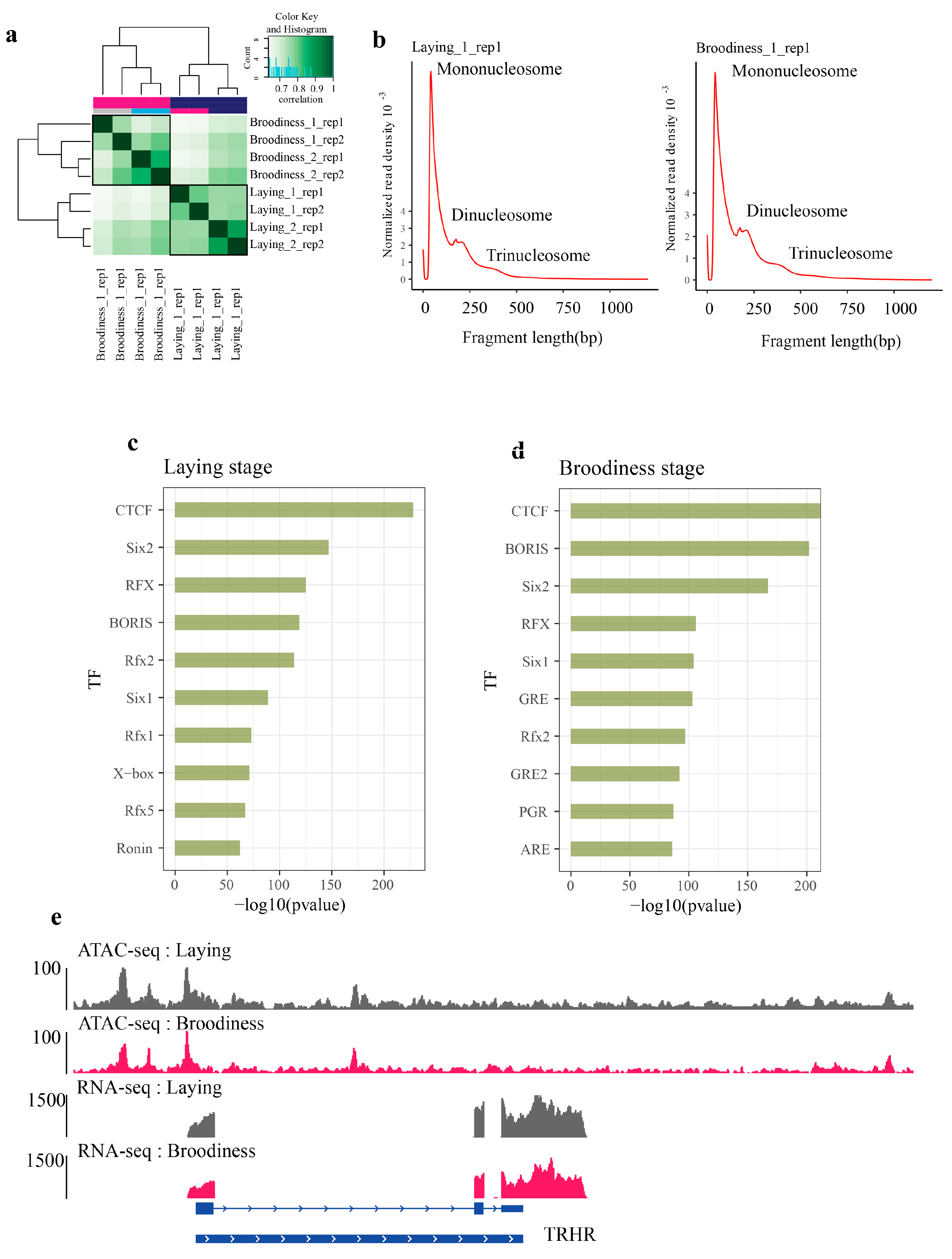
3.2. Characteristics of Chromatin Accessibility in Goose Pituitary Underlying Two Reproductive Stages
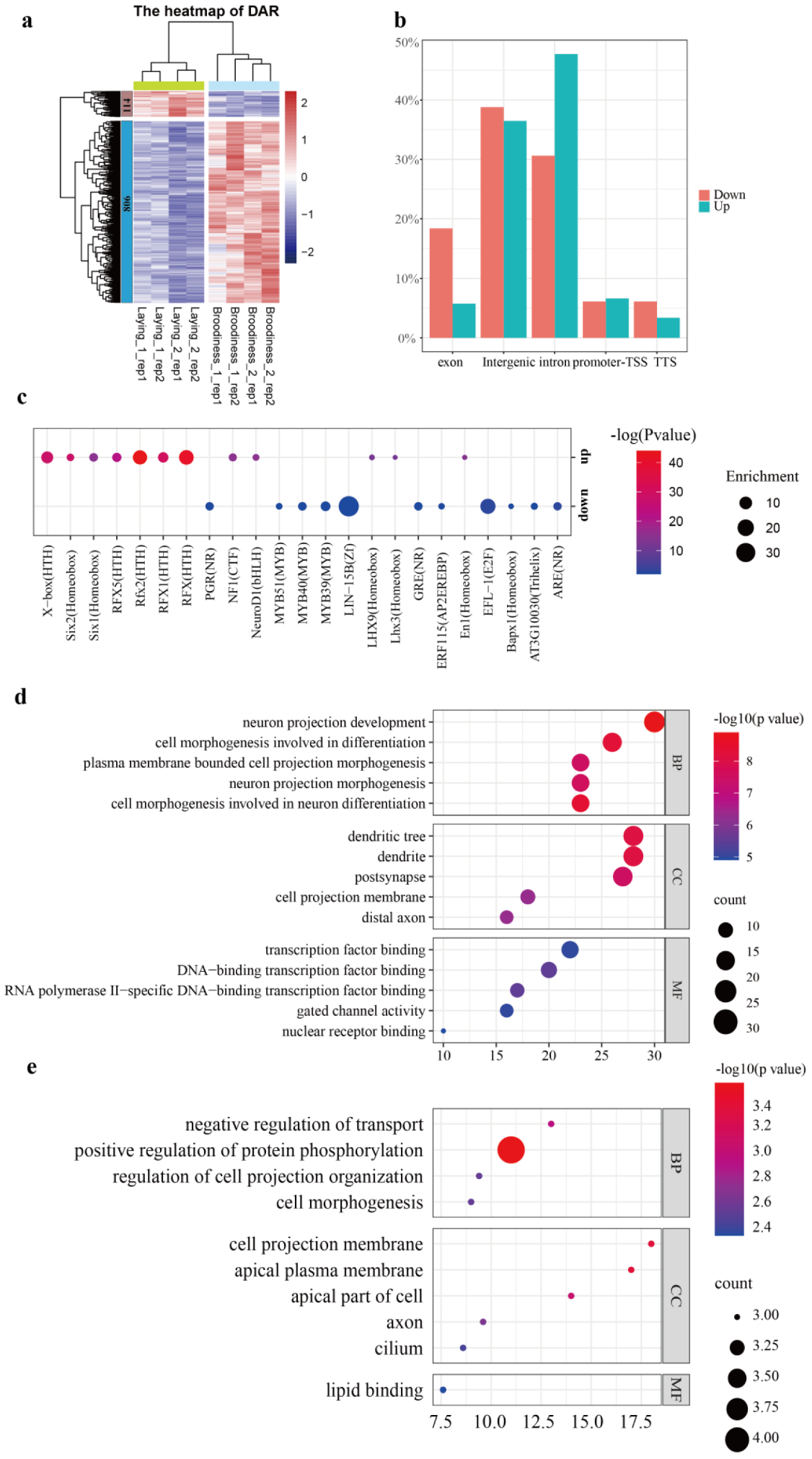
3.3. Differential Chromatin Accessibility Regions between Two Reproductive Stages
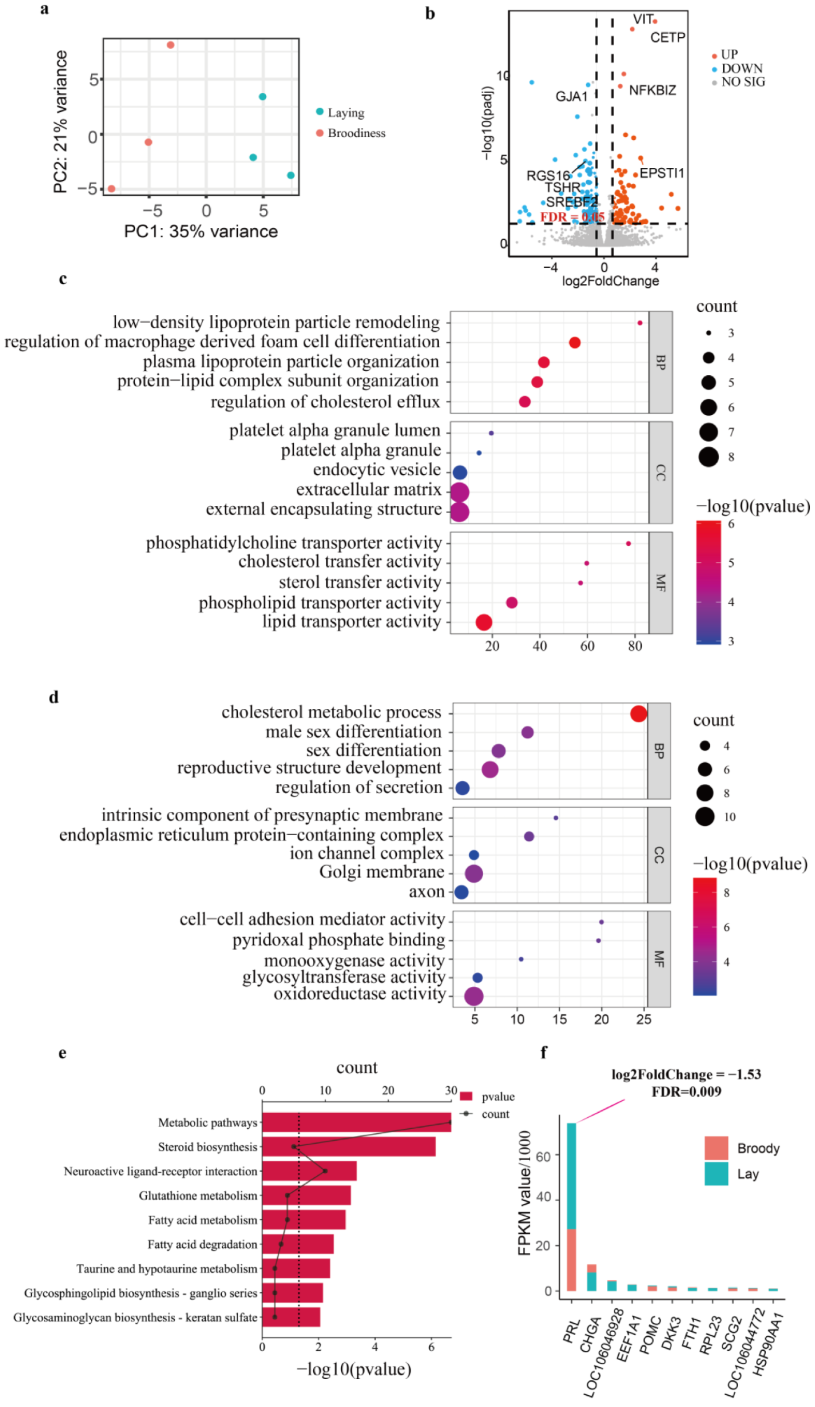
3.4. Transcriptome Change in Pituitary Underlying Two Reproductive Stages
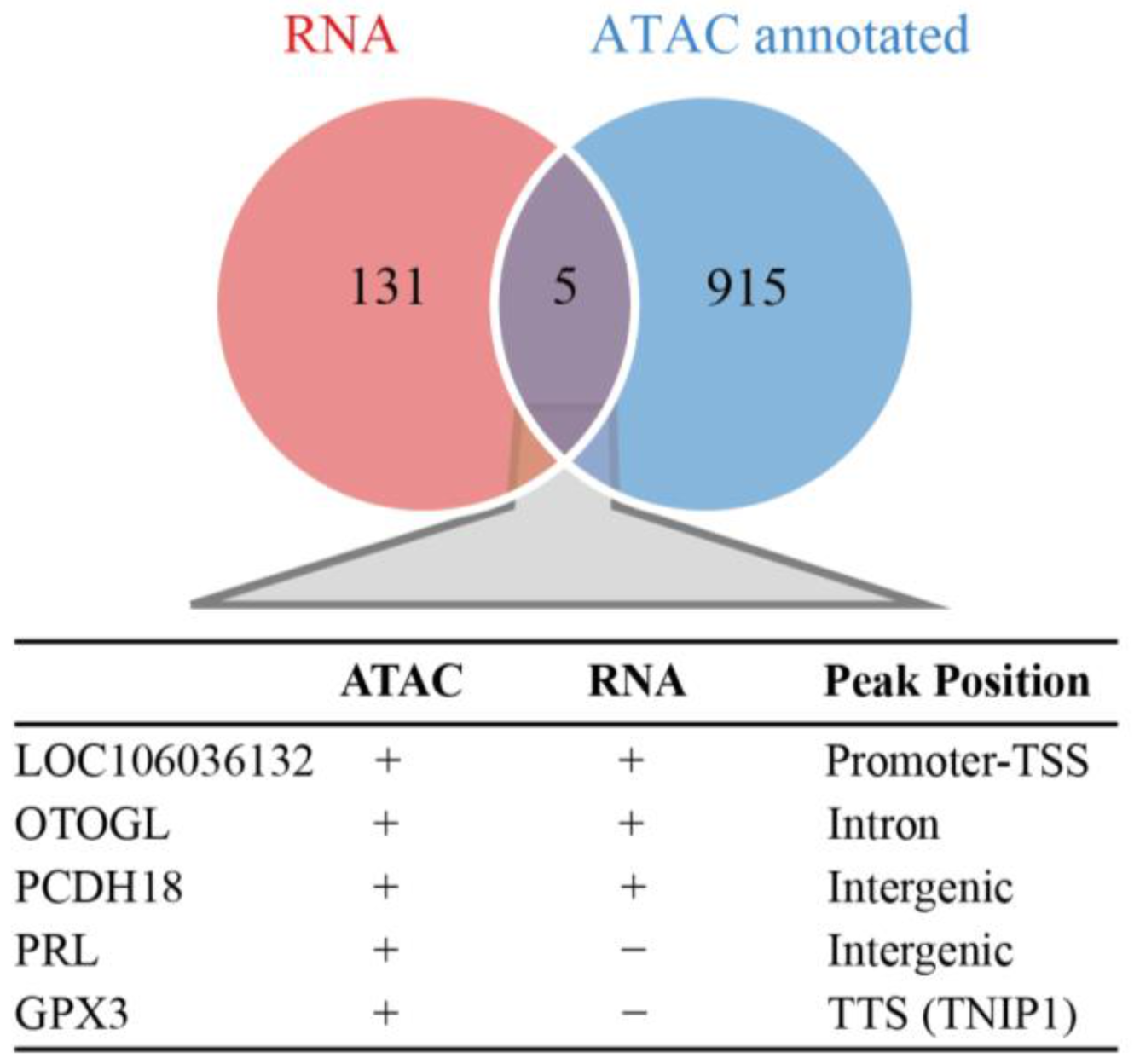
3.5. Expression Change in DAR-Related Genes to Goose Incubation Behavior
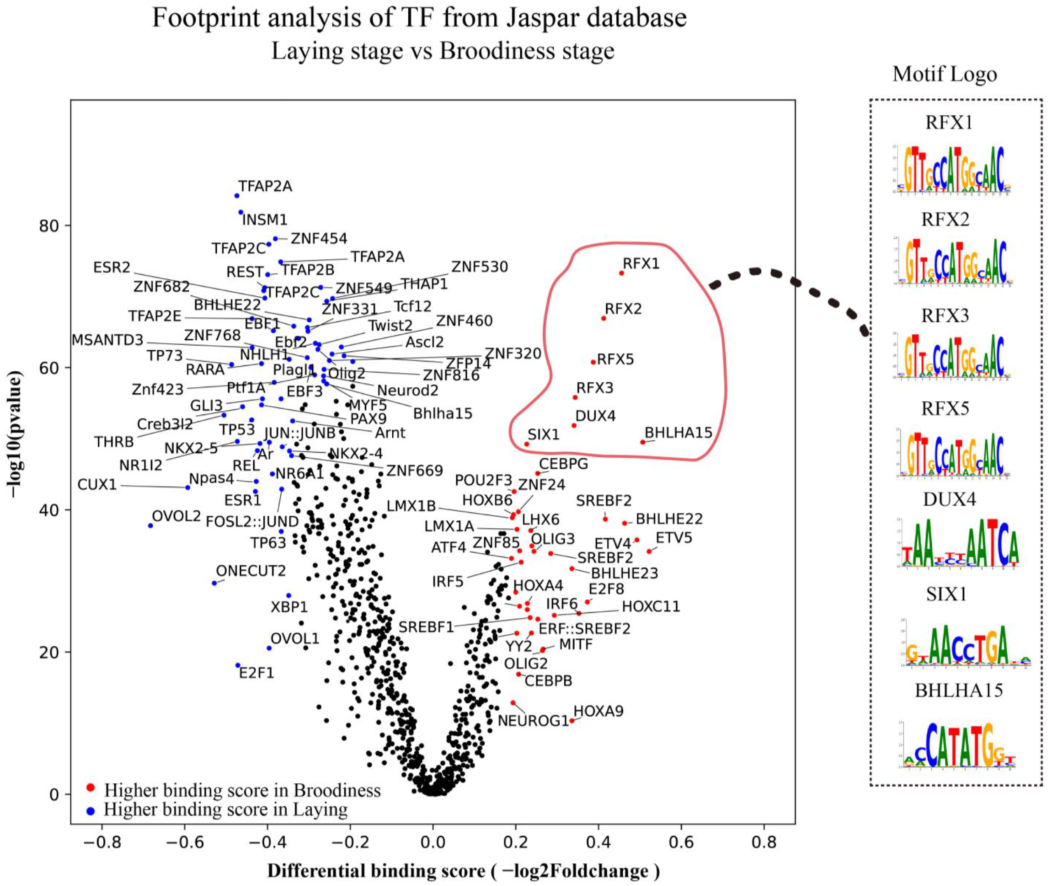
3.6. Key Transcription Factors Were Explored by Footprinting Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scanes, C.G.; Butler, L.D.; Kidd, M.T. Reproductive management of poultry. In Animal Agriculture; Elsevier: Amsterdam, The Netherlands, 2020; pp. 349–366. [Google Scholar] [CrossRef]
- Mauro, L.J.; Elde, R.P.; Youngren, O.M.; Phillips, R.E.; El Halawani, M.E. Alterations in hypothalamic vasoactive intestinal peptide-like immunoreactivity are associated with reproduction and prolactin release in the female turkey. Endocrinology 1989, 125, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- El Halawani, M.E.; Rozenboim, I. The Ontogeny and Control of Incubation Behavior in Turkeys. Poult. Sci. 1993, 72, 906–911. [Google Scholar] [CrossRef]
- Dunn, I.C.; Miao, Y.W.; Morris, A.; Romanov, M.N.; Wilson, P.W.; Waddington, D. A study of association between genetic markers in candidate genes and reproductive traits in one generation of a commercial broiler breeder hen population. Heredity 2004, 92, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.M.; Shi, Z.D.; Liu, Z.; Liu, Y.; Li, X.W. Endocrine regulations of reproductive seasonality, follicular development and incubation in Magang geese. Anim. Reprod. Sci. 2008, 104, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Donham, R.S. Annual cycle of plasma luteinizing hormone and sex hormones in male and female mallards (Anas platyrhynchos). Biol Reprod. 1979, 21, 1273–1285. [Google Scholar] [CrossRef]
- Ramsey, S.M.; Goldsmith, A.; Silver, R. Stimulus requirements for prolactin and LH secretion in incubating ring doves. Gen. Comp. Endocrinol. 1985, 59, 246–256. [Google Scholar] [CrossRef]
- El Halawani, M.E.; Silsby, J.L.; Behnke, E.J.; Fehrer, S.C. Hormonal induction of incubation behavior in ovariectomized female turkeys (Meleagris gallopavo). Biol. Reprod. 1986, 35, 59–67. [Google Scholar] [CrossRef]
- Hoglund, A.; Henriksen, R.; Fogelholm, J.; Churcher, A.M.; Guerrero-Bosagna, C.M.; Martinez-Barrio, A.; Johnsson, M.; Jensen, P.; Wright, D. The methylation landscape and its role in domestication and gene regulation in the chicken. Nat. Ecol. Evol 2020, 4, 1713–1724. [Google Scholar] [CrossRef]
- Bélteky, J.; Agnvall, B.; Bektic, L.; Höglund, A.; Jensen, P.; Guerrero-Bosagna, C. Epigenetics and early domestication: Differences in hypothalamic DNA methylation between red junglefowl divergently selected for high or low fear of humans. Genet. Sel. Evol. 2018, 50, 13. [Google Scholar] [CrossRef] [Green Version]
- Frankl-Vilches, C.; Gahr, M. Androgen and estrogen sensitivity of bird song: A comparative view on gene regulatory levels. J. Comp. Physiol. A Neuroethol. Sens. Neural. Behav. Physiol. 2018, 204, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Deplancke, B.; Alpern, D.; Gardeux, V. The Genetics of Transcription Factor DNA Binding Variation. Cell 2016, 166, 538–554. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lee, M.; Davis, B.W.; Lamichhaney, S.; Dorshorst, B.J.; Siegel, P.B.; Andersson, L. Mutations Upstream of the TBX5 and PITX1 Transcription Factor Genes Are Associated with Feathered Legs in the Domestic Chicken. Mol. Biol. Evol. 2020, 37, 2477–2486. [Google Scholar] [CrossRef]
- Castiglione, G.M.; Xu, Z.; Zhou, L.; Duh, E.J. Adaptation of the master antioxidant response connects metabolism, lifespan and feather development pathways in birds. Nat. Commun. 2020, 11, 2476. [Google Scholar] [CrossRef]
- Shen, X.; Bai, X.; Xu, J.; Zhou, M.; Xu, H.; Nie, Q.; Lu, X.; Zhang, X. Transcriptome sequencing reveals genetic mechanisms underlying the transition between the laying and brooding phases and gene expression changes associated with divergent reproductive phenotypes in chickens. Mol. Biol. Rep. 2016, 43, 977–989. [Google Scholar] [CrossRef]
- Ye, P.; Ge, K.; Li, M.; Yang, L.; Jin, S.; Zhang, C.; Chen, X.; Geng, Z. Egg-laying and brooding stage-specific hormonal response and transcriptional regulation in pituitary of Muscovy duck (Cairina moschata). Poult. Sci. 2019, 98, 5287–5296. [Google Scholar] [CrossRef]
- Ruf-Zamojski, F.; Zhang, Z.; Zamojski, M.; Smith, G.R.; Mendelev, N.; Liu, H.; Nudelman, G.; Moriwaki, M.; Pincas, H.; Castanon, R.G.; et al. Single nucleus multi-omics regulatory landscape of the murine pituitary. Nat. Commun. 2021, 12, 2677. [Google Scholar] [CrossRef]
- Fallahshahroudi, A.; Løtvedt, P.; Bélteky, J.; Altimiras, J.; Jensen, P. Changes in pituitary gene expression may underlie multiple domesticated traits in chickens. Heredity 2019, 122, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, J.; Li, L.; Han, C.; He, H.; Xu, H. Transcriptome analysis revealed the possible regulatory pathways initiating female geese broodiness within the hypothalamic-pituitary-gonadal axis. PLoS ONE 2018, 13, e0191213. [Google Scholar] [CrossRef] [Green Version]
- Lopez, M.E.; Gregory, C.C.; Porter, T. Cellular basis for elevated prolactin secretion during incubation behavior in Bantam chickens: Analysis by reverse hemolytic plaque assay. Biol. Reprod. 1996, 54, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, R.; Proudman, J.A.; Kuenzel, W.J. Changes in pituitary somatotroph and lactotroph distribution in laying and incubating turkey hens. Gen. Comp. Endocrinol. 1996, 104, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, R.; Solow, R.; Proudman, J.A.; Kuenzel, W.J. Identification of mammosomatotrophs in the turkey hen pituitary: Increased abundance during hyperprolactinemia. Endocrinology 1998, 139, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zamojski, M.; Smith, G.R.; Willis, T.L.; Yianni, V.; Mendelev, N.; Pincas, H.; Seenarine, N.; Amper, M.A.S.; Vasoya, M.; et al. Single nucleus transcriptome and chromatin accessibility of postmortem human pituitaries reveal diverse stem cell regulatory mechanisms. Cell Rep. 2022, 38, 110467. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Chen, A.F.; Favaro, P.; Reynolds, W.; Mueller, F.; Borges, L.; Jiang, S.; Park, H.S.; Kool, E.T.; Greenleaf, W.J.; et al. Integrating transcription-factor abundance with chromatin accessibility in human erythroid lineage commitment. Cell Rep. Methods 2022, 2, 100188. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Russell, T.; Hurley, L.; Jameson, J.L. Pituitary transcription factor-1 induces transient differentiation of adult hepatic stem cells into prolactin-producing cells in vivo. Mol. Endocrinol 2005, 19, 964–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017, 14, 959–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Broad Institute. Picard Toolkit; Broad Institute: Cambridge, MA, USA, 2019; Available online: https://github.com/broadinstitute/picard (accessed on 22 March 2023).
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Liu, T. Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol. Biol. 2014, 1150, 81–95. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, R.; Brown, G. DiffBind: Differential Binding Analysis of ChIP-Seq Peak Data. 2012. Available online: https://bioconductor.org/packages/release/bioc/html/DiffBind.html (accessed on 22 March 2023).
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Lemma, R.B.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Manosalva Pérez, N.; et al. JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022, 50, D165–D173. [Google Scholar] [CrossRef] [PubMed]
- Bentsen, M.; Goymann, P.; Schultheis, H.; Klee, K.; Petrova, A.; Wiegandt, R.; Fust, A.; Preussner, J.; Kuenne, C.; Braun, T.; et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun. 2020, 11, 4267. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Xu, H.; Zeng, H.; Luo, C.; Zhang, D.; Wang, Q.; Sun, L.; Yang, L.; Zhou, M.; Nie, Q.; Zhang, X. Genetic effects of polymorphisms in candidate genes and the QTL region on chicken age at first egg. BMC Genet. 2011, 12, 33. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Lei, M.; Rao, Y.; Nie, Q.; Zeng, H.; Xia, M.; Liang, F.; Zhang, D.; Zhang, X. Polymorphisms of vasoactive intestinal peptide receptor-1 gene and their genetic effects on broodiness in chickens. Poult. Sci. 2008, 87, 893–903. [Google Scholar] [CrossRef]
- Namken, S.; Sinpru, P.; Kamkrathok, B.; Sartsoongnoen, N.; Chaiseha, Y. Role of vasoactive intestinal peptide during the transition from incubation behavior to rearing behavior in the female native Thai chicken. Poult. Sci. 2017, 96, 3768–3774. [Google Scholar] [CrossRef]
- Romanov, M.N.; Talbot, R.T.; Wilson, P.; Sharp, P.J. Genetic Control of Incubation Behavior in the Domestic Hen. Poult. Sci. 2002, 81, 928–931. [Google Scholar] [CrossRef]
- Liu, L.; Xiao, Q.; Gilbert, E.R.; Cui, Z.; Zhao, X.; Wang, Y.; Yin, H.; Li, D.; Zhang, H.; Zhu, Q. Whole-transcriptome analysis of atrophic ovaries in broody chickens reveals regulatory pathways associated with proliferation and apoptosis. Sci. Rep. 2018, 8, 7231. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Mi, L.; Xiong, J.; Orchard, P.; Yu, Q.; Yu, L.; Zhao, X.Y.; Meng, Z.X.; Parker, S.C.J.; Lin, J.D.; et al. BAF60a deficiency uncouples chromatin accessibility and cold sensitivity from white fat browning. Nat. Commun. 2020, 11, 2379. [Google Scholar] [CrossRef]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef] [Green Version]
- Schubert, M.; Brunet, F.; Paris, M.; Bertrand, S.; Benoit, G.; Laudet, V. Nuclear hormone receptor signaling in amphioxus. Dev. Genes Evol. 2008, 218, 651–665. [Google Scholar] [CrossRef]
- Whirledge, S.; Cidlowski, J.A. Glucocorticoids and Reproduction: Traffic Control on the Road to Reproduction. Trends Endocrinol. Metab. 2017, 28, 399–415. [Google Scholar] [CrossRef]
- Son, Y.L.; Ubuka, T.; Narihiro, M.; Fukuda, Y.; Hasunuma, I.; Yamamoto, K.; Belsham, D.D.; Tsutsui, K. Molecular basis for the activation of gonadotropin-inhibitory hormone gene transcription by corticosterone. Endocrinology 2014, 155, 1817–1826. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.; Qi, J.; Filipp, F.V. Refinement of the androgen response element based on ChIP-Seq in androgen-insensitive and androgen-responsive prostate cancer cell lines. Sci. Rep. 2016, 6, 32611. [Google Scholar] [CrossRef]
- Schauwaers, K.; De Gendt, K.; Saunders, P.T.; Atanassova, N.; Haelens, A.; Callewaert, L.; Moehren, U.; Swinnen, J.V.; Verhoeven, G.; Verrijdt, G.; et al. Loss of androgen receptor binding to selective androgen response elements causes a reproductive phenotype in a knockin mouse model. Proc. Natl. Acad. Sci. USA 2007, 104, 4961–4966. [Google Scholar] [CrossRef] [Green Version]
- Akison, L.K.; Robker, R.L. The critical roles of progesterone receptor (PGR) in ovulation, oocyte developmental competence and oviductal transport in mammalian reproduction. Reprod. Domest. Anim. 2012, 47 (Suppl. S4), 288–296. [Google Scholar] [CrossRef]
- Dinh, D.T.; Breen, J.; Akison, L.K.; DeMayo, F.J.; Brown, H.M.; Robker, R.L.; Russell, D.L. Tissue-specific progesterone receptor-chromatin binding and the regulation of progesterone-dependent gene expression. Sci. Rep. 2019, 9, 11966. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.H.; Kwan, R.S.; Tsai, Z.T.; Lin, T.C.; Tsai, H.K. Borders of Cis-Regulatory DNA Sequences Preferentially Harbor the Divergent Transcription Factor Binding Motifs in the Human Genome. Front. Genet. 2018, 9, 571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choksi, S.P.; Lauter, G.; Swoboda, P.; Roy, S. Switching on cilia: Transcriptional networks regulating ciliogenesis. Development 2014, 141, 1427–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiaman-Trapman, D.; Vitezic, M.; Jouhilahti, E.M.; Mathelier, A.; Lauter, G.; Misra, S.; Daub, C.O.; Kere, J.; Swoboda, P. Characterization of the human RFX transcription factor family by regulatory and target gene analysis. BMC Genom. 2018, 19, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, H.K.; Nakayama, T.; Lai, J.; Zhao, B.; Argyrou, N.; Gubbels, C.S.; Soucy, A.; Genetti, C.A.; Suslovitch, V.; Rodan, L.H.; et al. Disruption of RFX family transcription factors causes autism, attention-deficit/hyperactivity disorder, intellectual disability, and dysregulated behavior. Genet. Med. 2021, 23, 1028–1040. [Google Scholar] [CrossRef]
- Her, Y.R.; Wang, L.; Chepelev, I.; Manterola, M.; Berkovits, B.; Cui, K.; Zhao, K.; Wolgemuth, D.J. Genome-wide chromatin occupancy of BRDT and gene expression analysis suggest transcriptional partners and specific epigenetic landscapes that regulate gene expression during spermatogenesis. Mol. Reprod. Dev. 2021, 88, 141–157. [Google Scholar] [CrossRef]
- Wu, Y.; Hu, X.; Li, Z.; Wang, M.; Li, S.; Wang, X.; Lin, X.; Liao, S.; Zhang, Z.; Feng, X.; et al. Transcription Factor RFX2 Is a Key Regulator of Mouse Spermiogenesis. Sci. Rep. 2016, 6, 20435. [Google Scholar] [CrossRef] [Green Version]
- Kistler, W.S.; Baas, D.; Lemeille, S.; Paschaki, M.; Seguin-Estevez, Q.; Barras, E.; Ma, W.; Duteyrat, J.L.; Morle, L.; Durand, B.; et al. RFX2 Is a Major Transcriptional Regulator of Spermiogenesis. PLoS Genet. 2015, 11, e1005368. [Google Scholar] [CrossRef]
- Meurer, L.; Ferdman, L.; Belcher, B.; Camarata, T. The SIX Family of Transcription Factors: Common Themes Integrating Developmental and Cancer Biology. Front. Cell Dev. Biol. 2021, 9, 707854. [Google Scholar] [CrossRef]
- Taylor, S.M.; Alvarez-Delfin, K.; Saade, C.J.; Thomas, J.L.; Thummel, R.; Fadool, J.M.; Hitchcock, P.F. The bHLH Transcription Factor NeuroD Governs Photoreceptor Genesis and Regeneration Through Delta-Notch Signaling. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7496–7515. [Google Scholar] [CrossRef]
- Liu, H.; Etter, P.; Hayes, S.; Jones, I.; Nelson, B.; Hartman, B.; Forrest, D.; Reh, T.A. NeuroD1 regulates expression of thyroid hormone receptor 2 and cone opsins in the developing mouse retina. J. Neurosci. 2008, 28, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Cherrington, B.D.; Bailey, J.S.; Diaz, A.L.; Mellon, P.L. NeuroD1 and Mash1 temporally regulate GnRH receptor gene expression in immortalized mouse gonadotrope cells. Mol. Cell. Endocrinol. 2008, 295, 106–114. [Google Scholar] [CrossRef] [Green Version]
- West, B.E.; Parker, G.E.; Savage, J.J.; Kiratipranon, P.; Toomey, K.S.; Beach, L.R.; Colvin, S.C.; Sloop, K.W.; Rhodes, S.J. Regulation of the follicle-stimulating hormone beta gene by the LHX3 LIM-homeodomain transcription factor. Endocrinology 2004, 145, 4866–4879. [Google Scholar] [CrossRef] [Green Version]
- Birk, O.S.; Casiano, D.E.; Wassif, C.A.; Cogliati, T.; Zhao, L.; Zhao, Y.; Grinberg, A.; Huang, S.; Kreidberg, J.A.; Parker, K.L.; et al. The LIM homeobox gene Lhx9 is essential for mouse gonad formation. Nature 2000, 403, 909–913. [Google Scholar] [CrossRef]
- Norquay, L.D.; Jin, Y.; Surabhi, R.M.; Gietz, R.D.; Tanese, N.; Cattini, P.A. A member of the nuclear factor-1 family is involved in the pituitary repression of the human placental growth hormone genes. Biochem. J. 2001, 354, 387–395. [Google Scholar] [CrossRef]
- Xiao, Y.; Kim, M.; Lazar, M.A. Nuclear receptors and transcriptional regulation in non-alcoholic fatty liver disease. Mol. Metab. 2021, 50, 101119. [Google Scholar] [CrossRef]
- O’Hara, L.; Curley, M.; Tedim Ferreira, M.; Cruickshanks, L.; Milne, L.; Smith, L.B. Pituitary androgen receptor signalling regulates prolactin but not gonadotrophins in the male mouse. PLoS ONE 2015, 10, e0121657. [Google Scholar] [CrossRef]
- Ryan, G.E.; Bohaczuk, S.C.; Cassin, J.; Witham, E.A.; Shojaei, S.; Ho, E.V.; Thackray, V.G.; Mellon, P.L. Androgen receptor positively regulates gonadotropin-releasing hormone receptor in pituitary gonadotropes. Mol. Cell. Endocrinol. 2021, 530, 111286. [Google Scholar] [CrossRef]
- Frank, F.; Ortlund, E.A.; Liu, X. Structural insights into glucocorticoid receptor function. Biochem. Soc. Trans. 2021, 49, 2333–2343. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, T.T.; Jiang, K.; Hong, W.S.; Chen, S.X. GFP expression pattern in pituitary and gonads under the control of nuclear progesterone receptor promoter in transgenic zebrafish. Dev. Dyn. 2020, 249, 1365–1376. [Google Scholar] [CrossRef]
- Yao, Y.; Yang, Y.Z.; Gu, T.T.; Cao, Z.F.; Zhao, W.M.; Qin, H.R.; Xu, Q.; Chen, G.H. Comparison of the broody behavior characteristics of different breeds of geese. Poult. Sci. 2019, 98, 5226–5233. [Google Scholar] [CrossRef]
- El Halawani, M.E.; Burke, W.H.; Dennison, P.T. Effect of nest-deprivation on serum prolactin level in nesting female turkeys. Biol. Reprod. 1980, 23, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Pitts, G.R.; Foster, D.N.; El Halawani, M.E. Transcriptional and post-transcriptional regulation of prolactin during the turkey reproductive cycle. J. Mol. Endocrinol. 1997, 18, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Roh, W.; Sullivan, R.J.; Wong, K.H.K.; Wittner, B.S.; Guo, H.; Dubash, T.D.; Sade-Feldman, M.; Wesley, B.; Horwitz, E.; et al. The Lipogenic Regulator SREBP2 Induces Transferrin in Circulating Melanoma Cells and Suppresses Ferroptosis. Cancer Discov. 2021, 11, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.; Kim, C.Y.; Lee, J.M.; Ryu, B.; Kim, J.; Bang, J.; Ahn, N.; Park, J.H. Loss of glutathione peroxidase 3 induces ROS and contributes to prostatic hyperplasia in Nkx3.1 knockout mice. Andrology 2020, 8, 1486–1493. [Google Scholar] [CrossRef]
- Kasnauskiene, J.; Ciuladaite, Z.; Preiksaitiene, E.; Matulevičienė, A.; Alexandrou, A.; Koumbaris, G.; Sismani, C.; Pepalytė, I.; Patsalis, P.C.; Kučinskas, V. A single gene deletion on 4q28.3: PCDH18-A new candidate gene for intellectual disability? Eur. J. Med. Genet. 2012, 55, 274–277. [Google Scholar] [CrossRef]
- Yariz, K.O.; Duman, D.; Zazo Seco, C.; Dallman, J.; Huang, M.; Peters, T.A.; Sirmaci, A.; Lu, N.; Schraders, M.; Skromne, I.; et al. Mutations in OTOGL, encoding the inner ear protein otogelin-like, cause moderate sensorineural hearing loss. Am. J. Hum. Genet. 2012, 91, 872–882. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.; Fan, D.; Liu, J.; Xu, Y.; Huang, X.; Tian, Y.; Xu, J.; Huang, Y.; Ruan, J.; Shen, X. Transcriptomic and Chromatin Landscape Analysis Reveals That Involvement of Pituitary Level Transcription Factors Modulate Incubation Behaviors of Magang Geese. Genes 2023, 14, 815. https://doi.org/10.3390/genes14040815
Chang J, Fan D, Liu J, Xu Y, Huang X, Tian Y, Xu J, Huang Y, Ruan J, Shen X. Transcriptomic and Chromatin Landscape Analysis Reveals That Involvement of Pituitary Level Transcription Factors Modulate Incubation Behaviors of Magang Geese. Genes. 2023; 14(4):815. https://doi.org/10.3390/genes14040815
Chicago/Turabian StyleChang, Jianye, Di Fan, Jiaxin Liu, Yanglong Xu, Xuefei Huang, Yunbo Tian, Jin Xu, Yunmao Huang, Jue Ruan, and Xu Shen. 2023. "Transcriptomic and Chromatin Landscape Analysis Reveals That Involvement of Pituitary Level Transcription Factors Modulate Incubation Behaviors of Magang Geese" Genes 14, no. 4: 815. https://doi.org/10.3390/genes14040815