
Implementation of Exome Sequencing in Clinical Practice for Neurological Disorders
 , , , ,
, , , ,
Abstract
:1. Background
2. Material and Methods
2.1. Patients
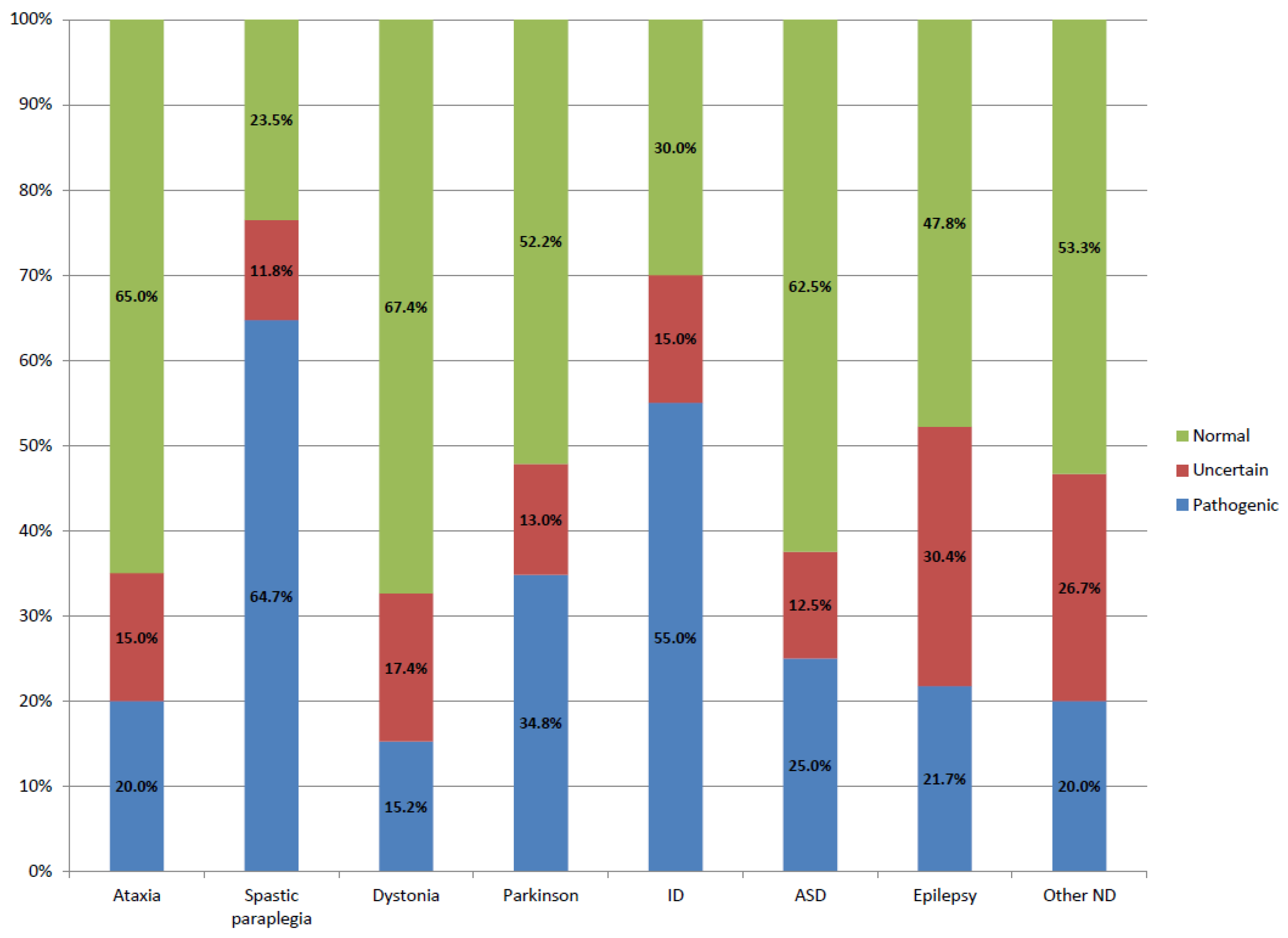
- Movement disorders cohort: 40 patients with ataxia ruled out microsatellite expansions (SCA1, SCA2, SCA3, SCA6, SCA7, DRPLA, and FXTAS), 38 patients with spastic paraplegia, 46 patients with dystonia, and 23 patients with Parkinson’s disease.
- NDD cohort: 20 patients with ID, 8 patients with autism spectrum disorder (ASD), and 23 patients with seizures. FMR1 expansion and CNVs were previously discarded in all patients.
- Other disorders: 15 patients with other neurological conditions including microcephaly, leukodystrophy, neurological channelopathies, familial hemiplegic migraine, within others.
2.2. Sequencing and Bioinformatic Analysis
3. Results and Discussion
3.1. Variant Classification
3.2. Diagnostic Yield
3.3. Variants of Unknown Significance
3.4. Genes with Incomplete Penetrance and Variable Expressivity
3.5. Secondary Findings
3.6. Variants Nomenclature
3.7. WES Limitations and New Diagnostic Strategies
3.8. Genetic Counselling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hartman, P.; Beckman, K.; Silverstein, K.; Yohe, S.; Schomaker, M.; Henzler, C.; Onsongo, G.; Lam, H.C.; Munro, S.; Daniel, J.; et al. Next generation sequencing for clinical diagnostics: Five year experience of an academic laboratory. Mol. Genet. Metab. Rep. 2019, 19, 100464. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Chowdhury, S.; Taft, R.J.; Lebo, M.S.; Buchan, J.G.; Harrison, S.M.; Rowsey, R.; Klee, E.W.; Liu, P.; Worthey, E.A.; et al. Best practices for the analytical validation of clinical whole-genome sequencing intended for the diagnosis of germline disease. NPJ Genom. Med. 2020, 5, 47. [Google Scholar] [CrossRef]
- Matthijs, G.; Souche, E.; Alders, M.; Corveleyn, A.; Eck, S.; Feenstra, I.; Race, V.; Sistermans, E.; Sturm, M.; Weiss, M.; et al. Guidelines for diagnostic next-generation sequencing. Eur. J. Hum. Genet. 2016, 24, 2–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, R.E.; Procopio-Allen, A.M.; Schroer, R.J.; Collins, J.S. Genetic syndromes among individuals with mental retardation. Am. J. Med. Genet. 2003, 123A, 29–32. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Manickam, K.; McClain, M.R.; Demmer, L.A.; Biswas, S.; Kearney, H.M.; Malinowski, J.; Massingham, L.J.; Miller, D.; Yu, T.W.; Hisama, F.M. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: An evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Anesthesia Analg. 2021, 23, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Juhos, S.; Larsson, M.; Olason, P.I.; Martin, M.; Eisfeldt, J.; DiLorenzo, S.; Sandgren, J.; Díaz De Ståhl, T.; Ewels, P.; et al. Sarek: A portable workflow for whole-genome sequencing analysis of germline and somatic variants. F1000Res 2020, 29, 63. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2015, 44, D862–D868. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesthesia Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- lvarez-Mora, M.I.; Sánchez, A.; Rodríguez-Revenga, L.; Corominas, J.; Rabionet, R.; Puig, S.; Madrigal, I. Diagnostic yield of next-generation sequencing in 87 families with neurodevelopmental disorders. Orphanet J. Rare Dis. 2022, 17, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Rexach, J.; Lee, H.; Martinez-Agosto, J.A.; Nemeth, A.; Fogel, B.L. Clinical application of next-generation sequencing to the practice of neurology. Lancet Neurol. 2019, 18, 492–503. [Google Scholar] [CrossRef]
- Ngo, K.J.; Rexach, J.E.; Lee, H.; Petty, L.E.; Perlman, S.; Valera, J.M.; Deignan, J.L.; Mao, Y.; Aker, M.; Posey, J.E.; et al. A diagnostic ceiling for exome sequencing in cerebellar ataxia and related neurological disorders. Hum. Mutat. 2019, 41, 487–501. [Google Scholar] [CrossRef]
- Eratne, D.; Schneider, A.; Lynch, E.; Martyn, M.; Velakoulis, D.; Fahey, M.; Kwan, P.; Leventer, R.; Rafehi, H.; Chong, B.; et al. The clinical utility of exome sequencing and extended bioinformatic analyses in adolescents and adults with a broad range of neurological phenotypes: An Australian perspective. J. Neurol. Sci. 2020, 420, 117260. [Google Scholar] [CrossRef]
- Sun, H.; Shen, X.-R.; Fang, Z.-B.; Jiang, Z.-Z.; Wei, X.-J.; Wang, Z.-Y.; Yu, X.-F. Next-Generation Sequencing Technologies and Neurogenetic Diseases. Life 2021, 11, 361. [Google Scholar] [CrossRef] [PubMed]
- Zech, M.; Jech, R.; Boesch, S.; Škorvánek, M.; Weber, S.; Wagner, M.; Zhao, C.; Jochim, A.; Necpál, J.; Dincer, Y.; et al. Monogenic variants in dystonia: An exome-wide sequencing study. Lancet Neurol. 2020, 19, 908–918. [Google Scholar] [CrossRef]
- Bullich, G.; Matalonga, L.; Pujadas, M.; Papakonstantinou, A.; Piscia, D.; Tonda, R.; Artuch, R.; Gallano, P.; Garrabou, G.; González, J.R.; et al. Systematic Collaborative Reanalysis of Genomic Data Improves Diagnostic Yield in Neurologic Rare Diseases. J. Mol. Diagn. 2022, 24, 529–542. [Google Scholar] [CrossRef]
- Trinh, J.; Farrer, M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013, 9, 445–454. [Google Scholar] [CrossRef]
- Seo, G.H.; Lee, H.; Lee, J.; Han, H.; Cho, Y.K.; Kim, M.; Choi, Y.; Choi, J.; Choi, I.H.; Rhie, S.; et al. Diagnostic performance of automated, streamlined, daily updated exome analysis in patients with neurodevelopmental delay. Mol. Med. 2022, 28, 38. [Google Scholar] [CrossRef]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Anesthesia Analg. 2016, 18, 696–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochtus, A.; Olson, H.E.; Smith, L.; Keith, L.G.; El Achkar, C.; Taylor, A.; Mahida, S.; Park, M.; Kelly, M.; Shain, C.; et al. Genetic diagnoses in epilepsy: The impact of dynamic exome analysis in a pediatric cohort. Epilepsia 2020, 61, 249–258. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.; Study, D.D.D.; Clayton-Smith, J. Sibling recurrence of total anomalous pulmonary venous drainage. Eur. J. Med. Genet. 2017, 60, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.R.; Davis, R.L.; Tchan, M.C.; Wali, G.; Mahant, N.; Ng, K.; Kotschet, K.; Siow, S.-F.; Gu, J.; Walls, Z.; et al. Whole genome sequencing for the genetic diagnosis of heterogenous dystonia phenotypes. Park. Relat. Disord. 2019, 69, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Crawford, H.; Scerif, G.; Wilde, L.; Beggs, A.; Stockton, J.; Sandhu, P.; Shelley, L.; Oliver, C.; McCleery, J. Genetic modifiers in rare disorders: The case of fragile X syndrome. Eur. J. Hum. Genet. 2020, 29, 173–183. [Google Scholar] [CrossRef]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Gordon, A.S.; Amendola, L.M.; Adelman, K.; Bale, S.J.; Chung, W.K.; Gollob, M.H.; Harrison, S.M.; Herman, G.E.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Anesthesia Analg. 2021, 23, 1391–1398. [Google Scholar] [CrossRef]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [Green Version]
- de Wert, G.; Dondorp, W.; Clarke, A.; Dequeker, E.M.C.; Cordier, C.; Deans, Z.; van El, C.G.; Fellmann, F.; Hastings, R.; Hentze, S.; et al. Opportunistic genomic screening. Recommendations of the European Society of Human Genetics. Eur. J. Hum. Genet. 2021, 29, 365–377. [Google Scholar] [CrossRef]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS recommendations for the description of sequence variants: 2016 update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Mora, M.; Corominas, J.; Gilissen, C.; Sanchez, A.; Madrigal, I.; Rodriguez-Revenga, L. Novel Compound Heterozygous Mutation in TRAPPC9 Gene: The Relevance of Whole Genome Sequencing. Genes 2021, 12, 557. [Google Scholar] [CrossRef] [PubMed]
- Maddirevula, S.; Kuwahara, H.; Ewida, N.; Shamseldin, H.E.; Patel, N.; Alzahrani, F.; Alsheddi, T.; Alobeid, E.; Alenazi, M.; AlSaif, H.S.; et al. Analysis of transcript-deleterious variants in Mendelian disorders: Implications for RNA-based diagnostics. Genome Biol. 2020, 21, 145. [Google Scholar] [CrossRef] [PubMed]
- Murdock, D.R.; Dai, H.; Burrage, L.C.; Rosenfeld, J.A.; Ketkar, S.; Müller, M.F.; Yépez, V.A.; Gagneur, J.; Liu, P.; Chen, S.; et al. Transcriptome-directed analysis for Mendelian disease diagnosis overcomes limitations of conventional genomic testing. J. Clin. Investig. 2021, 131, 1–13. [Google Scholar] [CrossRef]
- Yépez, V.A.; Gusic, M.; Kopajtich, R.; Mertes, C.; Smith, N.H.; Alston, C.L.; Ban, R.; Beblo, S.; Berutti, R.; Blessing, H.; et al. Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med. 2022, 14, 38. [Google Scholar] [CrossRef]
- Lee, H.; Huang, A.Y.; Wang, L.-K.; Yoon, A.J.; Renteria, G.; Eskin, A.; Signer, R.H.; Dorrani, N.; Nieves-Rodriguez, S.; Wan, J.; et al. Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Anesthesia Analg. 2019, 22, 490–499. [Google Scholar] [CrossRef]
- Sobreira, N.; Schiettecatte, F.; Valle, D.; Hamosh, A. GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene. Hum. Mutat. 2015, 36, 928–930. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Tools for Variant Interpretation | Description | Examples |
|---|---|---|
| Databases of genomic variants | They report gene variants with information on their clinical involvement or bibliographic sources in which they are mentioned. | ClinVar, dbSNP 1, HGMD 2, LOVD 3, DGV 4, or LitVar |
| Predictive programs or in silico studies | These programs include the importance of the alteration both at the nucleotide level and at the amino acid level. They are divided in two groups: (1) prediction whether the change is detrimental to the function or structure of the resulting protein, and (2) prediction if splicing is altered. | PolyPhen2, SIFT, Alamut, or Mutation Taster GeneSplicer, Human Splice Finder, Alamut, REVEL 5 CADD 6, or varSEAK |
| Evaluation of the frequency of the variant in the control population | Databases of exome and genome sequencing data from a wide variety of large-scale sequencing projects. These databases describe and analyze human genetic variation. | gnomAD 7, 1000 Genomes, or ESP 8 |
| Decision support software | These tools integrate information from several databases and combine it to carry out a classification according to the 2015 ACMG/AMP clinical guidelines | Franklin or Varsome |
| Disease | Number of Patients Analyzed | Our Cohort % P/PP Variants | Other Reports % P/PP Variants |
|---|---|---|---|
| Movement disorders cohort | |||
| Ataxia | 40 | 20% (8/40) | 13–52% [12,13,14,15,16] |
| Spastic paraplegia | 34 | 64.7% (22/34) | 40% [15] |
| Dystonia | 46 | 15.2% (7/46) | 8–37% [13,15,17,18] |
| Parkinson | 23 | 34.8% (8/23) | 11–14% [15,19] |
| Total | 31.5% (45/143) | ||
| Neurodevelopmental disorders cohort | |||
| ID | 20 | 55% (11/20) | 22–48% [11,13,18,20] |
| ASD | 8 | 25% (2/8) | 9–21% [12,13,16,18,21] |
| Epilepsy | 23 | 21.7% (5/23) | 15–40% [12,13,16,18,22] |
| Total | 35.3% (18/51) | ||
| Other disorders | |||
| Other | 15 | 20% (3/15) | |
| ALL COHORTS | 209 | 31.57% (66/209) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez-Mora, M.I.; Rodríguez-Revenga, L.; Jodar, M.; Potrony, M.; Sanchez, A.; Badenas, C.; Oriola, J.; Villanueva-Cañas, J.L.; Muñoz, E.; Valldeoriola, F.; et al. Implementation of Exome Sequencing in Clinical Practice for Neurological Disorders. Genes 2023, 14, 813. https://doi.org/10.3390/genes14040813
Alvarez-Mora MI, Rodríguez-Revenga L, Jodar M, Potrony M, Sanchez A, Badenas C, Oriola J, Villanueva-Cañas JL, Muñoz E, Valldeoriola F, et al. Implementation of Exome Sequencing in Clinical Practice for Neurological Disorders. Genes. 2023; 14(4):813. https://doi.org/10.3390/genes14040813
Chicago/Turabian StyleAlvarez-Mora, María Isabel, Laia Rodríguez-Revenga, Meritxell Jodar, Miriam Potrony, Aurora Sanchez, Celia Badenas, Josep Oriola, José Luis Villanueva-Cañas, Esteban Muñoz, Francesc Valldeoriola, and et al. 2023. "Implementation of Exome Sequencing in Clinical Practice for Neurological Disorders" Genes 14, no. 4: 813. https://doi.org/10.3390/genes14040813