
Improving Hereditary Hemorrhagic Telangiectasia Molecular Diagnosis: A Referral Center Experience
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects of Study
2.2. Gene Sequencing Panel
2.3. Panel Analysis
2.4. RNA Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shovlin, C.L.; Guttmacher, A.E.; Buscarini, E.; Faughnan, M.E.; Hyland, R.H.; Westermann, C.J.J.; Kjeldsen, A.D.; Plauchu, H. Diagnostic Criteria for Hereditary Hemorrhagic Telangiectasia (Rendu- Osler-Weber Syndrome). Am. J. Med. Genet. 2000, 91, 66–67. [Google Scholar] [CrossRef]
- Donaldson, J.W.; McKeever, T.M.; Hall, I.P.; Hubbard, R.B.; Fogarty, A.W. The UK Prevalence of Hereditary Haemorrhagic Telangiectasia and Its Association with Sex, Socioeconomic Status and Region of Residence: A Population-Based Study. Thorax 2014, 69, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Kjeldsen, A.D.; Vase, P.; Green, A. Hereditary Haemorrhagic Telangiectasia: A Population-Based Study of Prevalence and Mortality in Danish Patients. J. Intern. Med. 1999, 245, 31–39. [Google Scholar] [CrossRef]
- NCBI. Hereditary Hemorrhagic Telangiectasia—GeneReviews®. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1351/ (accessed on 5 February 2023).
- Faughnan, M.E.; Palda, V.A.; Garcia-Tsao, G.; Geisthoff, U.W.; McDonald, J.; Proctor, D.D.; Spears, J.; Brown, D.H.; Buscarini, E.; Chesnutt, M.S.; et al. International Guidelines for the Diagnosis and Management of Hereditary Haemorrhagic Telangiectasia. J. Med. Genet. 2011, 48, 73–87. [Google Scholar] [CrossRef]
- Prigoda, N.L.; Savas, S.; Abdalla, S.A.; Piovesan, B.; Rushlow, D.; Vandezande, K.; Zhang, E.; Ozcelik, H.; Gallie, B.L.; Letarte, M. Hereditary Haemorrhagic Telangiectasia: Mutation Detection, Test Sensitivity and Novel Mutations. J. Med. Genet. 2006, 43, 722–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, J.; Wooderchak-Donahue, W.; VanSant Webb, C.; Whitehead, K.; Stevenson, D.A.; Bayrak-Toydemir, P. Hereditary Hemorrhagic Telangiectasia: Genetics and Molecular Diagnostics in a New Era. Front. Genet. 2015, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallione, C.J.; Repetto, G.M.; Legius, E.; Rustgi, A.K.; Schelley, S.L.; Tejpar, S.; Mitchell, G.; Drouin, É.; Westermann, C.J.J.; Marchuk, D.A. A Combined Syndrome of Juvenile Polyposis and Hereditary Haemorrhagic Telangiectasia Associated with Mutations in MADH4 (SMAD4). Lancet 2004, 363, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Iriarte, A.; Figueras, A.; Cerdà, P.; Mora, J.M.; Jucglà, A.; Penín, R.; Viñals, F.; Riera-Mestre, A. PI3K (Phosphatidylinositol 3-Kinase) Activation and Endothelial Cell Proliferation in Patients with Hemorrhagic Hereditary Telangiectasia Type 1. Cells 2019, 8, 971. [Google Scholar] [CrossRef] [Green Version]
- Alsina-Sanchis, E.; Garcia-Ibanez, Y.; Figueiredo, A.M.; Riera-Domingo, C.; Figueras, A.; Matias-Guiu, X.; Casanovas, O.; Botella, L.M.; Pujana, M.A.; Riera-Mestre, A.; et al. ALK1 Loss Results in Vascular Hyperplasia in Mice and Humans through PI3K Activation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1216–1229. [Google Scholar] [CrossRef] [Green Version]
- Tillet, E.; Bailly, S. Emerging Roles of BMP9 and BMP10 in Hereditary Hemorrhagic Telangiectasia. Front. Genet. 2014, 5, 456. [Google Scholar] [CrossRef] [Green Version]
- Shovlin, C.L.; Buscarini, E.; Kjeldsen, A.D.; Mager, H.J.; Sabba, C.; Droege, F.; Geisthoff, U.; Ugolini, S.; Dupuis-Girod, S. European Reference Network for Rare Vascular Diseases (VASCERN) Outcome Measures for Hereditary Haemorrhagic Telangiectasia (HHT). Orphanet. J. Rare Dis. 2018, 13, 136. [Google Scholar] [CrossRef] [PubMed]
- Braverman, I.M.; Keh, A.; Jacobson, B.S. Ultrastructure and Three-Dimensional Organization of the Telanglectases of Hereditary Hemorrhagic Telangiectasia. J. Investig. Dermatol. 1990, 95, 422–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riera-Mestre, A.; Mora Luján, J.M.; Sanchez Martínez, R.; Torralba Cabeza, M.A.; Patier de la Peña, J.L.; Juyol Rodrigo, M.C.; Lopez Wolf, D.; Ojeda Sosa, A.; Monserrat, L.; López Rodríguez, M.; et al. Computerized Registry of Patients with Hemorrhagic Hereditary Telangiectasia (RiHHTa Registry) in Spain: Objectives, Methods, and Preliminary Results. Rev. Clin. Esp. 2018, 218, 468–476. [Google Scholar] [CrossRef]
- Hurvitz, N.; Azmanov, H.; Kesler, A.; Ilan, Y. Establishing a Second-Generation Artificial Intelligence-Based System for Improving Diagnosis, Treatment, and Monitoring of Patients with Rare Diseases. Eur. J. Hum. Genet. 2021, 29, 1485–1490. [Google Scholar] [CrossRef]
- Lesca, G.; Olivieri, C.; Burnichon, N.; Pagella, F.; Carette, M.F.; Gilbert-Dussardier, B.; Goizet, C.; Roume, J.; Rabilloud, M.; Saurin, J.C.; et al. Genotype-Phenotype Correlations in Hereditary Hemorrhagic Telangiectasia: Data from the French-Italian HHT Network. Genet. Med. 2007, 9, 14–22. [Google Scholar] [CrossRef] [Green Version]
- McDonald, J.; Bayrak-Toydemir, P.; Pyeritz, R.E. Hereditary Hemorrhagic Telangiectasia: An Overview of Diagnosis, Management, and Pathogenesis. Genet. Med. 2011, 13, 607–616. [Google Scholar] [CrossRef] [Green Version]
- van Gent, M.W.F.; Velthuis, S.; Post, M.C.; Snijder, R.J.; Westermann, C.J.J.; Letteboer, T.G.W.; Mager, J.J. Hereditary Hemorrhagic Telangiectasia: How Accurate Are the Clinical Criteria? Am. J. Med. Genet. A 2013, 161, 461–466. [Google Scholar] [CrossRef] [PubMed]
- ARUP Scientific Resource for Research and Education: ACVRL1 Database—University of Utah. Available online: https://arup.utah.edu/database/ACVRL1/ACVRL1_welcome.php (accessed on 5 February 2023).
- ARUP Scientific Resource for Research and Education: ENG Database—University of Utah. Available online: https://arup.utah.edu/database/ENG/ENG_welcome.php (accessed on 5 February 2023).
- Anna, A.; Monika, G. Splicing Mutations in Human Genetic Disorders: Examples, Detection, and Confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Lord, J.; Baralle, D. Splicing in the Diagnosis of Rare Disease: Advances and Challenges. Front. Genet. 2021, 12, 689892. [Google Scholar] [CrossRef]
- Krawczak, M.; Thomas, N.S.T.; Hundrieser, B.; Mort, M.; Wittig, M.; Hampe, J.; Cooper, D.N. Single Base-Pair Substitutions in Exon-Intron Junctions of Human Genes: Nature, Distribution, and Consequences for MRNA Splicing. Hum. Mutat. 2007, 28, 150–158. [Google Scholar] [CrossRef]
- Baralle, D.; Baralle, M. Splicing in Action: Assessing Disease Causing Sequence Changes. J. Med. Genet. 2005, 42, 737–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mora-Luján, J.M.; Iriarte, A.; Alba, E.; Sánchez-Corral, M.A.; Cerdà, P.; Cruellas, F.; Ordi, Q.; Corbella, X.; Ribas, J.; Castellote, J.; et al. Gender Differences in Hereditary Hemorrhagic Telangiectasia Severity. Orphanet. J. Rare Dis. 2020, 15, 63. [Google Scholar] [CrossRef] [Green Version]
- Barzilai, B.; Waggoner, A.D.; Spessert, C.; Picus, D.; Goodenberger, D. Two-Dimensional Contrast Echocardiography in the Detection and Follow-up of Congenital Pulmonary Arteriovenous Malformations. Am. J. Cardiol. 1991, 68, 1507–1510. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VarSEAK. Shared Experience and Knowledge. Available online: https://varseak.bio/ (accessed on 5 February 2023).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Bokhove, M.; Croci, R.; Zamora-Caballero, S.; Han, L.; Letarte, M.; de Sanctis, D.; Jovine, L. Structural Basis of the Human Endoglin-BMP9 Interaction: Insights into BMP Signaling and HHT1. Cell Rep. 2017, 19, 1917–1928. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; Schwartz, G.B.; Farh, K.K.H. Clinical Impact of Splicing in Neurodevelopmental Disorders. Genome Med. 2020, 12, 36. [Google Scholar] [CrossRef] [Green Version]
- Blakes, A.J.M.; Wai, H.A.; Davies, I.; Moledina, H.E.; Ruiz, A.; Thomas, T.; Bunyan, D.; Thomas, N.S.; Burren, C.P.; Greenhalgh, L.; et al. A Systematic Analysis of Splicing Variants Identifies New Diagnoses in the 100,000 Genomes Project. Genome Med. 2022, 14, 79. [Google Scholar] [CrossRef]
- Rivas, M.A.; Pirinen, M.; Conrad, D.F.; Lek, M.; Tsang, E.K.; Karczewski, K.J.; Maller, J.B.; Kukurba, K.R.; DeLuca, D.S.; Fromer, M.; et al. Effect of Predicted Protein-Truncating Genetic Variants on the Human Transcriptome. Science 2015, 348, 666–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvelo, A.; Hallegger, M.; Smith, C.W.J.; Eyras, E. Genome-Wide Association between Branch Point Properties and Alternative Splicing. PLoS Comput. Biol. 2010, 6, e1001016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shovlin, C.L.; Hughes, J.M.B.; Scott, J.; Seidman, C.E.; Seidman, J.G. Characterization of Endoglin and Identification of Novel Mutations in Hereditary Hemorrhagic Telangiectasia. Am. J. Hum. Genet. 1997, 61, 68–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchuk, D.A. Genetic Abnormalities in Hereditary Hemorrhagic Telangiectasia. Curr. Opin. Hematol. 1998, 5, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Brnich, S.E.; Abou Tayoun, A.N.; Couch, F.J.; Cutting, G.R.; Greenblatt, M.S.; Heinen, C.D.; Kanavy, D.M.; Luo, X.; McNulty, S.M.; Starita, L.M.; et al. Recommendations for Application of the Functional Evidence PS3/BS3 Criterion Using the ACMG/AMP Sequence Variant Interpretation Framework. Genome Med. 2019, 12, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesca, G.; Plauchu, H.; Coulet, F.; Lefebvre, S.; Plessis, G.; Odent, S.; Rivière, S.; Leheup, B.; Goizet, C.; Carette, M.F.; et al. Molecular Screening of ALK1/ACVRL1 and ENG Genes in Hereditary Hemorrhagic Telangiectasia in France. Hum. Mutat. 2004, 23, 289–299. [Google Scholar] [CrossRef]
- Richards-Yutz, J.; Grant, K.; Chao, E.C.; Walther, S.E.; Ganguly, A. Update on Molecular Diagnosis of Hereditary Hemorrhagic Telangiectasia. Hum. Genet. 2010, 128, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Mcdonald, J.; Damjanovich, K.; Millson, A.; Wooderchak, W.; Chibuk, J.M.; Stevenson, D.A.; Gedge, F.; Bayrak-Toydemir, P. Molecular Diagnosis in Hereditary Hemorrhagic Telangiectasia: Findings in a Series Tested Simultaneously by Sequencing and Deletion/Duplication Analysis. Clin. Genet. 2011, 79, 335–344. [Google Scholar] [CrossRef]
- Tørring, P.M.; Brusgaard, K.; Ousager, L.B.; Andersen, P.E.; Kjeldsen, A.D. National Mutation Study among Danish Patients with Hereditary Haemorrhagic Telangiectasia. Clin. Genet. 2014, 86, 123–133. [Google Scholar] [CrossRef]
- Zhang, M.Q. Statistical Features of Human Exons and Their Flanking Regions. Hum. Mol. Genet. 1998, 7, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Yeo, G.; Burge, C.B. Maximum Entropy Modeling of Short Sequence Motifs with Applications to RNA Splicing Signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A New Computational Method for Splice Site Prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reese, M.G. Improved Splice Site Detection in Genie. J. Comput. Biol. 1997, 4, 232–240. [Google Scholar] [CrossRef]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Bëroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An Online Bioinformatics Tool to Predict Splicing Signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Argyriou, L.; Twelkemeyer, S.; Panchulidze, I.; Wehner, L.E.; Teske, U.; Engel, W.; Nayernia, K. Novel Mutations in the ENG and ACVRL1 Genes Causing Hereditary Hemorrhagic Teleangiectasia. Int. J. Mol. Med. 2006, 17, 655–659. [Google Scholar] [CrossRef] [Green Version]
- Patrick, C.; McIntyre, K.; Ramidial, J.; Joa, S.; Dinsukhlal Zaveri, V.; Hansra, D. Novel ACLV1 Mutation Identified in Late Onset Hereditary Hemorrhagic Telangiectasia. Int. J. Otolaryngol. Head Neck Surg. 2016, 5, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Bossler, A.D.; Richards, J.; George, C.; Godmilow, L.; Ganguly, A. Novel Mutations in ENG and ACVRL1 Identified in a Series of 200 Individuals Undergoing Clinical Genetic Testing for Hereditary Hemorrhagic Telangiectasia (HHT): Correlation of Genotype with Phenotype. Hum. Mutat. 2006, 27, 667–675. [Google Scholar] [CrossRef]
- Jarvik, G.P.; Browning, B.L. Consideration of Cosegregation in the Pathogenicity Classification of Genomic Variants. Am. J. Hum. Genet. 2016, 98, 1077–1081. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Martínez, R.; Iriarte, A.; Mora-Luján, J.M.; Patier, J.L.; López-Wolf, D.; Ojeda, A.; Torralba, M.A.; Juyol, M.C.; Gil, R.; Añón, S.; et al. Current HHT Genetic Overview in Spain and Its Phenotypic Correlation: Data from RiHHTa Registry. Orphanet J. Rare Dis. 2020, 15, 138. [Google Scholar] [CrossRef] [PubMed]
- Pahl, K.S.; Choudhury, A.; Wusik, K.; Hammill, A.; White, A.; Henderson, K.; Pollak, J.; Kasthuri, R.S. Applicability of the Curaçao Criteria for the Diagnosis of Hereditary Hemorrhagic Telangiectasia in the Pediatric Population. J. Pediatr. 2018, 197, 207–213. [Google Scholar] [CrossRef] [Green Version]



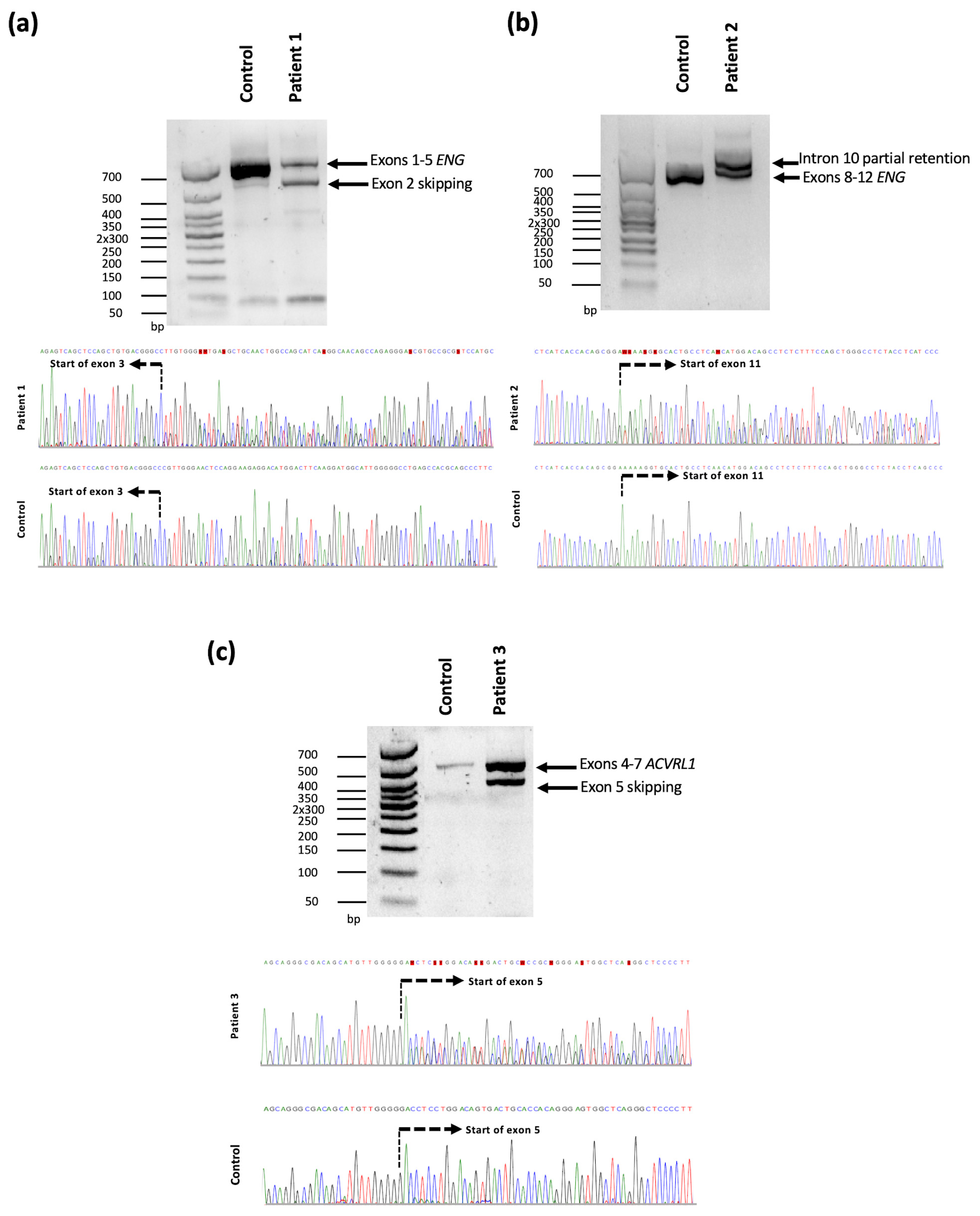{kind=link}
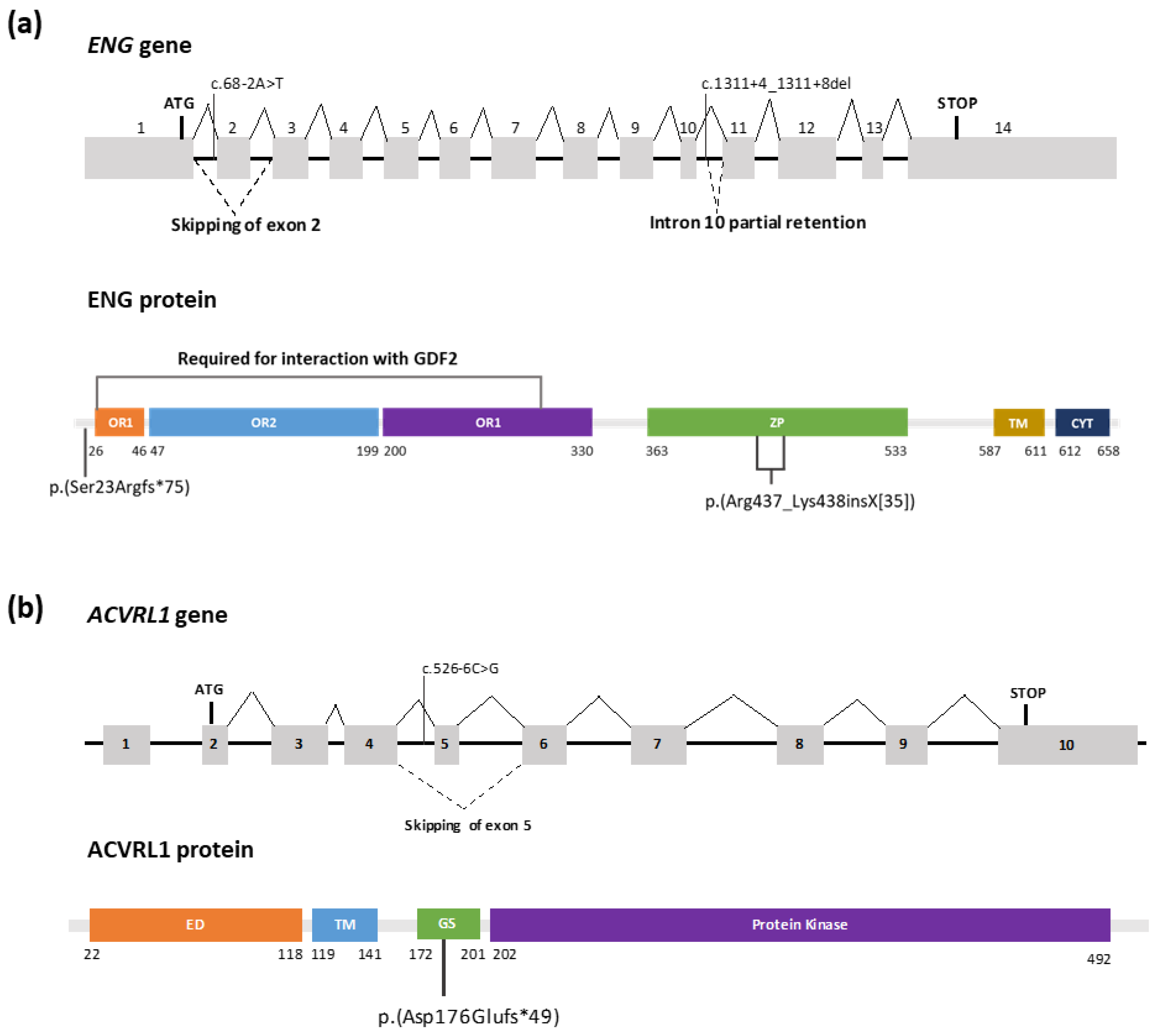{kind=link}
| ID | Sex | Age | Curaçao Criteria | Clinical Status | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FH | Epistaxis | Telangiectasia | Visceral VMS | Min Hb (g/L) | CI (L/min/m2) | HHT Specific Treatment | |||||||||
| F | L | T | N | GI | Lu | Li | Br | ||||||||
| 1 | F | 66 | + | + | + | + | − | + | + | − | + | − | 87 | 2.6 | BVZ |
| 2 | F | 74 | + | + | + | + | − | + | n/a | − | − | n/a | 118 | 3.78 | Oral iron |
| 3 | F | 54 | + | + | + | − | + | + | + | − | + | − | 140 | 2.76 | Oral iron |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilera, C.; Padró-Miquel, A.; Esteve-Garcia, A.; Cerdà, P.; Torres-Iglesias, R.; Llecha, N.; Riera-Mestre, A. Improving Hereditary Hemorrhagic Telangiectasia Molecular Diagnosis: A Referral Center Experience. Genes 2023, 14, 772. https://doi.org/10.3390/genes14030772
Aguilera C, Padró-Miquel A, Esteve-Garcia A, Cerdà P, Torres-Iglesias R, Llecha N, Riera-Mestre A. Improving Hereditary Hemorrhagic Telangiectasia Molecular Diagnosis: A Referral Center Experience. Genes. 2023; 14(3):772. https://doi.org/10.3390/genes14030772
Chicago/Turabian StyleAguilera, Cinthia, Ariadna Padró-Miquel, Anna Esteve-Garcia, Pau Cerdà, Raquel Torres-Iglesias, Núria Llecha, and Antoni Riera-Mestre. 2023. "Improving Hereditary Hemorrhagic Telangiectasia Molecular Diagnosis: A Referral Center Experience" Genes 14, no. 3: 772. https://doi.org/10.3390/genes14030772