
Next-Generation Sequencing (NGS) Analysis Illustrates the Phenotypic Variability of Collagen Type IV Nephropathies
 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Methods
2.1. Clinical Information
2.2. Molecular Analysis
2.3. Variants Interpretation
2.4. Patient Selection
3. Results
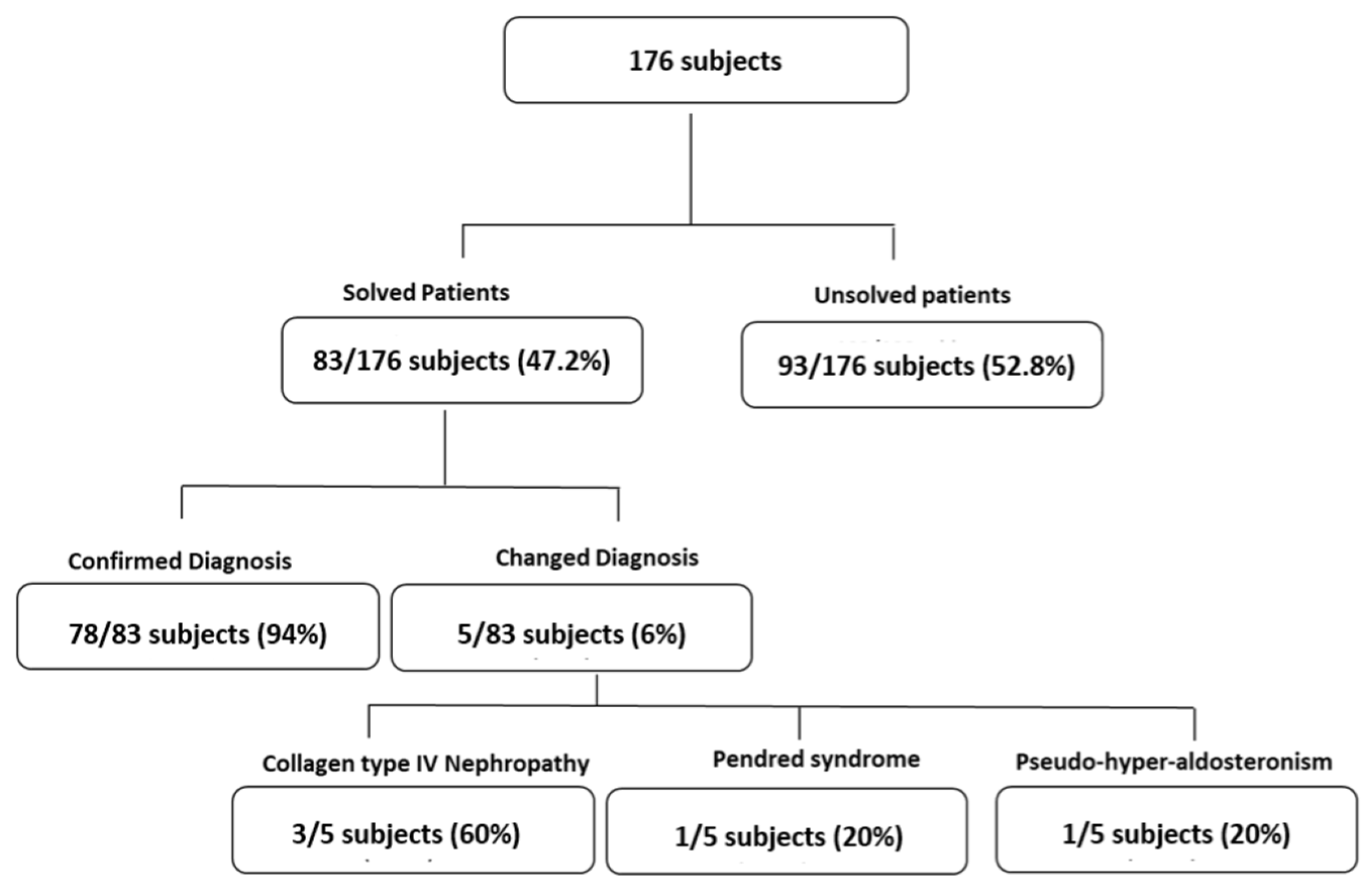
3.1. Patient Cohort
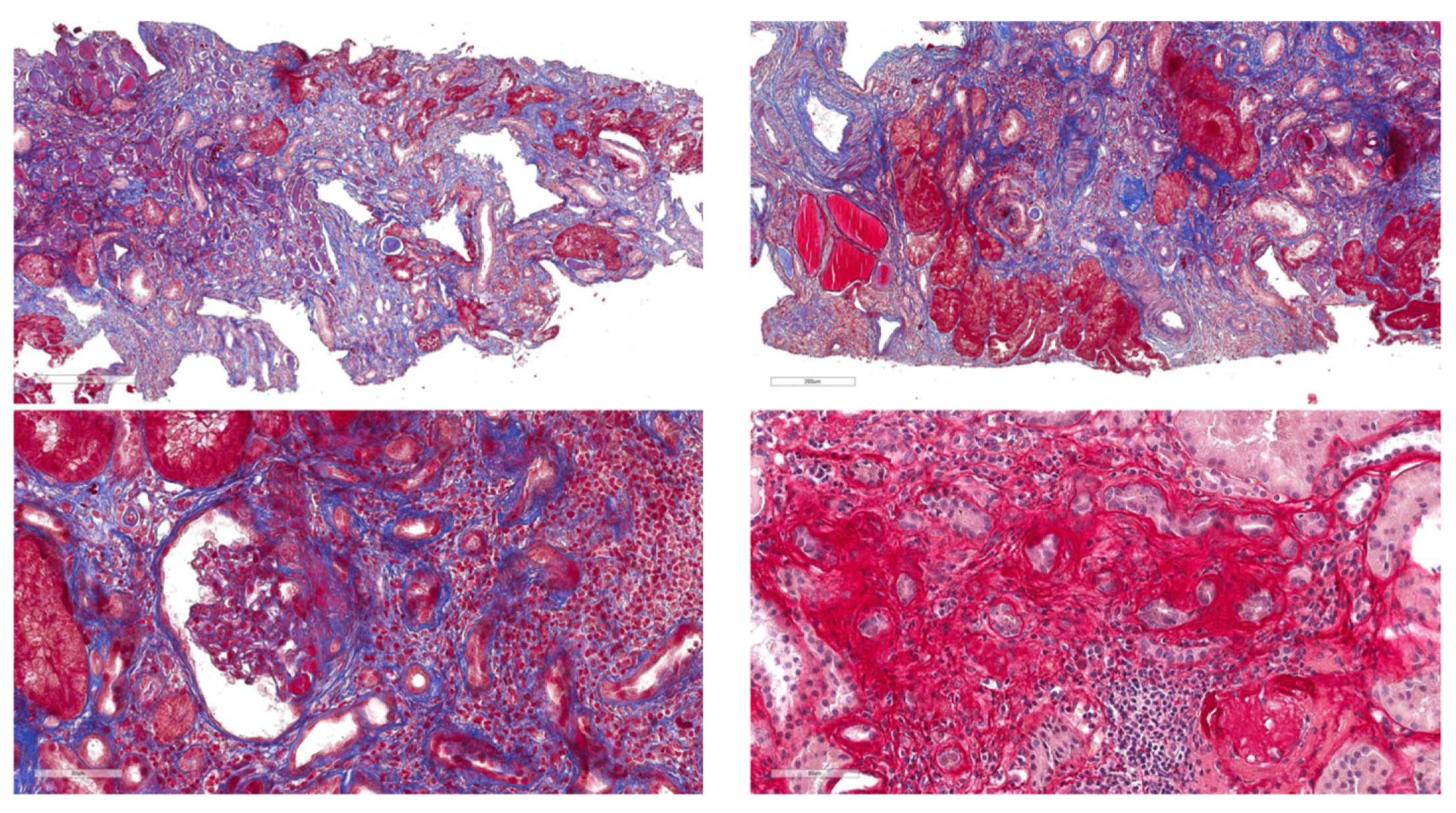
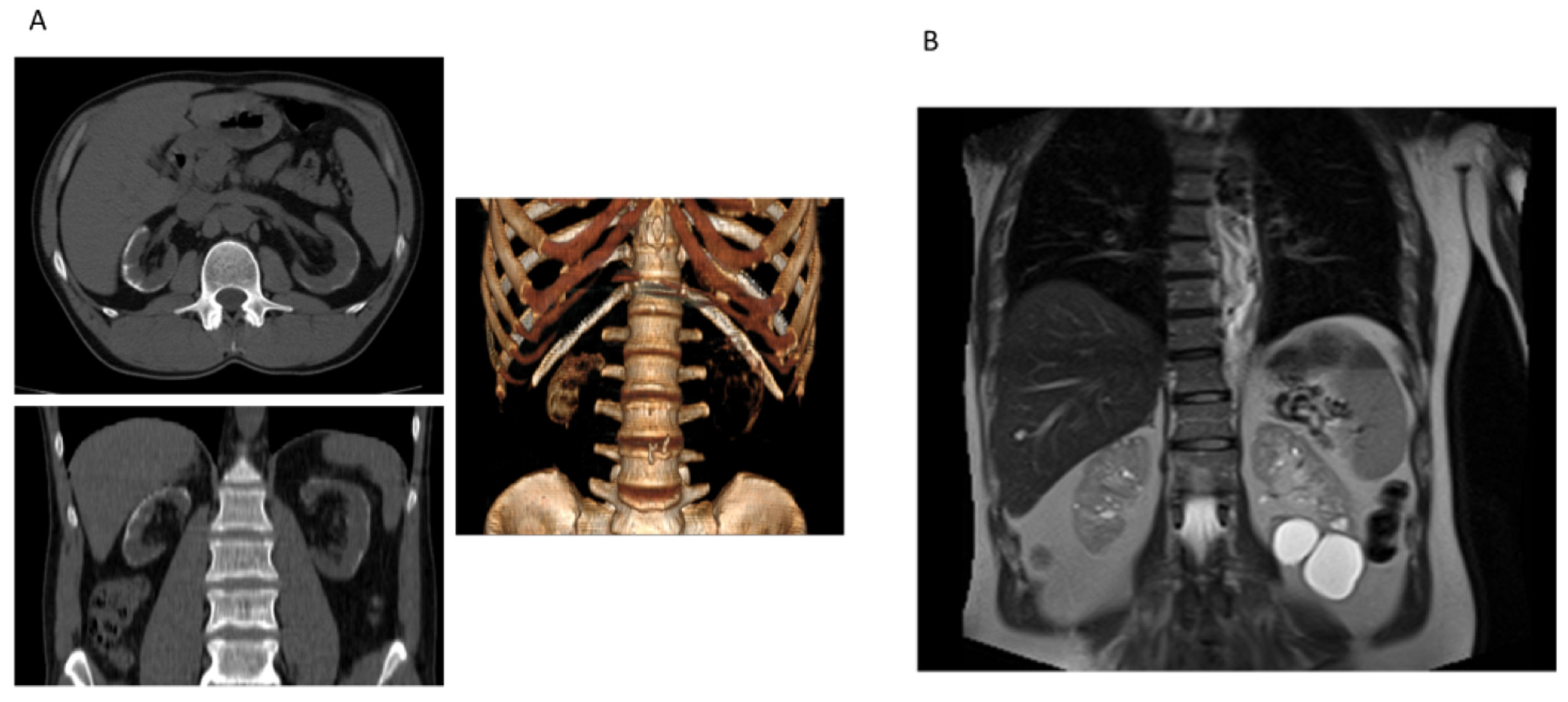
3.2. Clinical Features of COL4 Patients
3.3. Genetics Analysis of Patients
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miner, J.H. Type IV Collagen and Diabetic Kidney Disease. Nat. Rev. Nephrol. 2020, 16, 3–4. [Google Scholar] [CrossRef]
- Zacchia, M.; Blanco, F.D.V.; Trepiccione, F.; Blasio, G.; Torella, A.; Melluso, A.; Capolongo, G.; Pollastro, R.M.; Piluso, G.; Di Iorio, V.; et al. Nephroplex: A Kidney-Focused NGS Panel Highlights the Challenges of PKD1 Sequencing and Identifies a Founder BBS4 Mutation. J. Nephrol. 2021, 34, 1855–1874. [Google Scholar] [CrossRef] [PubMed]
- Flinter, F.A.; Cameron, J.S.; Chantler, C.; Houston, I.; Bobrow, M. Genetics of Classic Alport’s Syndrome. Lancet 1988, 2, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Savige, J.; Rana, K.; Tonna, S.; Buzza, M.; Dagher, H.; Wang, Y.Y. Thin Basement Membrane Nephropathy. Kidney Int. 2003, 64, 1169–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashtan, C.E.; Ding, J.; Garosi, G.; Heidet, L.; Massella, L.; Nakanishi, K.; Nozu, K.; Renieri, A.; Rheault, M.; Wang, F.; et al. Alport Syndrome: A Unified Classification of Genetic Disorders of Collagen IV α345: A Position Paper of the Alport Syndrome Classification Working Group. Kidney Int. 2018, 93, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Obaji, J.; Dupuis, A.; Paterson, A.D.; Magistroni, R.; Dicks, E.; Parfrey, P.; Cramer, B.; Coto, E.; Torra, R.; et al. Unified Criteria for Ultrasonographic Diagnosis of ADPKD. J. Am. Soc. Nephrol. 2009, 20, 205–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savige, J.; Lipska-Zietkiewicz, B.S.; Watson, E.; Hertz, J.M.; Deltas, C.; Mari, F.; Hilbert, P.; Plevova, P.; Byers, P.; Cerkauskaite, A.; et al. Guidelines for Genetic Testing and Management of Alport Syndrome. Clin. J. Am. Soc. Nephrol. 2022, 17, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Zacchia, M.; Blanco, F.D.V.; Torella, A.; Raucci, R.; Blasio, G.; Onore, M.E.; Marchese, E.; Trepiccione, F.; Vitagliano, C.; Iorio, V.D.; et al. Urine Concentrating Defect as Presenting Sign of Progressive Renal Failure in Bardet-Biedl Syndrome Patients. Clin. Kidney J. 2021, 14, 1545–1551. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC Browser: Displaying Reference Data Information from over 60 000 Exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K. The Genome Aggregation Database (gnomAD). Available online: https://ibg.colorado.edu/cdrom2019/nealeB/Gnomad/boulder_190307.pdf (accessed on 13 March 2023).
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions Using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of Protein Stability Changes for Single-Site Mutations Using Support Vector Machines. Proteins 2006, 62, 1125–1132. [Google Scholar] [CrossRef]
- Calabrese, R.; Capriotti, E.; Fariselli, P.; Martelli, P.L.; Casadio, R. Functional Annotations Improve the Predictive Score of Human Disease-Related Mutations in Proteins. Hum. Mutat. 2009, 30, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Uniprot; Surhone, L.M.; Tennoe, M.T.; Henssonow, S.F. (Eds.) Betascript Publishing: Beau Bassin, Mauritius, 2010; ISBN 9786134665261. [Google Scholar]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the Insurgence of Human Genetic Diseases Associated to Single Point Protein Mutations with Support Vector Machines and Evolutionary Information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Thomas, P.D. PANTHER-PSEP: Predicting Disease-Causing Genetic Variants Using Position-Specific Evolutionary Preservation. Bioinformatics 2016, 32, 2230–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Testa, F.; Zacchia, M.; Crispo, A.A.; Di Iorio, V.; Capolongo, G.; Rinaldi, L.; D’Antonio, M.; Fioretti, T.; Iadicicco, P.; et al. Genetic Characterization of Italian Patients with Bardet-Biedl Syndrome and Correlation to Ocular, Renal and Audio-Vestibular Phenotype: Identification of Eleven Novel Pathogenic Sequence Variants. BMC Med. Genet. 2017, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- SpliceAI Lookup. Available online: https://spliceailookup.broadinstitute.org/ (accessed on 13 March 2023).
- Savige, J.; Storey, H.; Watson, E.; Hertz, J.M.; Deltas, C.; Renieri, A.; Mari, F.; Hilbert, P.; Plevova, P.; Byers, P.; et al. Consensus Statement on Standards and Guidelines for the Molecular Diagnostics of Alport Syndrome: Refining the ACMG Criteria. Eur. J. Hum. Genet. 2021, 29, 1186–1197. [Google Scholar] [CrossRef]
- Veit, G.; Kobbe, B.; Keene, D.R.; Paulsson, M.; Koch, M.; Wagener, R. Collagen XXVIII, a Novel von Willebrand Factor A Domain-Containing Protein with Many Imperfections in the Collagenous Domain. J. Biol. Chem. 2006, 281, 3494–3504. [Google Scholar] [CrossRef] [Green Version]
- Khoshnoodi, J.; Pedchenko, V.; Hudson, B.G. Mammalian Collagen IV. Microsc. Res. Technol. 2008, 71, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Hudson, B.G.; Tryggvason, K.; Sundaramoorthy, M.; Neilson, E.G. Alport’s Syndrome, Goodpasture’s Syndrome, and Type IV Collagen. N. Engl. J. Med. 2003, 348, 2543–2556. [Google Scholar] [CrossRef] [PubMed]
- Savige, J. Alport Syndrome: Deducing the Mode of Inheritance from the Presence of Haematuria in Family Members. Pediatr. Nephrol. 2020, 35, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, B.A.; Feldman, D.L.; Fornoni, A.; Gross, O.; Kashtan, C.E.; Lagas, S.; Lennon, R.; Miner, J.H.; Rheault, M.N.; Simon, J.F.; et al. Clinical Trial Recommendations for Potential Alport Syndrome Therapies. Kidney Int. 2020, 97, 1109–1116. [Google Scholar] [CrossRef]
- Malone, A.F.; Phelan, P.J.; Hall, G.; Cetincelik, U.; Homstad, A.; Alonso, A.S.; Jiang, R.; Lindsey, T.B.; Wu, G.; Sparks, M.A.; et al. Rare Hereditary COL4A3/COL4A4 Variants May Be Mistaken for Familial Focal Segmental Glomerulosclerosis. Kidney Int. 2014, 86, 1253–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miner, J.H. Pathology vs. Molecular Genetics: (re)defining the Spectrum of Alport Syndrome. Kidney Int. 2014, 86, 1081–1083. [Google Scholar] [CrossRef] [Green Version]
- Pierides, A.; Voskarides, K.; Athanasiou, Y.; Ioannou, K.; Damianou, L.; Arsali, M.; Zavros, M.; Pierides, M.; Vargemezis, V.; Patsias, C.; et al. Clinico-Pathological Correlations in 127 Patients in 11 Large Pedigrees, Segregating One of Three Heterozygous Mutations in the COL4A3/COL4A4 Genes Associated with Familial Haematuria and Significant Late Progression to Proteinuria and Chronic Kidney Disease from Focal Segmental Glomerulosclerosis. Nephrol. Dial. Transplant 2009, 24, 2721–2729. [Google Scholar]
- Sevillano, A.M.; Gutierrez, E.; Morales, E.; Hernandez, E.; Molina, M.; Gonzalez, E.; Praga, M. Multiple Kidney Cysts in Thin Basement Membrane Disease with Proteinuria and Kidney Function Impairment. Clin. Kidney J. 2014, 7, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Gulati, A.; Sevillano, A.M.; Praga, M.; Gutierrez, E.; Alba, I.; Dahl, N.K.; Besse, W.; Choi, J.; Somlo, S. Collagen IV Gene Mutations in Adults with Bilateral Renal Cysts and CKD. Kidney Int. Rep. 2020, 5, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public Archive of Interpretations of Clinically Relevant Variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Khan, M.A.; Capasso, G.; Zacchia, M. Computational and Structural Analysis to Assess the Pathogenicity of Bardet-Biedl Syndrome Related Missense Variants Identified in Bardet-Biedl Syndrome 10 Gene (BBS10). ACS Omega 2022, 7, 37654–37662. [Google Scholar] [CrossRef]
- Melton, S.D.; Brackhahn, E.A.E.; Orlin, S.J.; Jin, P.; Chenoweth, D.M. Rules for the Design of Aza-Glycine Stabilized Triple-Helical Collagen Peptides. Chem. Sci. 2020, 11, 10638–10646. [Google Scholar] [CrossRef] [PubMed]
- Plant, K.E.; Green, P.M.; Vetrie, D.; Flinter, F.A. Detection of Mutations in COL4A5 in Patients with Alport Syndrome. Hum. Mutat. 1999, 13, 124–132. [Google Scholar] [CrossRef]
- Hashimura, Y.; Nozu, K.; Kaito, H.; Nakanishi, K.; Fu, X.J.; Ohtsubo, H.; Hashimoto, F.; Oka, M.; Ninchoji, T.; Ishimori, S.; et al. Milder Clinical Aspects of X-Linked Alport Syndrome in Men Positive for the Collagen IV α5 Chain. Kidney Int. 2014, 85, 1208–1213. [Google Scholar] [CrossRef] [Green Version]
- Shavit, L.; Jaeger, P.; Unwin, R.J. What Is Nephrocalcinosis? Kidney Int. 2015, 88, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirangelo, I.; Borriello, M.; Liccardo, M.; Scafuro, M.; Russo, P.; Iannuzzi, C. Hydroxytyrosol Selectively Affects Non-Enzymatic Glycation in Human Insulin and Protects by AGEs Cytotoxicity. Antioxidants 2021, 10, 1127. [Google Scholar] [CrossRef] [PubMed]
- Agnello, F.; Albano, D.; Micci, G.; Di Buono, G.; Agrusa, A.; Salvaggio, G.; Pardo, S.; Sparacia, G.; Bartolotta, T.V.; Midiri, M.; et al. CT and MR Imaging of Cystic Renal Lesions. Insights Imaging 2020, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Zacchia, M.; Marchese, E.; Trani, E.M.; Caterino, M.; Capolongo, G.; Perna, A.; Ruoppolo, M.; Capasso, G. Proteomics and Metabolomics Studies Exploring the Pathophysiology of Renal Dysfunction in Autosomal Dominant Polycystic Kidney Disease and Other Ciliopathies. Nephrol. Dial. Transplant 2020, 35, 1853–1861. [Google Scholar] [CrossRef]
- Caterino, M.; Zacchia, M.; Costanzo, M.; Bruno, G.; Arcaniolo, D.; Trepiccione, F.; Siciliano, R.A.; Mazzeo, M.F.; Ruoppolo, M.; Capasso, G. Urine Proteomics Revealed a Significant Correlation Between Urine-Fibronectin Abundance and Estimated-GFR Decline in Patients with Bardet-Biedl Syndrome. Kidney Blood Press. Res. 2018, 43, 389–405. [Google Scholar] [CrossRef]
- Ninomiya, Y.; Kagawa, M.; Iyama, K.; Naito, I.; Kishiro, Y.; Seyer, J.M.; Sugimoto, M.; Oohashi, T.; Sado, Y. Differential Expression of Two Basement Membrane Collagen Genes, COL4A6 and COL4A5, Demonstrated by Immunofluorescence Staining Using Peptide-Specific Monoclonal Antibodies. J. Cell Biol. 1995, 130, 1219–1229. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total of Patients Analyzed | 176 |
|---|---|
| Patients Analyzed with Nephroplex | 107 |
| Patients Analyzed with Clinical Exome Sequencing | 69 |
| Males | 82 |
| Females | 94 |
| Average Age (years) | 40.4 |
| Minimum Age (years) | 18 |
| Maximum Age (years) | 79 |
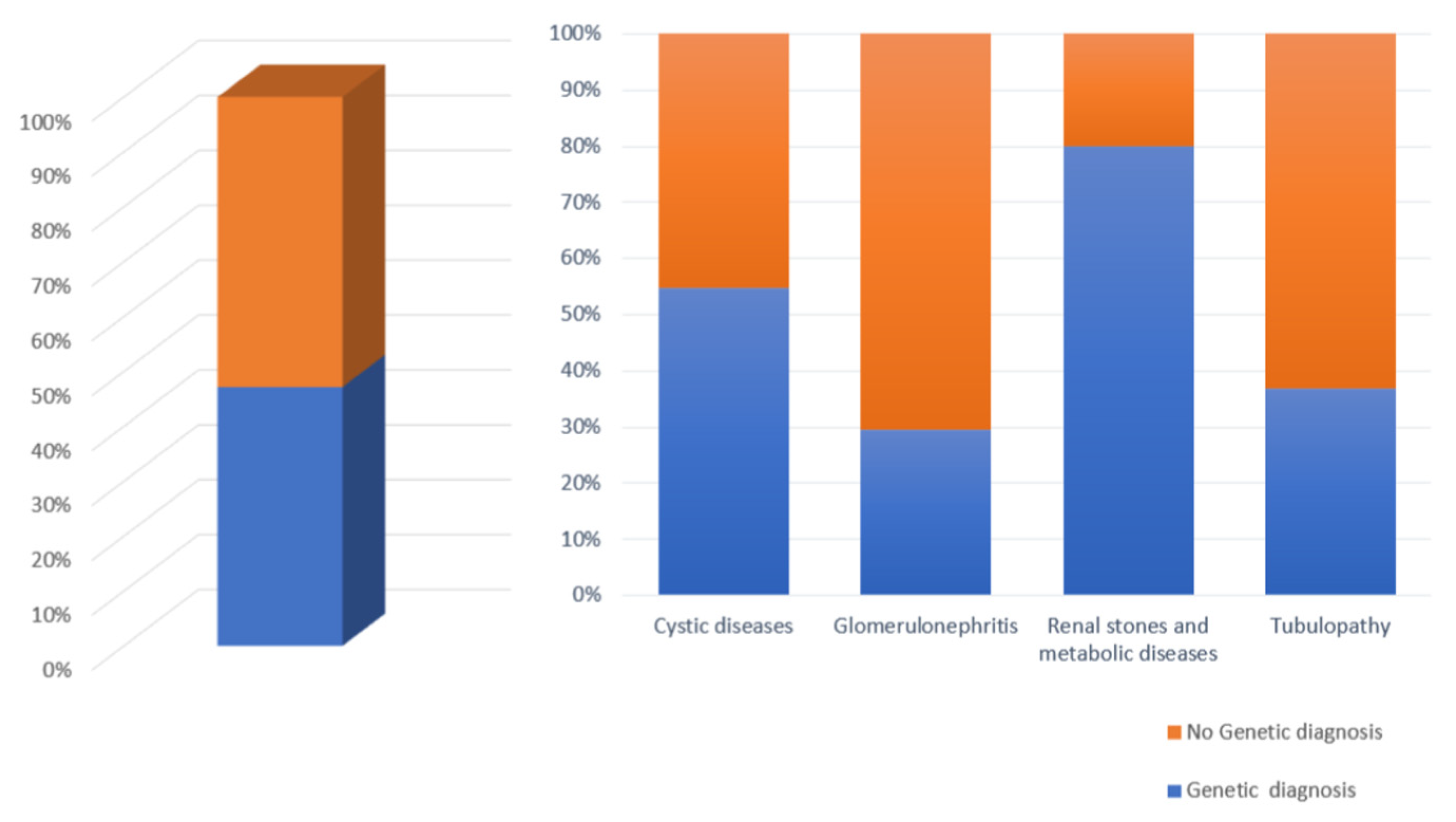
| Patients with Cystic Disorders | 104 |
| Positive Genetic Analysis | 57 |
| Patients with Glomerulonephritis | 29 |
| Positive Genetic Analysis | 8 |
| Patients with Renal Stones and Metabolic Diseases | 5 |
| Positive Genetic Analysis | 4 |
| Patients with Tubulopathies | 38 |
| Positive Genetic Analysis | 14 |
| Proband | Gender | Age (Years) | Hematuria | Proteinuria | eGFR (mL/min/ 1.73 m2) | Family History | Renal Histology | Mode of Inheritance |
|---|---|---|---|---|---|---|---|---|
| Case 1 | male | 43 | + | + | 60 | + | FSGS | AD |
| Case 2 | male | 49 | - | - | 13 | - | Unavailable | Sporadic case |
| Case 3 | male | 59 | + | + | 39 | + | Unavailable | AD |
| GENE | VARIANT | AMINO ACID CHANGE | ACMG | CLINVAR | SIFT | Mu PRO | PANTHER | PhD-SNP | SNP n GO | SPLICE AI | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CASE 1 | COL4A4 | c.693+2T>C | None | Likely pathogenic | Not reported | Not applicable | Not applicable | Not applicable | Not applicable | Not applicable | 0.89 delta score as donor loss |
| CASE 2 | COL4A5 | c.991G>A | p.Gly331Ser | Pathogenic | Not reported | Affecting protein function (score 0.00) | Decrease Stability | Probably Damaging | Disease | Disease | / |
| CASE 3 | COL4A4 | c.1589G>A | p.Gly530Glu | Likely pathogenic | Not reported | Affecting protein function (score 0.00) | Decrease Stability | Probably Damaging | Disease | Disease | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zacchia, M.; Capolongo, G.; Del Vecchio Blanco, F.; Secondulfo, F.; Gupta, N.; Blasio, G.; Pollastro, R.M.; Cervesato, A.; Piluso, G.; Gigliotti, G.; et al. Next-Generation Sequencing (NGS) Analysis Illustrates the Phenotypic Variability of Collagen Type IV Nephropathies. Genes 2023, 14, 764. https://doi.org/10.3390/genes14030764
Zacchia M, Capolongo G, Del Vecchio Blanco F, Secondulfo F, Gupta N, Blasio G, Pollastro RM, Cervesato A, Piluso G, Gigliotti G, et al. Next-Generation Sequencing (NGS) Analysis Illustrates the Phenotypic Variability of Collagen Type IV Nephropathies. Genes. 2023; 14(3):764. https://doi.org/10.3390/genes14030764
Chicago/Turabian StyleZacchia, Miriam, Giovanna Capolongo, Francesca Del Vecchio Blanco, Floriana Secondulfo, Neha Gupta, Giancarlo Blasio, Rosa Maria Pollastro, Angela Cervesato, Giulio Piluso, Giuseppe Gigliotti, and et al. 2023. "Next-Generation Sequencing (NGS) Analysis Illustrates the Phenotypic Variability of Collagen Type IV Nephropathies" Genes 14, no. 3: 764. https://doi.org/10.3390/genes14030764