

Therapeutic Targeting of P53: A Comparative Analysis of APR-246 and COTI-2 in Human Tumor Primary Culture 3-D Explants
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissue Procurement and Tumor Cell Preparation
2.2. Drug Exposure and Measurement of Cell Viability
2.3. Delayed-Loss-of-Membrane Integrity
2.4. ATP Content
2.5. Statistical Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2009, 2, a001008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.; Strasser, A.; Kelly, G.L. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026062. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Simpson, E.R.; Brown, K.A. p53: Protection against Tumor Growth beyond Effects on Cell Cycle and Apoptosis. Cancer Res. 2015, 75, 5001–5007. [Google Scholar] [CrossRef] [Green Version]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2017, 25, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Zawacka-Pankau, J.; Grinkevich, V.V.; Hünten, S.; Nikulenkov, F.; Gluch, A.; Li, H.; Enge, M.; Kel, A.; Selivanova, G. Inhibition of glycolytic enzymes mediated by pharmacologically activated p53: Targeting Warburg effect to fight cancer. J. Biol. Chem. 2011, 286, 41600–41615. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 2019, 35, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Marques, M.A.; de Andrade, G.C.; Silva, J.L.; de Oliveira, G.A.P. Protein of a thousand faces: The tumor-suppressive and oncogenic responses of p53. Front. Mol. Biosci. 2022, 9, 944955. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 Opens the Mitochondrial Permeability Transition Pore to Trigger Necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nature 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, K.L.; Chen, X. The functional domains in p53 family proteins exhibit both common and distinct properties. Cell Death Differ. 2006, 13, 890–897. [Google Scholar] [CrossRef]
- Gencel-Augusto, J.; Lozano, G. p53 tetramerization: At the center of the dominant-negative effect of mutant p53. Genes Dev. 2020, 34, 1128–1146. [Google Scholar] [CrossRef]
- Li, X.; Sun, W. Applications of Recombinant Adenovirus-p53 Gene Therapy for Cancers in the Clinic in China. Curr. Gene Ther. 2020, 20, 127–141. [Google Scholar] [CrossRef]
- Meng, X.; Gao, J.Z.; Gomendoza, S.M.T.; Li, J.W.; Yang, S. Recent Advances of WEE1 Inhibitors and Statins in Cancers With p53 Mutations. Front. Med. 2021, 8, 737951. [Google Scholar] [CrossRef]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 703. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. Mutations of p53 associated with pancreatic cancer and therapeutic implications. Ann. Hepato-Biliary-Pancreatic Surg. 2021, 25, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Cymbalista, F.; Ghia, P.; Jäger, U.; Pospisilova, S.; Rosenquist, R.; Schuh, A.; Stilgenbauer, S. TP53 aberrations in chronic lymphocytic leukemia: An overview of the clinical implications of improved diagnostics. Haematologica 2018, 103, 1956–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef]
- Oduah, E.I.; Grossman, S.R. Harnessing the vulnerabilities of p53 mutants in lung cancer—Focusing on the proteasome: A new trick for an old foe? Cancer Biol. Ther. 2020, 21, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Bally, C.; Adès, L.; Renneville, A.; Sebert, M.; Eclache, V.; Preudhomme, C.; Mozziconacci, M.-J.; de The, H.; Lehmann-Che, J.; Fenaux, P. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk. Res. 2014, 38, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Craddock, C.F.; Houlton, A.E.; Quek, L.S.; Ferguson, P.; Gbandi, E.; Roberts, C.; Metzner, M.; Garcia-Martin, N.; Kennedy, A.; Hamblin, A.; et al. Outcome of Azacitidine Therapy in Acute Myeloid Leukemia Is not Improved by Concurrent Vorinostat Therapy but Is Predicted by a Diagnostic Molecular Signature. Clin. Cancer Res. 2017, 23, 6430–6440. [Google Scholar] [CrossRef] [Green Version]
- Short, N.J.; Kantarjian, H.M.; Loghavi, S.; Huang, X.; Qiao, W.; Borthakur, G.; Kadia, T.M.; Daver, N.; Ohanian, M.; Dinardo, C.D.; et al. Treatment with a 5-day versus a 10-day schedule of decitabine in older patients with newly diagnosed acute myeloid leukaemia: A randomised phase 2 trial. Lancet Haematol. 2019, 6, e29–e37. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Wang, R.; Wang, X.; Shallis, R.M.; Podoltsev, N.A.; Bewersdorf, J.P.; Huntington, S.F.; Neparidze, N.; Giri, S.; Gore, S.D.; et al. Clinical outcomes of older patients with AML receiving hypomethylating agents: A large population-based study in the United States. Blood Adv. 2020, 4, 2192–2201. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Zhang, Q.; Zhang, M.; Ceder, S.; Abrahmsen, L.; Wiman, K.G. Targeting of Mutant p53 and the Cellular Redox Balance by APR-246 as a Strategy for Efficient Cancer Therapy. Front. Oncol. 2016, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Zhang, M.-Q.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.N.; Arnér, E.S.J.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef] [Green Version]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.-H.; Bally, C.; et al. Synergistic effects of PRIMA-1Met (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2019, 105, 1539–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andrén, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in Vivo: A First-in-Human Study With p53-Targeting Compound APR-246 in Refractory Hematologic Malignancies and Prostate Cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef] [Green Version]
- Deneberg, S.; Cherif, H.; Lazarevic, V.; Andersson, P.-O.; von Euler, M.; Juliusson, G.; Lehmann, S. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016, 6, e447. [Google Scholar] [CrossRef] [Green Version]
- Salim, K.Y.; Vareki, S.M.; Danter, W.R.; San-Marina, S.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vareki, S.M.; Salim, K.Y.; Danter, W.R.; Koropatnick, J. Novel anti-cancer drug COTI-2 synergizes with therapeutic agents and does not induce resistance or exhibit cross-resistance in human cancer cell lines. PLoS ONE 2018, 13, e0191766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagourney, R.A.; Link, J.S.; Blitzer, J.B.; Forsthoff, C.; Evans, S.S. Gemcitabine Plus Cisplatin Repeating Doublet Therapy in Previously Treated, Relapsed Breast Cancer Patients. J. Clin. Oncol. 2000, 18, 2245–2249. [Google Scholar] [CrossRef]
- D’Amora, P.; Silva, I.D.C.; Tewari, K.S.; Bristow, R.E.; Cappuccini, F.; Evans, S.S.; Salzgeber, M.B.; Addis-Bernard, P.J.; Palma, A.M.; Marchioni, D.M.; et al. Platinum resistance in gynecologic malignancies: Response, disease free and overall survival are predicted by biochemical signature: A metabolomic analysis. Gynecol. Oncol. 2021, 163, 162–170. [Google Scholar] [CrossRef]
- Nagourney, R.A. Ex vivo programmed cell death and the prediction of response to chemotherapy. Curr. Treat. Options Oncol. 2006, 7, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Nagourney, R.A. Gemcitabine plus cisplatin in breast cancer. Oncology 2001, 15, 28–33. [Google Scholar] [PubMed]
- Nagourney, R.A.; Blitzer, J.B.; Shuman, R.L.; Asciuto, T.J.; Deo, E.A.; Paulsen, M.; Newcomb, R.L.; Evans, S.S. Functional profiling to select chemotherapy in untreated, advanced or metastatic non-small cell lung cancer. Anticancer Res. 2012, 32, 4453–4460. [Google Scholar]
- Chou, T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Feinstein, A.R. Zeta and delta: Critical descriptive boundaries in statistical analysis. J. Clin. Epidemiol. 1998, 51, 527–530. [Google Scholar] [CrossRef]
- Cheadle, C.; Vawter, M.P.; Freed, W.J.; Becker, K.G. Analysis of Microarray Data Using Z Score Transformation. J. Mol. Diagn. 2003, 5, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemkin, P.F.; Thornwall, G.C.; Walton, K.D.; Hennighausen, L. The Microarray Explorer tool for data mining of cDNA microarrays: Application for the mammary gland. Nucleic Acids Res. 2000, 28, 4452–4459. [Google Scholar] [CrossRef] [PubMed] [Green Version]





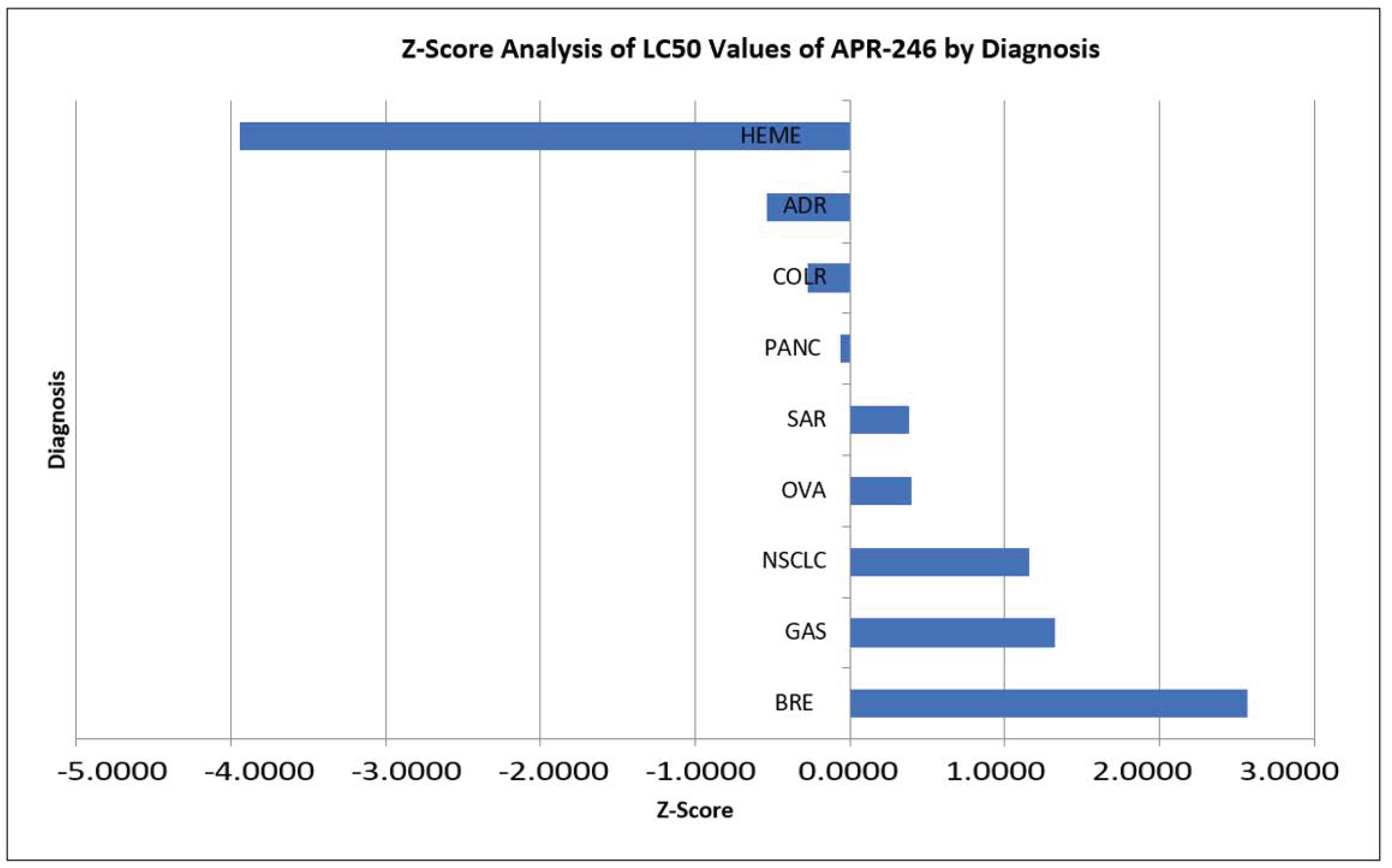{kind=link}
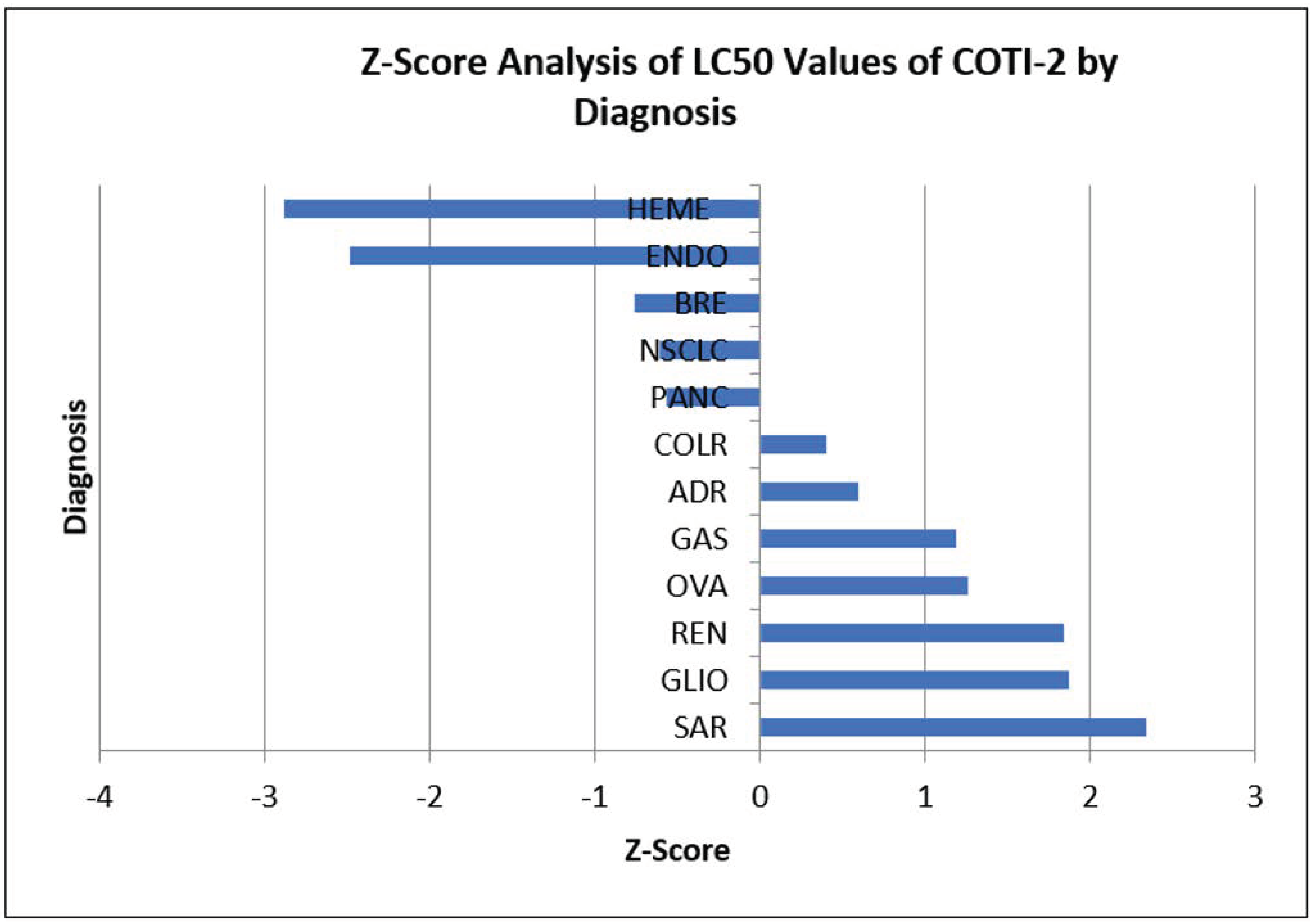{kind=link}
{kind=link}
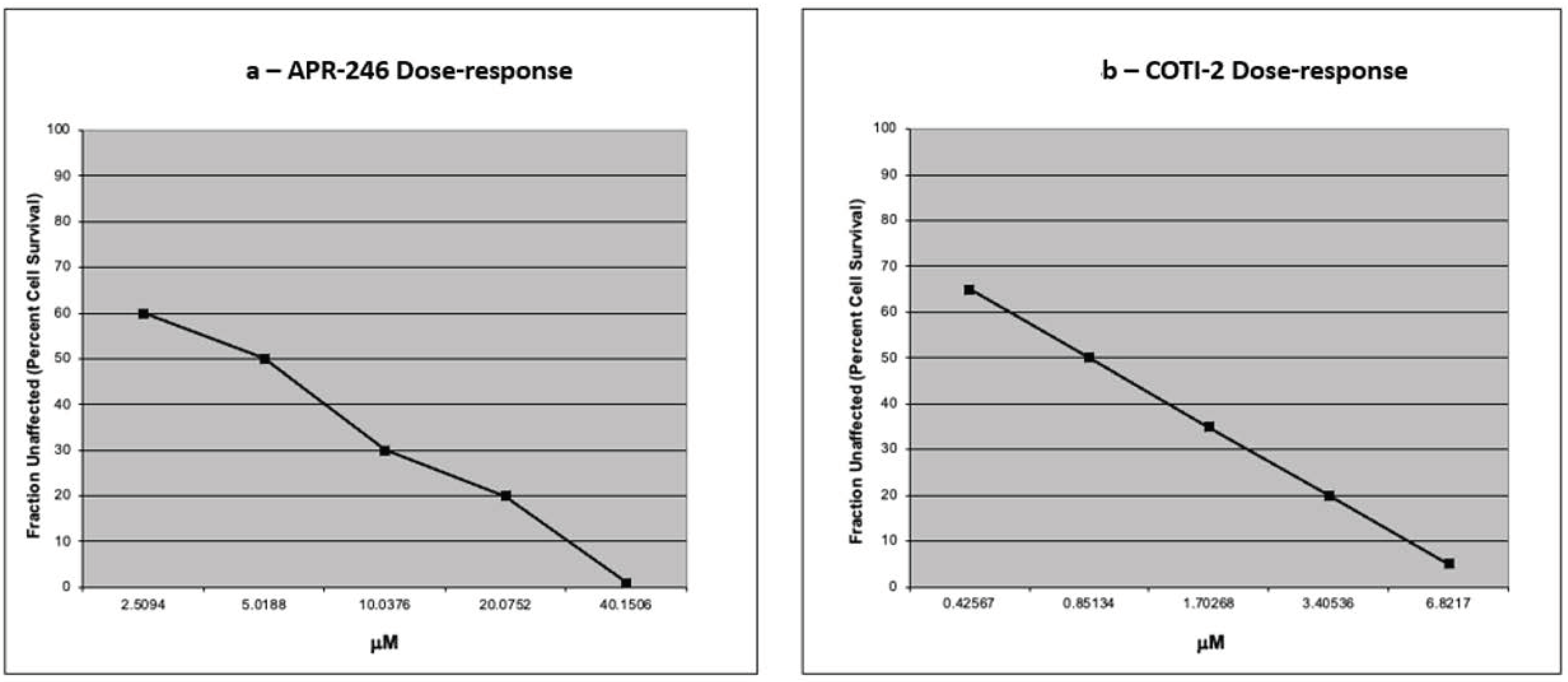{kind=link}
| Drugs Correlated with COTI-2 and APR-246 | R Value | p Value | Bonferroni Significance | |
|---|---|---|---|---|
| APR-246 | AZD6 | 0.005 | 0.978 | No |
| CISGEM | 0.112 | 0.450 | No | |
| DOX | 0.413 | 0.015 | No | |
| NM | 0.173 | 0.344 | No | |
| TAX | 0.194 | 0.278 | No | |
| CDDP | −0.258 | 0.213 | No | |
| COTI-2 | 0.103 | 0.381 | No | |
| COTI-2 | AZD6 | 0.321 | 0.00037 | Yes |
| CISGEM | 0.330 | 0.000012 | Yes | |
| DOX | 0.347 | 0.000752 | Yes | |
| NM | 0.449 | 0.000019 | Yes | |
| TAX | −0.036 | 0.726282 | No | |
| CDDP | 0.307 | 0.013542 | No | |
| APR-246 | 0.103 | 0.381 | No | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagourney, A.J.; Gipoor, J.B.; Evans, S.S.; D’Amora, P.; Duesberg, M.S.; Bernard, P.J.; Francisco, F.; Nagourney, R.A. Therapeutic Targeting of P53: A Comparative Analysis of APR-246 and COTI-2 in Human Tumor Primary Culture 3-D Explants. Genes 2023, 14, 747. https://doi.org/10.3390/genes14030747
Nagourney AJ, Gipoor JB, Evans SS, D’Amora P, Duesberg MS, Bernard PJ, Francisco F, Nagourney RA. Therapeutic Targeting of P53: A Comparative Analysis of APR-246 and COTI-2 in Human Tumor Primary Culture 3-D Explants. Genes. 2023; 14(3):747. https://doi.org/10.3390/genes14030747
Chicago/Turabian StyleNagourney, Adam J., Joshua B. Gipoor, Steven S. Evans, Paulo D’Amora, Max S. Duesberg, Paula J. Bernard, Federico Francisco, and Robert A. Nagourney. 2023. "Therapeutic Targeting of P53: A Comparative Analysis of APR-246 and COTI-2 in Human Tumor Primary Culture 3-D Explants" Genes 14, no. 3: 747. https://doi.org/10.3390/genes14030747