
Reduced OPA1, Mitochondrial Fragmentation and Increased Susceptibility to Apoptosis in Granular Corneal Dystrophy Type 2 Corneal Fibroblasts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies, Inhibitors and Treatments
2.2. Isolation and Culture of Corneal Fibroblasts
2.3. Cell Viability Assay
2.4. Preparation of Cell Lysates and Western Blot Analysis
2.5. Mitochondrial Fractionation
2.6. Quantification of mtDNA Copy Number and Deletion
2.7. Mitochondrial Network Imaging by Fluorescence and Electron Microscopy
2.8. Statistical Analysis
3. Results
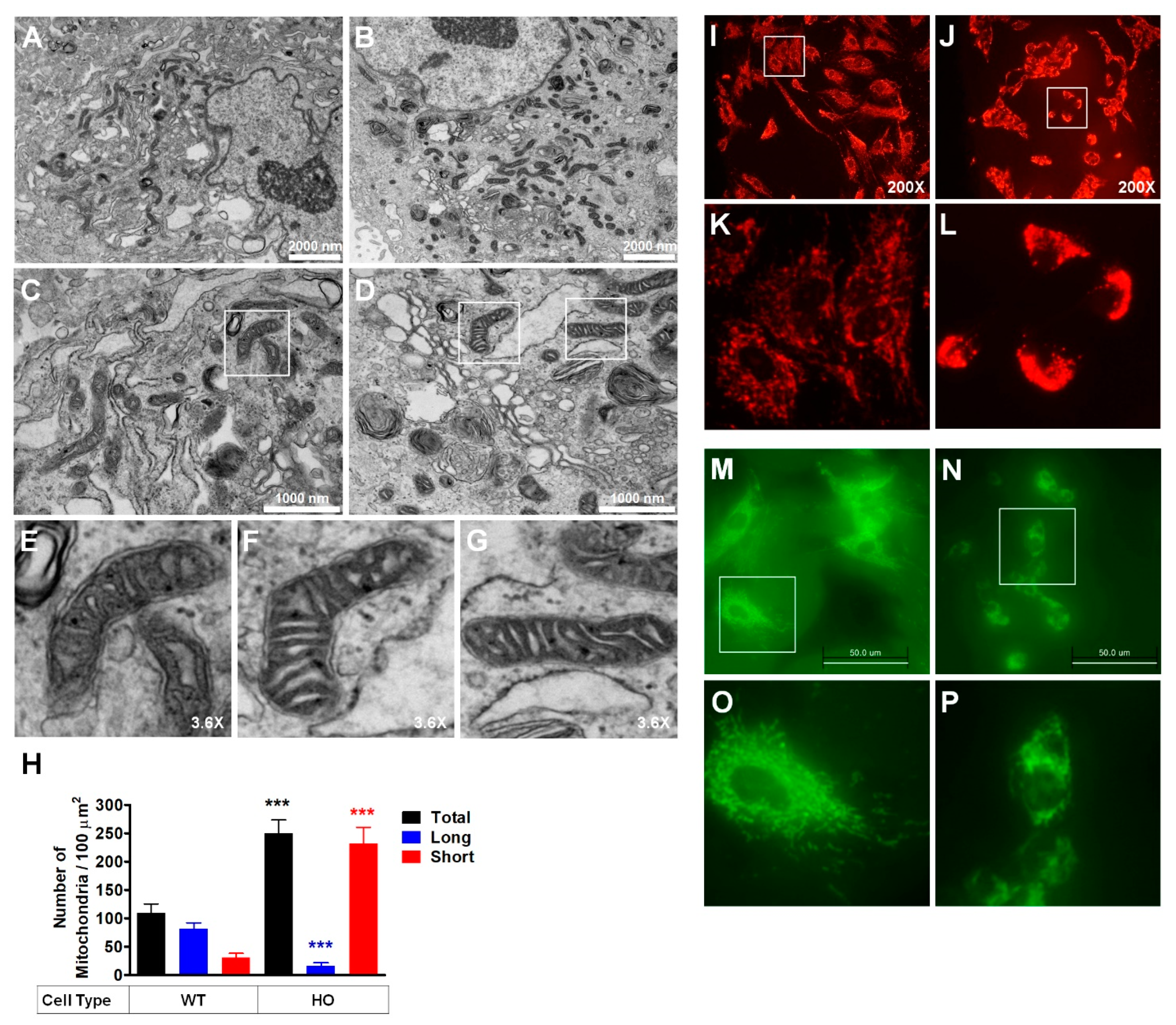
3.1. Abnormal Mitochondrial Morphology in GCD2 Corneal Fibroblasts
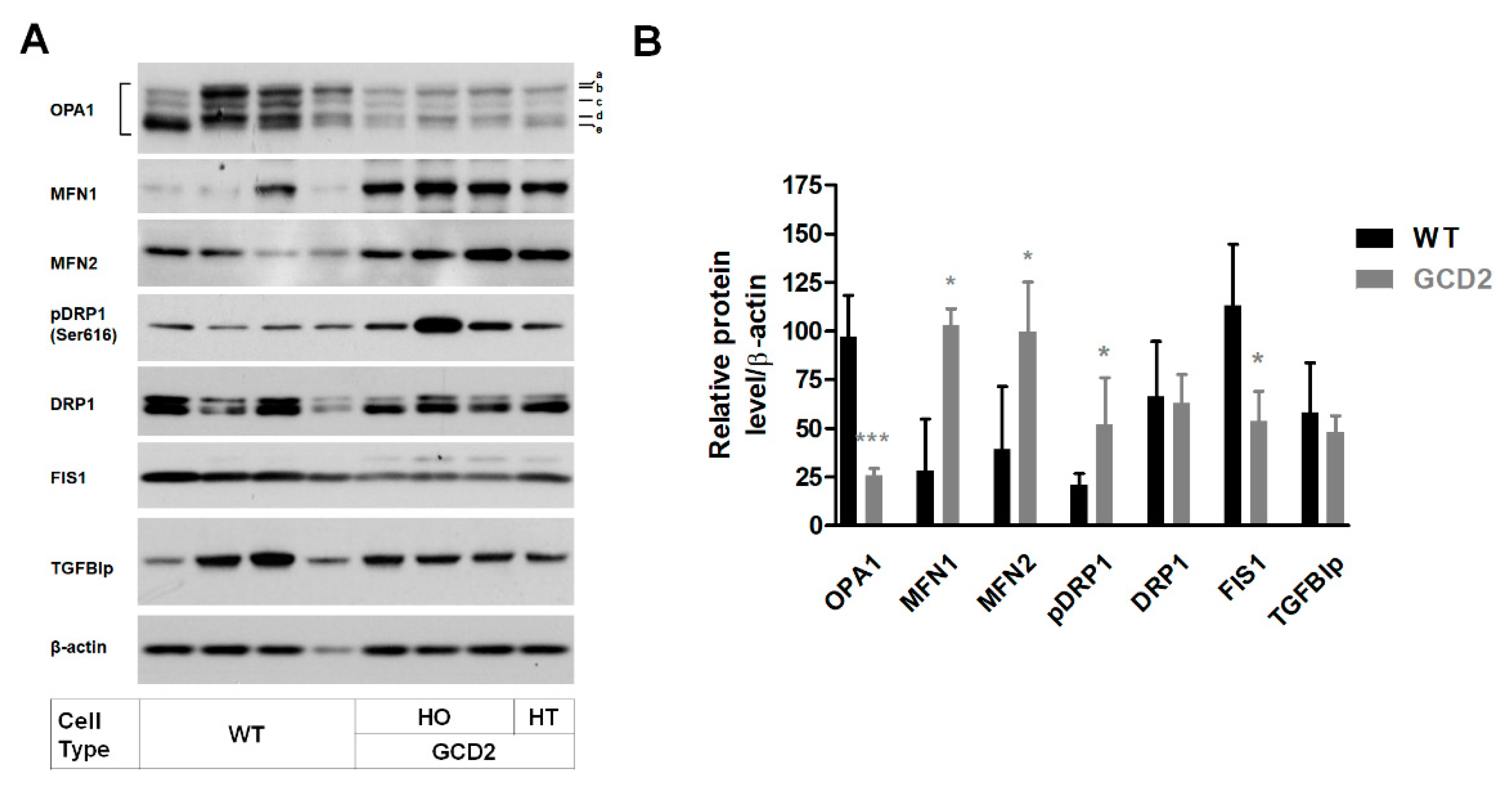
3.2. Altered Levels of Mitochondrial Dynamics-Related Proteins in GCD2 Corneal Fibroblasts
3.3. OPA1 Reduction as the Major Factor in Mitochondrial Abnormalities in GCD2 Corneal Fibroblasts
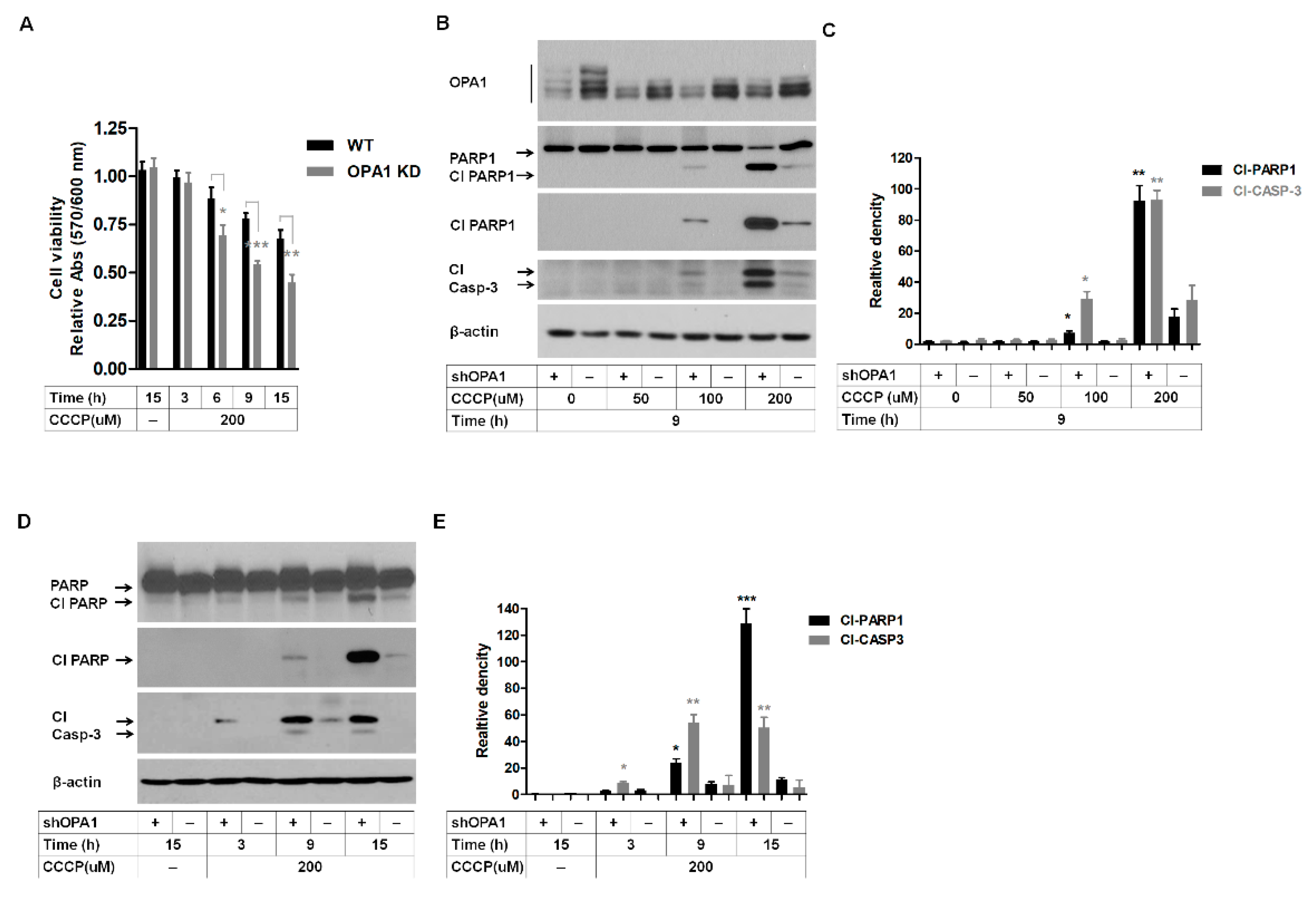
3.4. Reduction of OPA1 Level Increases the Susceptibility of Corneal Fibroblasts to Mitochondrial Stress-Induced Cell Death
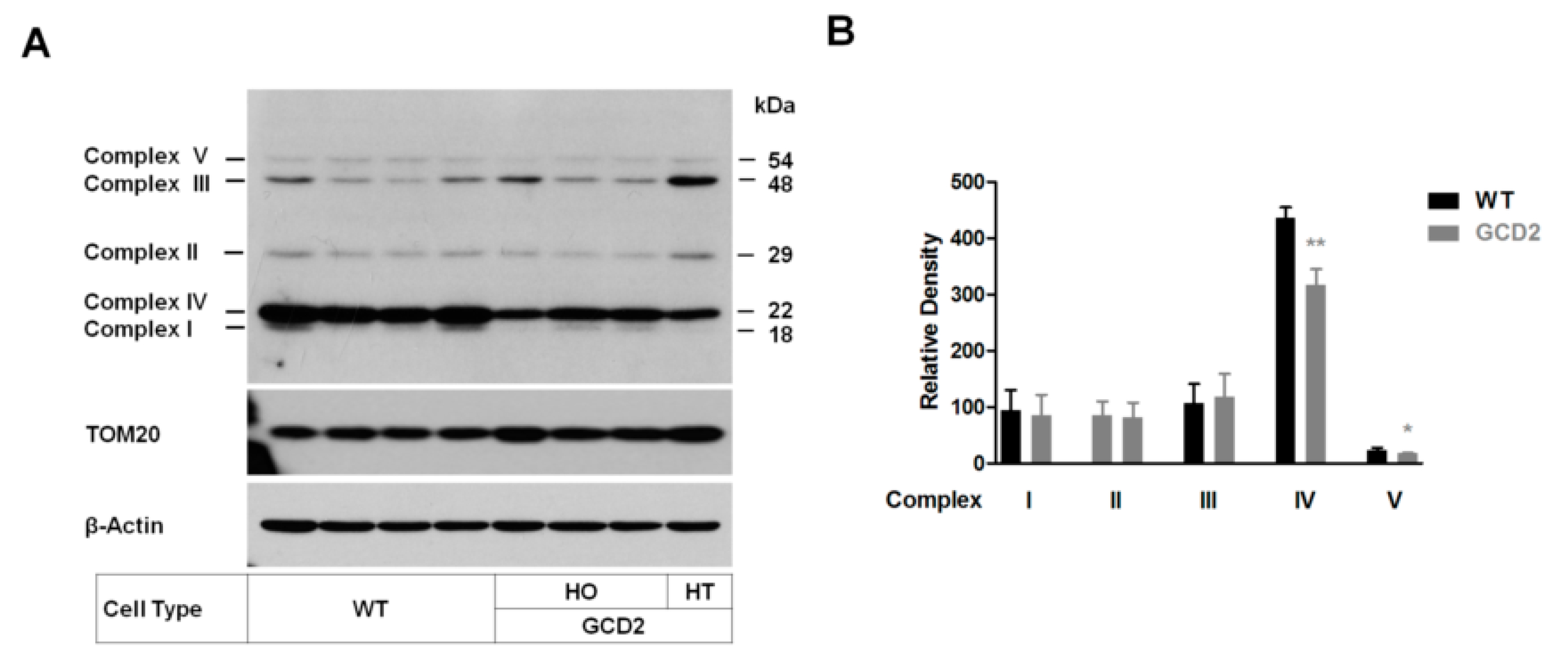
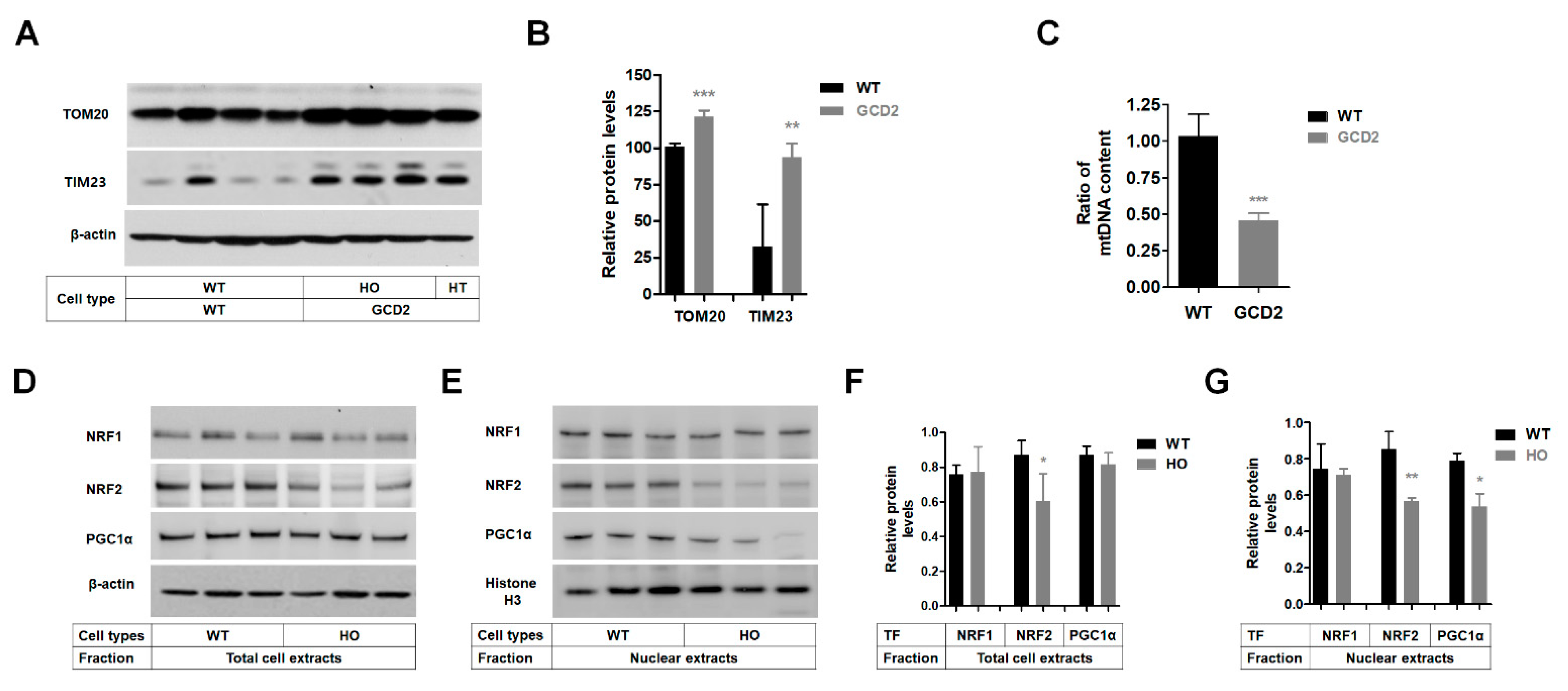
3.5. Altered Levels of OXPHOS Complex Proteins and mtDNA Content in GCD2 Corneal Fibroblasts
3.6. Mitochondrial Biogenesis Protein Levels Are Reduced in GCD2 Corneal Fibroblasts
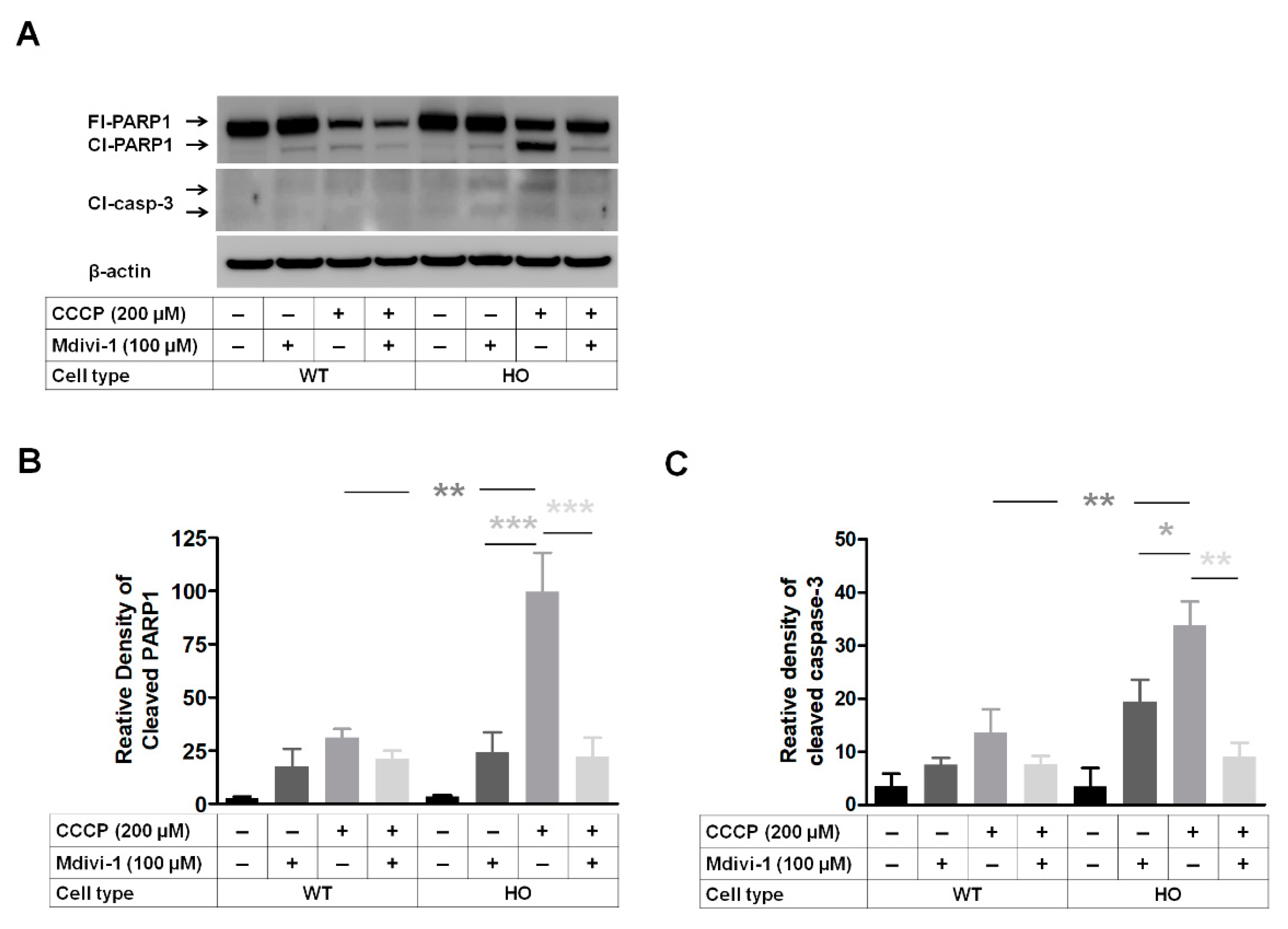
3.7. DRP1 Inhibitor Mdivi-1 Prevents Apoptosis Induced by Mitochondrial Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Munier, F.L.; Korvatska, E.; Djemai, A.; Le Paslier, D.; Zografos, L.; Pescia, G.; Schorderet, D.F. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat. Genet. 1997, 15, 247–251. [Google Scholar] [CrossRef]
- Skonier, J.; Neubauer, M.; Madisen, L.; Bennett, K.; Plowman, G.D.; Purchio, A.F. cDNA cloning and sequence analysis of beta ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol. 1992, 11, 511–522. [Google Scholar] [CrossRef]
- Klintworth, G.K. Advances in the molecular genetics of corneal dystrophies. Am. J. Ophthalmol. 1999, 128, 747–754. [Google Scholar] [CrossRef]
- Korvatska, E.; Henry, H.; Mashima, Y.; Yamada, M.; Bachmann, C.; Munier, F.L.; Schorderet, D.F. Amyloid and non-amyloid forms of 5q31-linked corneal dystrophy resulting from kerato-epithelin mutations at Arg-124 are associated with abnormal turnover of the protein. J. Biol. Chem. 2000, 275, 11465–11469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberto, D.; Garello, R. Corneal Sublayers Thickness Estimation Obtained by High-Resolution FD-OCT. Int. J. Biomed. Imaging 2013, 2013, 989624. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D. Development of the vertebrate cornea. Int. Rev. Cytol. 1980, 63, 263–322. [Google Scholar] [PubMed]
- Garcia, B.; Garcia-Suarez, O.; Merayo-Lloves, J.; Alcalde, I.; Alfonso, J.F.; Cueto, L.F.V.; Meana, A.; Vazquez, F.; Quiros, L.M. Differential Expression of Proteoglycans by Corneal Stromal Cells in Keratoconus. Investig. Ophth. Vis. Sci. 2016, 57, 2618–2628. [Google Scholar] [CrossRef] [Green Version]
- Hassell, J.R.; Kane, B.P.; Etheredge, L.T.; Valkov, N.; Birk, D.E. Increased stromal extracellular matrix synthesis and assembly by insulin activated bovine keratocytes cultured under agarose. Exp. Eye Res. 2008, 87, 604–611. [Google Scholar] [CrossRef] [Green Version]
- Hassell, J.R.; Birk, D.E. The molecular basis of corneal transparency. Exp. Eye Res. 2010, 91, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Bereiter-Hahn, J.; Voth, M. Dynamics of mitochondria in living cells: Shape changes, dislocations, fusion, and fission of mitochondria. Microsc. Res. Tech. 1994, 27, 198–219. [Google Scholar] [CrossRef] [PubMed]
- Frazier, A.E.; Kiu, C.; Stojanovski, D.; Hoogenraad, N.J.; Ryan, M.T. Mitochondrial morphology and distribution in mammalian cells. Biol. Chem. 2006, 387, 1551–1558. [Google Scholar] [CrossRef]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [Green Version]
- Hermann, G.J.; Shaw, J.M. Mitochondrial dynamics in yeast. Annu. Rev. Cell Dev. Biol. 1998, 14, 265–303. [Google Scholar] [CrossRef]
- Griparic, L.; van der Wel, N.N.; Orozco, I.J.; Peters, P.J.; van der Bliek, A.M. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 2004, 279, 18792–18798. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Herlan, M.; Vogel, F.; Bornhovd, C.; Neupert, W.; Reichert, A.S. Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J. Biol. Chem. 2003, 278, 27781–27788. [Google Scholar] [CrossRef] [Green Version]
- Elachouri, G.; Vidoni, S.; Zanna, C.; Pattyn, A.; Boukhaddaoui, H.; Gaget, K.; Yu-Wai-Man, P.; Gasparre, G.; Sarzi, E.; Delettre, C.; et al. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome. Res. 2011, 21, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [Green Version]
- Cipolat, S.; Rudka, T.; Hartmann, D.; Costa, V.; Serneels, L.; Craessaerts, K.; Metzger, K.; Frezza, C.; Annaert, W.; D′Adamio, L.; et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 2006, 126, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, R.; Lartigue, L.; Perkins, G.; Scott, R.T.; Dixit, A.; Kushnareva, Y.; Kuwana, T.; Ellisman, M.H.; Newmeyer, D.D. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol. Cell 2008, 31, 557–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnaune-Pelloquin, L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. Embo. Rep. 2010, 11, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Sanchis-Gomar, F.; Garcia-Gimenez, J.L.; Gomez-Cabrera, M.C.; Pallardo, F.V. Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches. Curr. Pharm. Des. 2014, 20, 5619–5633. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.I.; Kim, T.I.; Kim, K.S.; Kim, B.Y.; Ahn, S.Y.; Cho, H.J.; Lee, H.K.; Cho, H.S.; Kim, E.K. Decreased catalase expression and increased susceptibility to oxidative stress in primary cultured corneal fibroblasts from patients with granular corneal dystrophy type II. Am. J. Pathol. 2009, 175, 248–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.I.; Kim, H.; Lee, D.J.; Choi, S.I.; Kang, S.W.; Kim, E.K. Altered mitochondrial function in type 2 granular corneal dystrophy. Am. J. Pathol. 2011, 179, 684–692. [Google Scholar] [CrossRef]
- Choi, S.I.; Kim, B.Y.; Dadakhujaev, S.; Oh, J.Y.; Kim, T.I.; Kim, J.Y.; Kim, E.K. Impaired autophagy and delayed autophagic clearance of transforming growth factor beta-induced protein (TGFBI) in granular corneal dystrophy type 2. Autophagy 2012, 8, 1782–1797. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.I.; Maeng, Y.S.; Kim, T.I.; Lee, Y.; Kim, Y.S.; Kim, E.K. Lysosomal trafficking of TGFBIp via caveolae-mediated endocytosis. PLoS ONE 2015, 10, e0119561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, A.N.; Shahni, R.; Rodriguez-de-Ledesma, A.; Laftah, A.; Cunningham, P. Mitochondrial DNA as a non-invasive biomarker: Accurate quantification using real time quantitative PCR without co-amplification of pseudogenes and dilution bias. Biochem. Biophys Res. Commun. 2011, 412, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Pfanner, N.; Chacinska, A. The mitochondrial import machinery: Preprotein-conducting channels with binding sites for presequences. Biochim. Biophys Acta 2002, 1592, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissiere, A.; Campos, Y.; Rivera, H.; de la Aleja, J.G.; Carroccia, R.; et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 2008, 131, 338–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu-Wai-Man, P.; Sitarz, K.S.; Samuels, D.C.; Griffiths, P.G.; Reeve, A.K.; Bindoff, L.A.; Horvath, R.; Chinnery, P.F. OPA1 mutations cause cytochrome c oxidase deficiency due to loss of wild-type mtDNA molecules. Hum. Mol. Genet. 2010, 19, 3043–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Disatnik, M.H.; Shen, N.; Sobel, R.A.; Mochly-Rosen, D. Aberrant mitochondrial fission in neurons induced by protein kinase C{delta} under oxidative stress conditions in vivo. Mol. Biol. Cell 2011, 22, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Shurland, D.L.; Ryazantsev, S.N.; van der Bliek, A.M. A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 1998, 143, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olichon, A.; Landes, T.; Arnaune-Pelloquin, L.; Emorine, L.J.; Mils, V.; Guichet, A.; Delettre, C.; Hamel, C.; Amati-Bonneau, P.; Bonneau, D.; et al. Effects of OPA1 mutations on mitochondrial morphology and apoptosis: Relevance to ADOA pathogenesis. J. Cell Physiol. 2007, 211, 423–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Jeong, S.Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, C.; Ashley, N.; Diot, A.; Morten, K.; Phadwal, K.; Williams, A.; Fearnley, I.; Rosser, L.; Lowndes, J.; Fratter, C.; et al. Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology 2017, 88, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.I.; Woo, J.H.; Kim, E.K. Lysosomal dysfunction of corneal fibroblasts underlies the pathogenesis of Granular Corneal Dystrophy Type 2 and can be rescued by TFEB. J. Cell Mol. Med. 2020, 24, 10343–10355. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, A.; Alberdi, E.; Matute, C. Mitochondrial Division Inhibitor 1 (mdivi-1) Protects Neurons against Excitotoxicity through the Modulation of Mitochondrial Function and Intracellular Ca(2+) Signaling. Front. Mol. Neurosci. 2018, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.M.; Yi, Z.; Liu, S.Z.; Chang, J.H.; Dang, X.B.; Li, Q.Y.; Zhang, Y.L. The mitochondrial division inhibitor mdivi-1 attenuates spinal cord ischemia-reperfusion injury both in vitro and in vivo: Involvement of BK channels. Brain Res. 2015, 1619, 155–165. [Google Scholar] [CrossRef]
- Chakravarti, S.; Petroll, W.M.; Hassell, J.R.; Jester, J.V.; Lass, J.H.; Paul, J.; Birk, D.E. Corneal opacity in lumican-null mice: Defects in collagen fibril structure and packing in the posterior stroma. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3365–3373. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.-I.; Lee, G.-H.; Woo, J.-H.; Jun, I.; Kim, E.K. Reduced OPA1, Mitochondrial Fragmentation and Increased Susceptibility to Apoptosis in Granular Corneal Dystrophy Type 2 Corneal Fibroblasts. Genes 2023, 14, 566. https://doi.org/10.3390/genes14030566
Choi S-I, Lee G-H, Woo J-H, Jun I, Kim EK. Reduced OPA1, Mitochondrial Fragmentation and Increased Susceptibility to Apoptosis in Granular Corneal Dystrophy Type 2 Corneal Fibroblasts. Genes. 2023; 14(3):566. https://doi.org/10.3390/genes14030566
Chicago/Turabian StyleChoi, Seung-Il, Ga-Hyun Lee, Jong-Hwan Woo, Ikhyun Jun, and Eung Kweon Kim. 2023. "Reduced OPA1, Mitochondrial Fragmentation and Increased Susceptibility to Apoptosis in Granular Corneal Dystrophy Type 2 Corneal Fibroblasts" Genes 14, no. 3: 566. https://doi.org/10.3390/genes14030566