
Identification of Small RNAs Associated with Salt Stress in Chrysanthemums through High-Throughput Sequencing and Bioinformatics Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Salt Treatment
2.2. Small RNA Sequencing and miRNA Identification
2.3. Degradome Sequencing and Target Identification
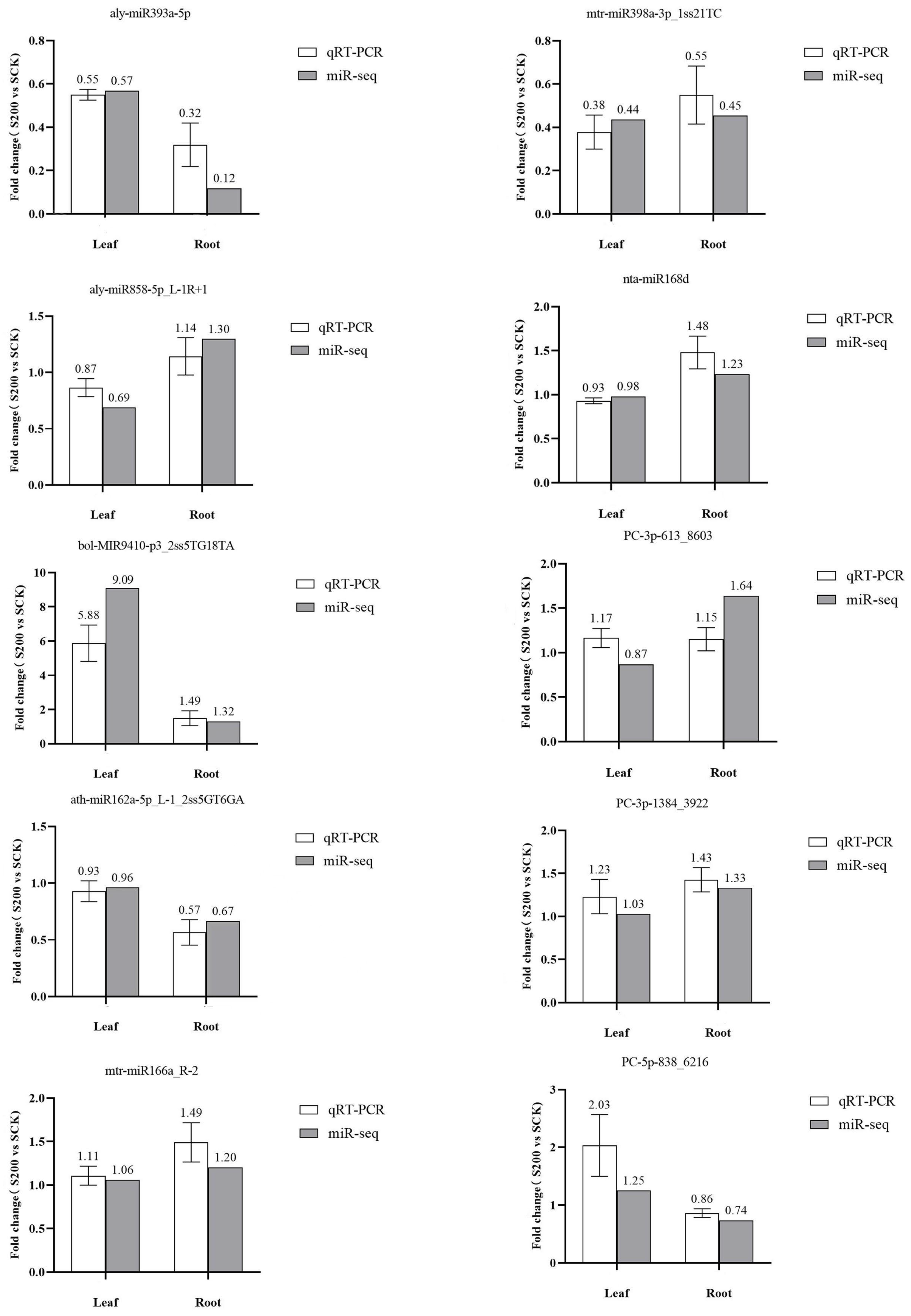
2.4. The Verification of Authenticity of High-throughput Sequencing Data via qRT–PCR
3. Results
3.1. MiRNA Sequence Analysis
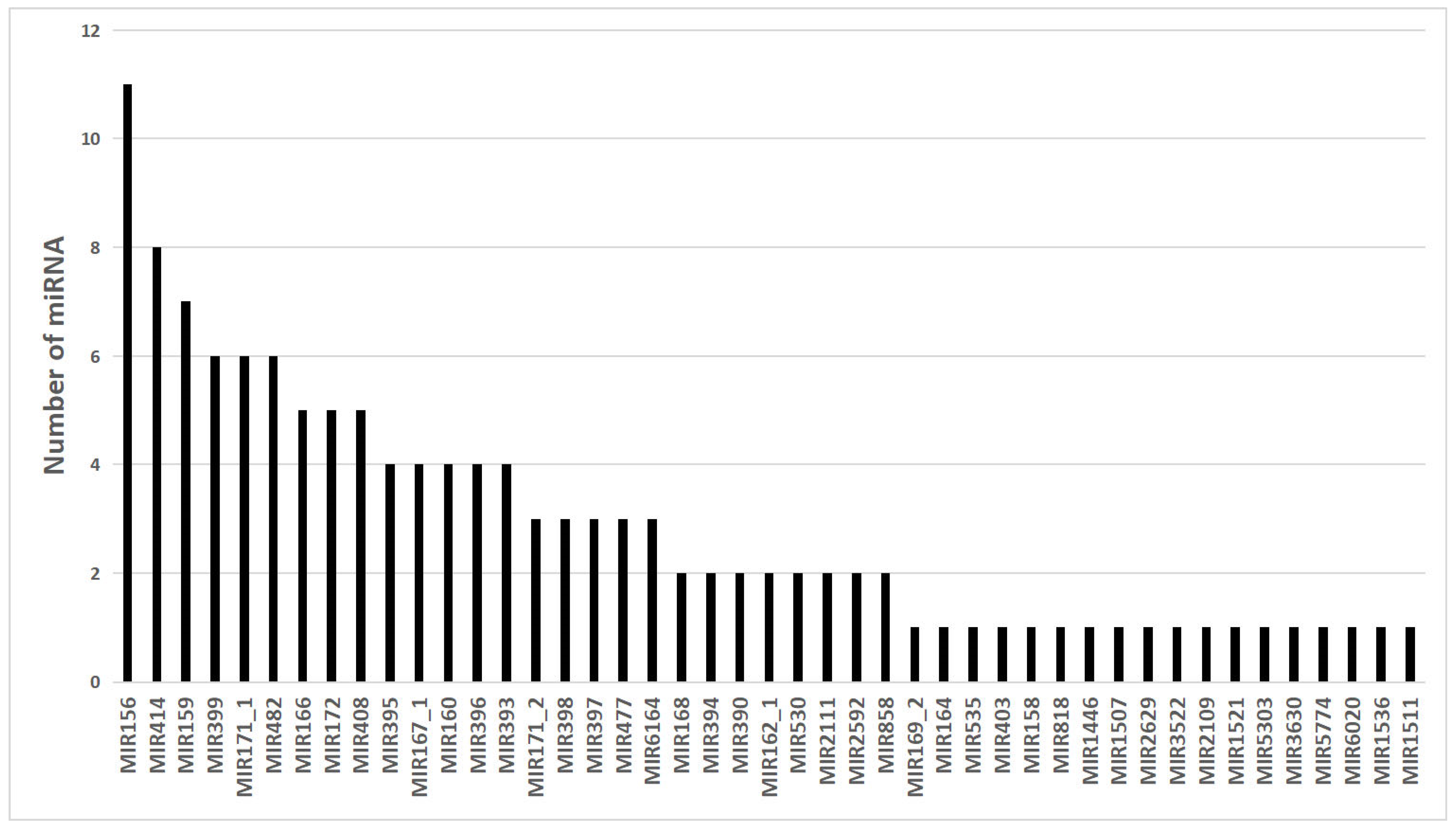
3.2. Identification of Conserved miRNAs
3.3. Identification of Novel miRNAs
3.4. Identification of Differentially Expressed miRNAs
3.5. Validation of miRNA Expression via qRT–PCR
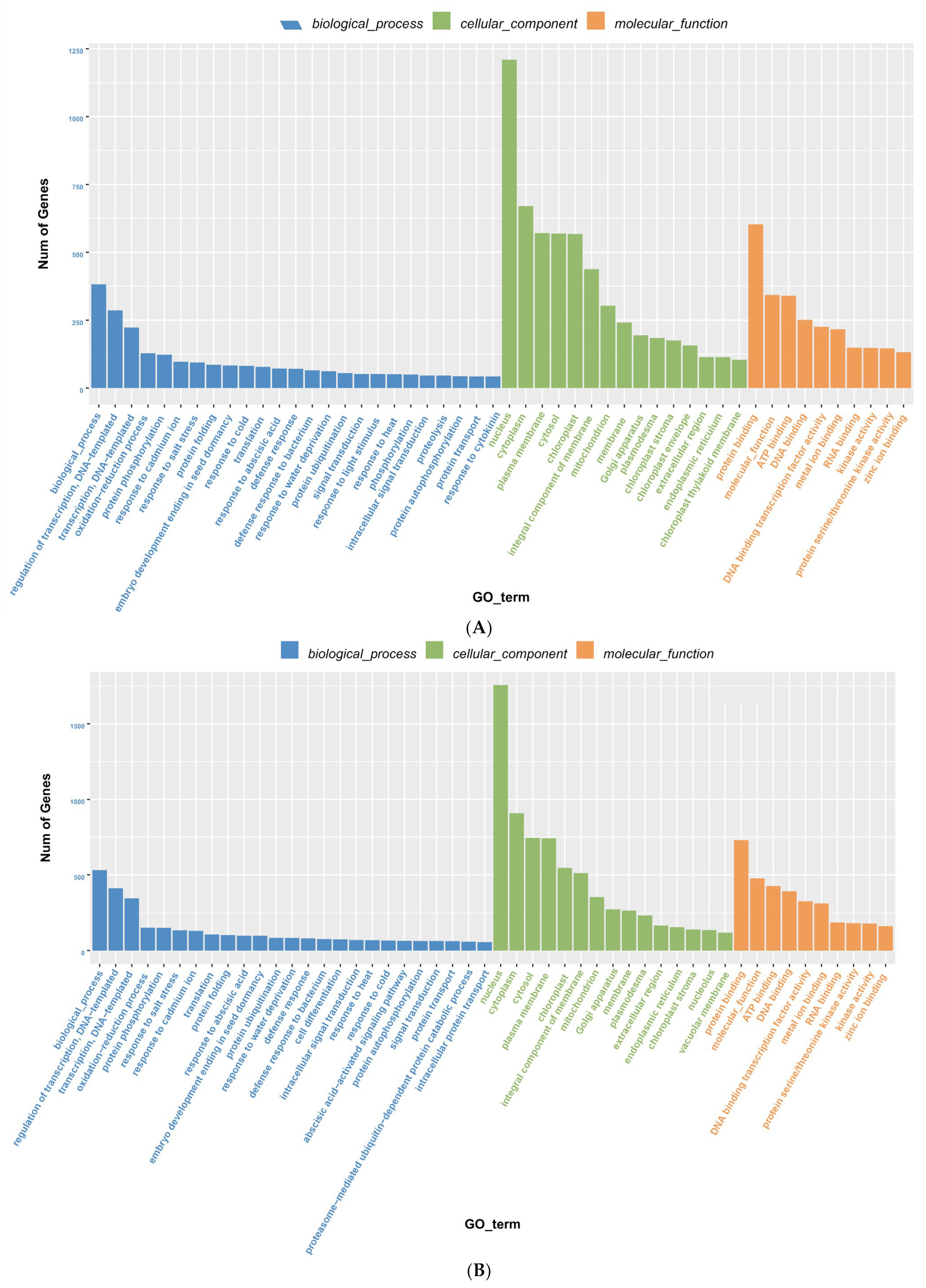
3.6. Degradome Sequencing and GO Enrichment Analysis
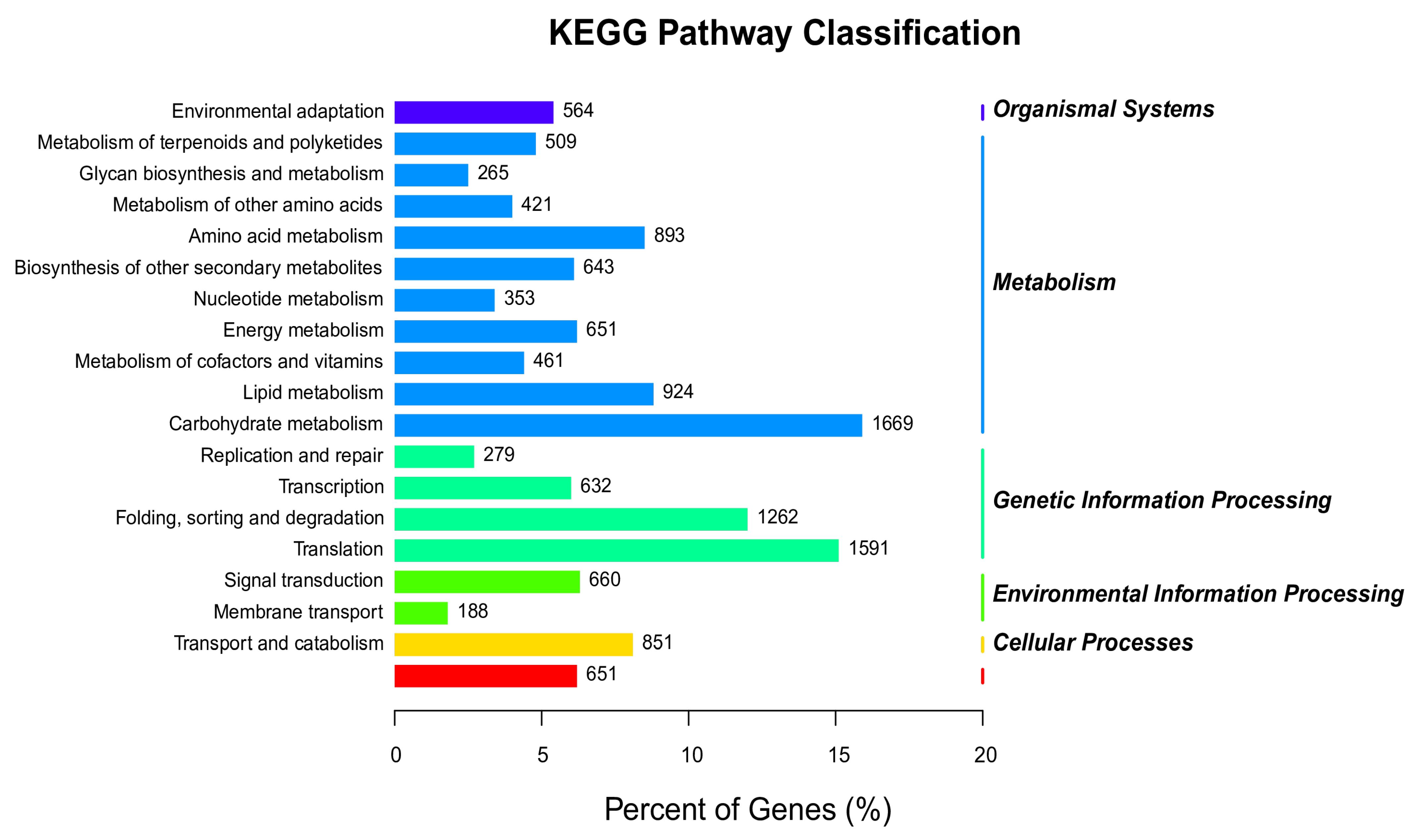
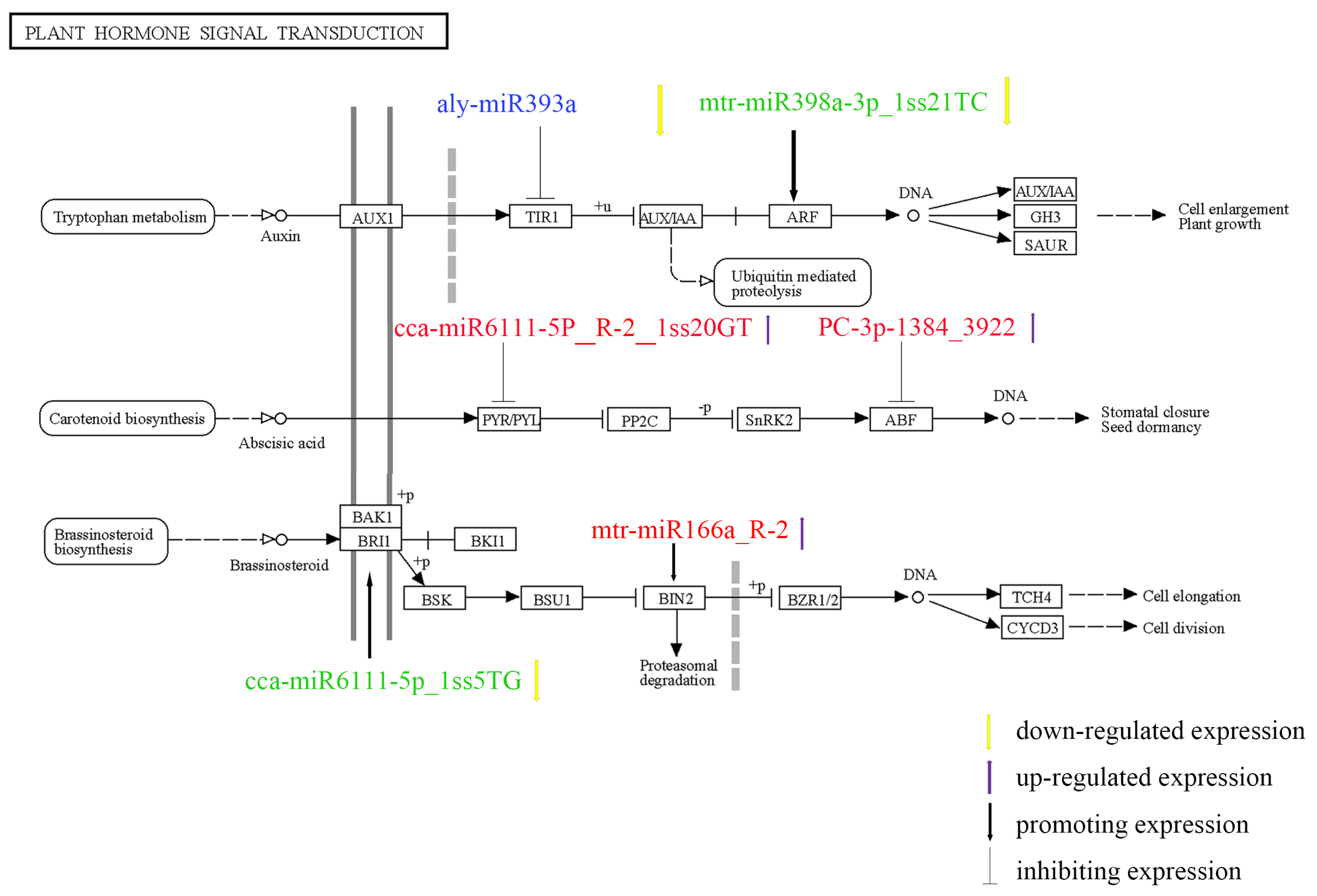
3.7. Analysis of the KEGG Pathway
3.8. Combined Analysis of Significant miRNA-Targets
4. Discussion
4.1. Prediction of Novel miRNAs in Ground-Grown Chrysanthemum Using High-Throughput Sequencing
4.2. Differential miRNA Profiling in Response to Salt Stress of the Ground-Grown Chrysanthemum
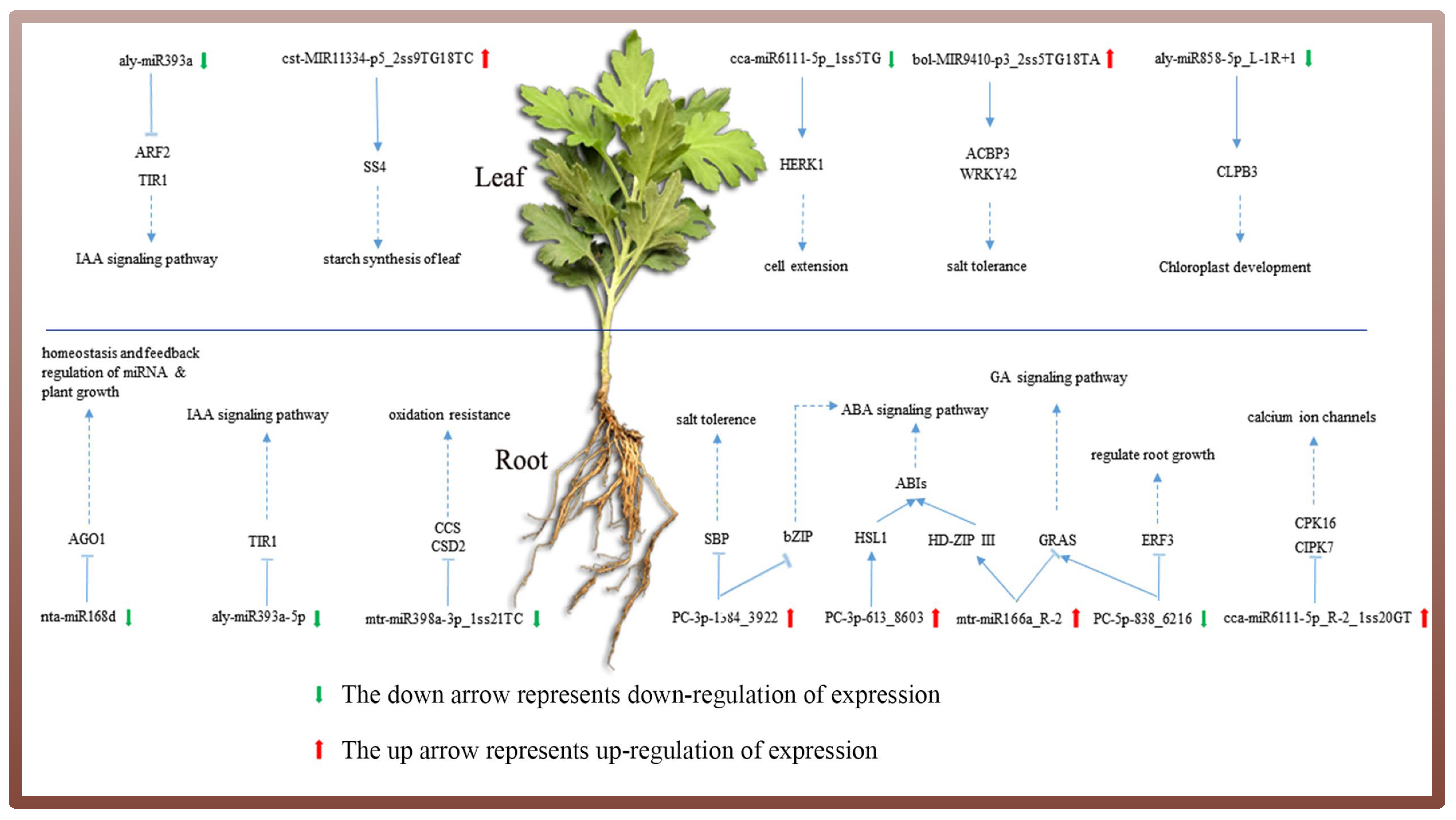
4.3. Combining sRNA and Degradome Sequencing to Analyze the Role of Ground-Grown Chrysanthemum miRNAs in Response to Salt Stress
4.4. MiRNA-Mediated Phytohormone Signal Transduction Pathway Involved in the Salt Stress Response
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Du, H.; Li, S.S.; Wu, Q.; Ji, K.X.; Wu, J.; Liu, Y.; Wang, L.S. Analysis of active compounds and antioxidant activity assessment of six popular Chinese Juhua teas. Nat. Prod. Commun. 2015, 10, 495–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zunqin, W.; Shouquan, Z.; Pei, Y. Saline Soils in China; Science Press: Beijing, China, 1993. [Google Scholar]
- Zhang, J.L.; Shi, H. Physiological and molecular mechanisms of plant salt tolerance. Photosynth. Res. 2013, 115, 1–22. [Google Scholar] [CrossRef]
- Bo, H.; Miao, H.; Bing, D.; Xiaofang, Z. Effects of space mutagenesis on dwarf characters of Chrysanthemum cultivated in open field. J. Plant Res. 2000, 2, 2–4. [Google Scholar]
- Ulrich, D.; Aaron, B.; Julian, I. Plant salt-tolerance mechanisms. Trends Plant Sci. 2014, 19, 371–379. [Google Scholar]
- Cohen, A.; Bray, E.A. Characterization of three mRNAs that accumulate in wilted tomato leaves in response to elevated levels of endogenous abscisic acid. Planta 1990, 182, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Genetic analysis of plant salt tolerance using Arabidopsis. Plant Physiol. 2000, 124, 941–948. [Google Scholar] [CrossRef] [Green Version]
- Sunkar, R.; Zhu, J.K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, F. Arabidopsis endogenous small RNAs: Highways and byways. Trends Plant Sci. 2006, 11, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, P.; Sakvarelidze-Achard, L.; Bruun-Rasmussen, M.; Dunoyer, P.; Yamamoto, Y.Y.; Sieburth, L.; Voinnet, O. Widespread translational inhibition by plant miRNAs and siRNAs. Science 2008, 320, 1185–1190. [Google Scholar] [CrossRef]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolata, J.; Bajczyk, M.; Bielewicz, D.; Niedojadlo, K.; Niedojadlo, J.; Pietrykowska, H.; Walczak, W.; Szweykowska-Kulinska, Z.; Jarmolowski, A. Salt stress reveals a new role for ARGONAUTE1 in miRNA biogenesis at the transcriptional and posttranscriptional levels. Plant Physiol. 2016, 172, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. Plant microRNA: A small regulatory molecule with big impact. Dev. Biol. 2006, 289, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ge, L.; Liang, R.; Li, W.; Ruan, K.; Lin, H.; Jin, Y. Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Mol. Biol. 2009, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Bai, X.; Yang, L.; Lv, D.; Pan, X.; Li, Y.; Cai, H.; Ji, W.; Chen, Q.; Zhu, Y. osa-MIR393: A salinity- and alkaline stress-related microRNA gene. Mol. Biol. Rep. 2011, 38, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, W.; Shen, H.; Zhu, X.; Zhai, L.; Xu, L.; Wang, R.; Gong, Y.; Limera, C.; Liu, L. Identification of radish (Raphanus sativus L.) miRNAs and their target genes to explore miRNA-mediated regulatory networks in lead (Pb) stress responses by high-throughput sequencing and degradome analysis. Plant Mol. Biol. Rep. 2015, 33, 358–376. [Google Scholar] [CrossRef]
- Xin, M.; Wang, Y.; Yao, Y.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol. 2010, 10, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruszka, K.; Pacak, A.; Swida-Barteczka, A.; Nuc, P.; Alaba, S.; Wroblewska, Z.; Karlowski, W.; Jarmolowski, A.; Szweykowska-Kulinska, Z. Transcriptionally and post-transcriptionally regulated microRNAs in heat stress response in barley. J. Exp. Bot. 2014, 65, 6123–6135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.H.; Tian, X.; Li, Y.J.; Wu, C.A.; Zheng, C.C. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 2008, 14, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nodine, M.D.; Bartel, D.P. MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev. 2010, 24, 2678–2692. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, A.; Margaret, Y.G.; Ken, W. An insight into microRNA-156 role in salinity stress responses of alfalfa. Front. Plant Sci. 2017, 8, 356–359. [Google Scholar]
- Ding, D.; Zhang, L.F.; Wang, H.; Liu, Z.; Zhang, Z.; Zheng, Y. Differential expression of miRNAs in response to salt stress in maize roots. Ann. Bot. 2009, 103, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, T.; Liu, Z.; Dai, X.; Xiang, F. Primary root growth in Arabidopsis thaliana is inhibited by the miR159 mediated repression of MYB33, MYB65 and MYB101. Plant Sci. 2017, 262, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, D.; Li, Z.; Hu, Q.; Yang, C.; Zhu, L.; Luo, H. Constitutive expression of a miR319 gene alters plant development and enhances salt and drought tolerance in transgenic creeping bentgrass. Plant Physiol. 2013, 161, 1375–1391. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.L.; Wang, Q.L.; Sun, R.R.; Zhang, B. Deep sequencing reveals important roles of microRNAs in response to drought and salinity stress in cotton. J. Exp. Bot. 2015, 66, 789–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naya, L.; Paul, S.; Valdés-López, O.; Mendoza-Soto, A.B.; Nova-Franco, B.; Sosa-Valencia, G.; Reyes, J.L.; Hernández, G. Regulation of copper homeostasis and biotic interactions by microRNA 398b in common bean. PLoS ONE 2014, 9, e84416. [Google Scholar] [CrossRef]
- Jia, X.Y.; Wang, W.X.; Ren, L.G.; Chen, Q.J.; Mendu, V.; Willcut, B.; Dinkins, R.; Tang, X.; Tang, G. Differential and dynamic regulation of miR398 in response to ABA and salt stress in Populus tremula and Arabidopsis thaliana. Plant Mol. Biol. 2009, 71, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Kapoor, A.; Zhu, J.K. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 2006, 18, 2051–2065. [Google Scholar] [CrossRef] [Green Version]
- Higashi, Y.; Takechi, K.; Takano, H.; Takio, S. Involvement of microRNA in copper deficiency-induced repression of chloroplastic CuZn-superoxide dismutase genes in the moss Physcomitrella patens. Plant Cell Physiol. 2013, 54, 1345–1355. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Kundu, A.; Pal, A. Identification and validation of conserved microRNAs along with their differential expression in roots of Vigna unguiculata grown under salt stress. Plant Cell Tissue Organ Cult. 2011, 105, 233–242. [Google Scholar] [CrossRef]
- Yang, R.; Zeng, Y.; Yi, X.; Zhao, L.; Zhang, Y. Small RNA deep sequencing reveals the important role of microRNAs in the halophyte Halostachys caspica. Plant Biotechnol. J. 2015, 13, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Sachin, A.G.; Birendra Prasad, S. Novel and conserved miRNAs in the halophyte Suaeda maritima identified by deep sequencing and computational predictions using the ESTs of two mangrove plants. BMC Plant Biol. 2015, 15, 1–18. [Google Scholar]
- Li, X.; Shahid, M.Q.; Wu, J.; Wang, L.; Liu, X.; Lu, Y. Comparative small RNA analysis of pollen development in autotetraploid and diploid rice. Int. J. Mol. Sci. 2016, 17, 499. [Google Scholar] [CrossRef] [Green Version]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2008, 25, 130–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real time quantitative PCR and the 2-ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, Y.; Lou, X.; Li, H.; Zhang, C. Identification of Blueberry miRNAs and Their Targets Based on High-Throughput Sequencing and Degradome Analyses. Int. J. Mol. Sci. 2018, 19, 983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Zhu, P.; Kang, H.; Liu, L.; Cao, Q.; Sun, J.; Dong, T.; Zhu, M.; Li, Z.; Xu, T. High-throughput deep sequencing reveals the important role that microRNAs play in the salt response in sweet potato (Ipomoea batatas L.). BMC Genom. 2020, 21, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyaraj, A.; Liu, S.; Zhang, X.; Zhang, R.; Shangguan, M.; Wei, G. Genome-wide identification of microRNAs responsive to Ectropis oblique feeding in tea plant (Camellia sinensis L.). Sci. Rep. 2017, 7, 13634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Dai, X.R.; Huang, X.; Xu, M.X.; Wang, Q.; Yan, X.J.; Sederoff, R.R.; Li, Q.Z. Single-cell RNA sequencing reveals a high-resolution cell atlas of xylem in Populus. J. Integr. Plant Biol. 2021, 63, 1906–1921. [Google Scholar] [CrossRef]
- Ning, L.; Du, W.; Song, H.; Shao, H.; Qi, W.; Sheteiwy, M.S.A.; Yu, D. Identification of responsive miRNAs involved in combination stresses of phosphate starvation and salt stress in soybean root. Environ. Exp. Bot. 2019, 167, 103823. [Google Scholar] [CrossRef]
- Moxon, S.; Jing, R.; Szittya, G.; Schwach, F.; Rusholme Pilcher, R.L.; Moulton, V.; Dalmay, T. Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res. 2008, 18, 1602–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Guo, J.; Cheng, J.; Jiang, Z.; Xu, N.; An, X.; Chen, Z.; Hao, J.; Yang, S.; Xu, Z.; et al. Identification of Uv-b radiation responsive microRNAs and their target genes in Chrysanthemum (chrysanthemum Morifolium Ramat) using high-throughput sequencing. Ind. Crops Prod. 2020, 151, 112484. [Google Scholar] [CrossRef]
- Brodersen, P.; Voinnet, O. The diversity of RNA silencing pathways in plants. Trends Genet. 2006, 22, 268–280. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, H.Y.; Zhang, Q.Q.; Zhang, J.; Ni, F.; Liu, C.; Qi, Y. DNA methylation mediated by a microRNA pathway. Mol. Cell. 2010, 38, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Cannon, C.H.; Cobb, G.P.; Anderson, T.A. Conservation and divergence of plant microRNA genes. Plant J. 2006, 46, 243–259. [Google Scholar] [CrossRef]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that miRNAs are different from other RNAs. Cell. Mol. Life Sci. 2006, 63, 246–254. [Google Scholar] [CrossRef]
- Joshi, R.; Gupta, P.; Singla-Pareek, S.L.; Pareek, A. Biomass production and salinity response in plants: Role of microRNAs. Indian J. Plant Physiol. 2017, 22, 448–457. [Google Scholar] [CrossRef]
- Jian, X.Y.; Zhang, L.; Li, G.L.; Zhang, L.; Wang, X.J.; Cao, X.F.; Fang, X.H.; Chen, F. Identification of novel stress-regulated microRNAs from Oryza sativa L. Genomics 2010, 95, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Frazier, T.P.; Sun, G.; Burklew, C.E.; Zhang, B. Salt and drought stresses induce the aberrant expression of microRNA genes in tobacco. Mol. Biotechnol. 2011, 49, 159–165. [Google Scholar] [CrossRef]
- Jia, X.; Ding, N.; Fan, W.; Yan, J.; Gu, Y.; Tang, X.; Li, R.; Tang, G. Functional plasticity of mir165/166 in plant development revealed by small tandem target mimic. Plant Sci. 2015, 233, 11–21. [Google Scholar] [CrossRef]
- Yan, J.; Zhao, C.; Zhou, J.; Yang, Y.; Wang, P.; Zhu, X.; Tang, G.; Bressan, R.A.; Zhu, J.K. The mir165/166 mediated regulatory module plays critical roles in aba homeostasis and response in Arabidopsis thaliana. PLoS Genet. 2016, 12, e1006416. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias, A.A.; Reyes, J.L. Post-transcriptional gene regulation of salinity and drought responses by plant microRNAs. Plant Cell Environ. 2010, 33, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Hu, L.Z.; Han, N.; Hu, J.Q.; Yang, Y.J.; Xiang, T.H.; Zhang, X.J.; Wang, L.L. Overexpression of a miR393-resistant form of transport inhibitor response Protein 1 (mTIR1) enhances salt tolerance by increased osmoregulation and Na+ exclusion in Arabidopsis thaliana. Plant Cell Physiol. 2015, 56, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Data on plant biotechnology discussed by researchers at Sichuan Agricultural University. Biotechnology-Plant Biotechnology; Biotech Week 2019.
- Zhao, J.; Yuan, S.; Zhou, M.; Yuan, N.; Li, Z.; Hu, Q.; Bethea, F.G., Jr.; Liu, H.; Li, S.; Luo, H. Transgenic creeping bentgrass overexpressing Osa-miR393a exhibits altered plant development and improved multiple stress tolerance. Plant Biotechnol. J. 2019, 17, 233–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauclair, L.; Yu, A.; Bouché, N. microRNA-directed cleavage and translational repression of the copper chaperone for superoxide dismutase mRNA in Arabidopsis. Plant J. 2010, 62, 454–462. [Google Scholar] [CrossRef]
- Lu, Y.Z.; Zhen, F.; Bian, L.Y.; Xie, H.; Liang, J.S. MiR398 regulation in rice of the responses to abiotic and biotic stresses depends on CSD1 and CSD2 expression. Funct. Plant Biol. 2010, 38, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, G.; Saini, A.; Sunkar, R. Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta 2009, 229, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Vaucheret, H.; Vazquez, F.; Crété, P.; Bartel, D.P. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 2004, 18, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Vaucheret, H. AGO1 homeostasis involves differential production of 21-nt and 22-nt miR168 Species by MIR168a and MIR168b. Plos One 2009, 4, e6442. [Google Scholar] [CrossRef] [PubMed]
- Boudsocq, M.; Sheen, J. CDPKs in immune and stress signaling. Trends Plant Sci. 2013, 18, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, A.N.; Kudla, J.; Sanders, D. The language of calcium signaling. Annu. Rev. Plant Biol. 2010, 61, 593–620. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.R.; Sha, L.V.L.; Cheng, J.; SI, J.N.; Zhao, R. The role of CDPKs in plant adaptation to abiotic stress. Plant Physiol. 2014, 51, 1387–1394. [Google Scholar]
- Sateesh, K.; Kevin, R. EAR motif-mediated transcriptional repression in plants: An underlying mechanism for epigenetic regulation of gene expression. Epigenetics 2011, 6, 141–146. [Google Scholar]
- Wei, R.; Lin, Q.; Aiyun, W.; Xingguo, Y.; Lipu, D.; Hongxia, L.; Xin, Z.; Zhang, Z. The ERF transcription factor TaERF3 promotes tolerance to salt and drought stresses in wheat. Plant Biotechnol. 2014, 12, 468–479. [Google Scholar]
- Sun, X.; Jones, W.T.; Rikkerink, E.H. GRAS proteins: The versatile roles of intrinsically disordered proteins in plant signalling. Biochem. J. 2012, 442, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.Y.; Wu, H.Y.; Li, X. Identification and expression of GRAS family genes in maize (Zea mays L.). PLoS ONE 2017, 12, e0185418. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Liu, J.; Yang, Z.E.; Chen, E.Y.; Zhang, C.J.; Zhang, X.Y. Genome-wide analysis of GRAS transcription factor gene family in Gossypium hirsutum L. BMC Genom. 2018, 19, 1–12. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, P.; Wu, S.; Lu, Y.; Sun, J.; Cao, Q.; Li, Z.; Xu, T. Identification and expression analysis of GRAS transcription factors in the wild relative of sweet potato Ipomoea trifida. BMC Genom. 2019, 20, 911. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Srivastava, A.K.; Suprasanna, P.; D’Souza, S.F. Identification and profiling of arsenic stress-induced microRNAs in Brassica juncea. J. Exp. Bot. 2013, 64, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Achard, P.; Herr, A.; Baulcombe, D.C.; Harberd, N.P. Modulation of floral development by a gibberellin-regulated microRNA. Development 2004, 131, 3357–3365. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, M.J.; Terrile, M.C.; Bartoli, C.G.; D’Ippólito, S.; Casalongué, C.A. Auxin signaling participates in the adaptative response against oxidative stress and salinity by interacting with redox metabolism in Arabidopsis. Plant Mol. Biol. 2010, 74, 215–222. [Google Scholar] [CrossRef]
- Chen, Z.H.; Bao, M.L.; Sun, Y.Z.; Yang, Y.J.; Xu, X.H.; Wang, J.H.; Han, N.; Bian, H.W.; Zhu, M.Y. Regulation of auxin response by miR393-targeted transport inhibitor response protein 1 is involved in normal development in Arabidopsis. Plant Mol. Biol. 2011, 77, 619–629. [Google Scholar] [CrossRef]
- Suzuki, N.; Bassil, E.; Hamilton, J.S.; Inupakutika, M.A.; Zandalinas, S.I.; Tripathy, D.; Luo, Y.; Dion, E.; Fukui, G.; Kumazaki, A. ABA is required for plant acclimation to a combination of salt and heat stress. PLoS ONE 2016, 11, e0147625. [Google Scholar] [CrossRef] [Green Version]
- Sripinyowanich, S.; Klomsakul, P.; Boonburapong, B.; Bangyeekhun, T.; Asami, T.; Gu, H.; Buaboocha, T.; Chadchawan, S. Exogenous ABA induces salt tolerance in indica rice (Oryza sativa L.): The role of OsP5CS1 and OsP5CR gene expression during salt stress. Environ. Exp. Bot. 2013, 86, 94–105. [Google Scholar] [CrossRef]
- Kim, S.; Kang, J.Y.; Cho, D.I.; Park, J.H.; Kim, S.Y. ABF2, an ABRE-binding bZIP factor, is an essential component of glucose signaling and its overexpression affects multiple stress tolerance. Plant J. 2004, 40, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Fujita, M.; Satoh, R.; Maruyama, K.; Parvez, M.M.; Seki, M.; Hiratsu, K.; Ohme-Takagi, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AREB1 is a transcription activator of novel ABRE-dependent ABA signaling that enhances drought stress tolerance in Arabidopsis. Plant Cell 2005, 17, 3470–3488. [Google Scholar] [CrossRef] [Green Version]
- YAN, H.J.; Zhao, Y.F.; Shi, H.; Li, J.; Wang, Y.C.; Tang, D.C. Brassinosteroid Signaling KINASE1 phosphorylates MAPKKK5 to regulate immunity in Arabidopsis. Plant Physiol. 2018, 176, 2991–3002. [Google Scholar] [CrossRef] [Green Version]
- Moumita, S.; Srivastava Anjil, K.; Beatriz, O.P.; Alberto, C.; Cunjin, Z.; Ari, S. SUMO conjugation to BZR1 enables brassinosteroid signaling to integrate environmental cues to shape plant growth. Curr. Biol. 2021, 31, 668–669. [Google Scholar]
- Liu, Y.Q.; Wang, H.R.; Jiang, Z.M.; Wang, W.; Xu, R.N.; Wang, Q.H.; Zhang, Z.H. Genomic basis of geographical adaptation to soil nitrogen in rice. Nature 2021, 590, 600–605. [Google Scholar] [CrossRef] [PubMed]
- She, J.; Han, Z.; Zhou, B.; Chai, J. Structural basis for differential recognition of brassinolide by its receptors. Protein Cell 2013, 4, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.H.; Wang, H.J.; Qiao, S.L.; Leng, L.N.; Wang, X.L. Histone deacetylase HDA6 enhances brassinosteroid signaling by inhibiting the BIN2 kinase. Proc. Natl. Acad. Sci. USA 2016, 113, 10418–10423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.Y.; Liu, J.J.; Wang, H.J.; Yang, C.J.; Chen, Y.X.; Li, Y.C. GSK3-like kinases positively modulate abscisic acid signaling through phosphorylating subgroup III SnRK2s in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, 9651–9656. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Raw Reads | Clean Reads | Clean Unique Reads | Unique miRNA |
|---|---|---|---|---|
| SCK_L1 | 13,448,770 | 10,942,269 | 3,279,444 (81.65%) | 192 |
| SCK_L2 | 11,410,041 | 8,833,876 | 2,595,425 (81.06%) | 183 |
| SCK_L3 | 13,279,293 | 10,968,789 | 2,897,994 (80.56%) | 194 |
| S200_L1 | 16,090,479 | 11,374,147 | 2,814,510 (75.59%) | 207 |
| S200_L2 | 15,997,714 | 12,464,350 | 3,203,303 (79.79%) | 204 |
| S200_L3 | 10,850,667 | 7,799,571 | 2,202,790 (74.78%) | 187 |
| SCK_R1 | 10,507,148 | 6,672,627 | 2,180,643 (65.43%) | 189 |
| SCK_R2 | 12,656,682 | 8,945,834 | 2,852,786 (71.38%) | 203 |
| SCK_R3 | 13,882,772 | 9,815,489 | 2,877,824 (69.29%) | 203 |
| S200_R1 | 11,337,238 | 7,882,658 | 2,593,661 (68.67%) | 188 |
| S200_R2 | 20,096,897 | 14,474,914 | 4,047,289 (71.22%) | 223 |
| S200_R3 | 17,508,315 | 11,602,778 | 3,128,276 (71.31%) | 210 |
| miR_name | miR_seq | Up/Down | log2 | p-Value | Expression Level | |
|---|---|---|---|---|---|---|
| 1 | aly-miR393a-5p | TCCAAAGGGATCGCATTGATCC | down | −0.81 | 5.70 × 10−3 | middle |
| 2 | bol-MIR9410-p3_2ss5TG18TA | CTTTGCAGACGACTTAAATA | up | 3.19 | 8.55 × 10−3 | middle |
| 3 | aly-miR393a-3p | ATCATGCTATCTCTTTGGATT | down | −0.94 | 8.88 × 10−3 | middle |
| 4 | cca-miR6111-5p_1ss5TG | TCTTGATGTCACGATGTATGAC | down | −3.01 | 2.03 × 10−2 | middle |
| 5 | cca-MIR6111-p3_2ss17GA19CT | TTATGAAGGTAGTCTAACTCAC | up | 0.62 | 3.90 × 10−2 | high |
| 6 | cst-MIR11334-p5_2ss9TG18TC | TAAGGAGTGTGTAACAAC | up | 2.03 | 4.09 × 10−2 | middle |
| 7 | aly-miR858-5p_L-1R+1 | TTCGTTGTCTGTTCGACCTTG | down | −0.53 | 4.56 × 10−2 | middle |
| miR_Name | miR_seq | Up/ Down | log2 | p-Value | Expression Level | |
|---|---|---|---|---|---|---|
| 1 | PC-5p-838_6216 | TAAACCTATCTATAACAACCT | down | −0.45 | 2.93 × 10−3 | middle |
| 2 | nta-miR168d | TCGCTTGGTGCAGGTCGGGAA | up | 0.31 | 5.02 × 10−3 | middle |
| 3 | cca-MIR6111-p3 | TTATGAAGGTAGTCTAGCCCAC | down | −0.18 | 5.85 × 10−3 | middle |
| 4 | aly-miR393a-5p | TCCAAAGGGATCGCATTGATCC | down | −3.08 | 8.06 × 10−3 | middle |
| 5 | PC-3p-153871_47 | TGGCTCATAAGTCTCTAACTTG | up | 3.33 | 1.11 × 10−2 | middle |
| 6 | mtr-miR398a-5p_2ss12GC21AT | GGAGTGACACTCAGAACACATG | down | −2.86 | 1.45 × 10−2 | middle |
| 7 | cca-miR6111-5p_R-2_1ss20GT | TCTTTATGTCACGATGTATT | up | 1.04 | 2.17 × 10−2 | middle |
| 8 | PC-3p-613_8603 | TTTAAGTAGTGGACAATTGGA | up | 0.7 | 2.32 × 10−2 | middle |
| 9 | mtr-miR166a_R-2 | TCGGACCAGGCTTCATTCC | up | 0.27 | 2.65 × 10−2 | middle |
| 10 | PC-3p-1384_3922 | TCCACTCTGCTTTCTCTGAGGT | up | 0.41 | 3.60 × 10−2 | middle |
| 11 | mtr-miR398a-3p_1ss21TC | TGTGTTCTCAGGTCACCCCTC | down | −1.14 | 4.50 × 10−2 | middle |
| 12 | ath-miR162a-5p_L-1_2ss5GT6GA | GGATACAGCGGTTCATCGATC | down | −0.58 | 4.97 × 10−2 | middle |
| Tissue | KEGG ID | KEGG Name | Gene Number |
|---|---|---|---|
| ko04075 | Plant hormone signal transduction | 11 | |
| ko04626 | Plant–pathogen interaction | 4 | |
| S200-L vs. CK-L | ko00260 | Glycine, serine, and threonine metabolism | 4 |
| ko04120 | Ubiquitin-mediated proteolysis | 4 | |
| ko04075 | Plant hormone signal transduction | 11 | |
| ko03013 | RNA transport | 6 | |
| S200-R vs. CK-R | ko03015 | mRNA surveillance pathway | 4 |
| ko04144 | Endocytosis | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nai, J.; Ma, T.; Liu, Y.; Zhou, Y. Identification of Small RNAs Associated with Salt Stress in Chrysanthemums through High-Throughput Sequencing and Bioinformatics Analysis. Genes 2023, 14, 561. https://doi.org/10.3390/genes14030561
Nai J, Ma T, Liu Y, Zhou Y. Identification of Small RNAs Associated with Salt Stress in Chrysanthemums through High-Throughput Sequencing and Bioinformatics Analysis. Genes. 2023; 14(3):561. https://doi.org/10.3390/genes14030561
Chicago/Turabian StyleNai, Jiefei, Tieming Ma, Yingjie Liu, and Yunwei Zhou. 2023. "Identification of Small RNAs Associated with Salt Stress in Chrysanthemums through High-Throughput Sequencing and Bioinformatics Analysis" Genes 14, no. 3: 561. https://doi.org/10.3390/genes14030561