
Understanding Drug Resistance of Wild-Type and L38HL Insertion Mutant of HIV-1 C Protease to Saquinavir
 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
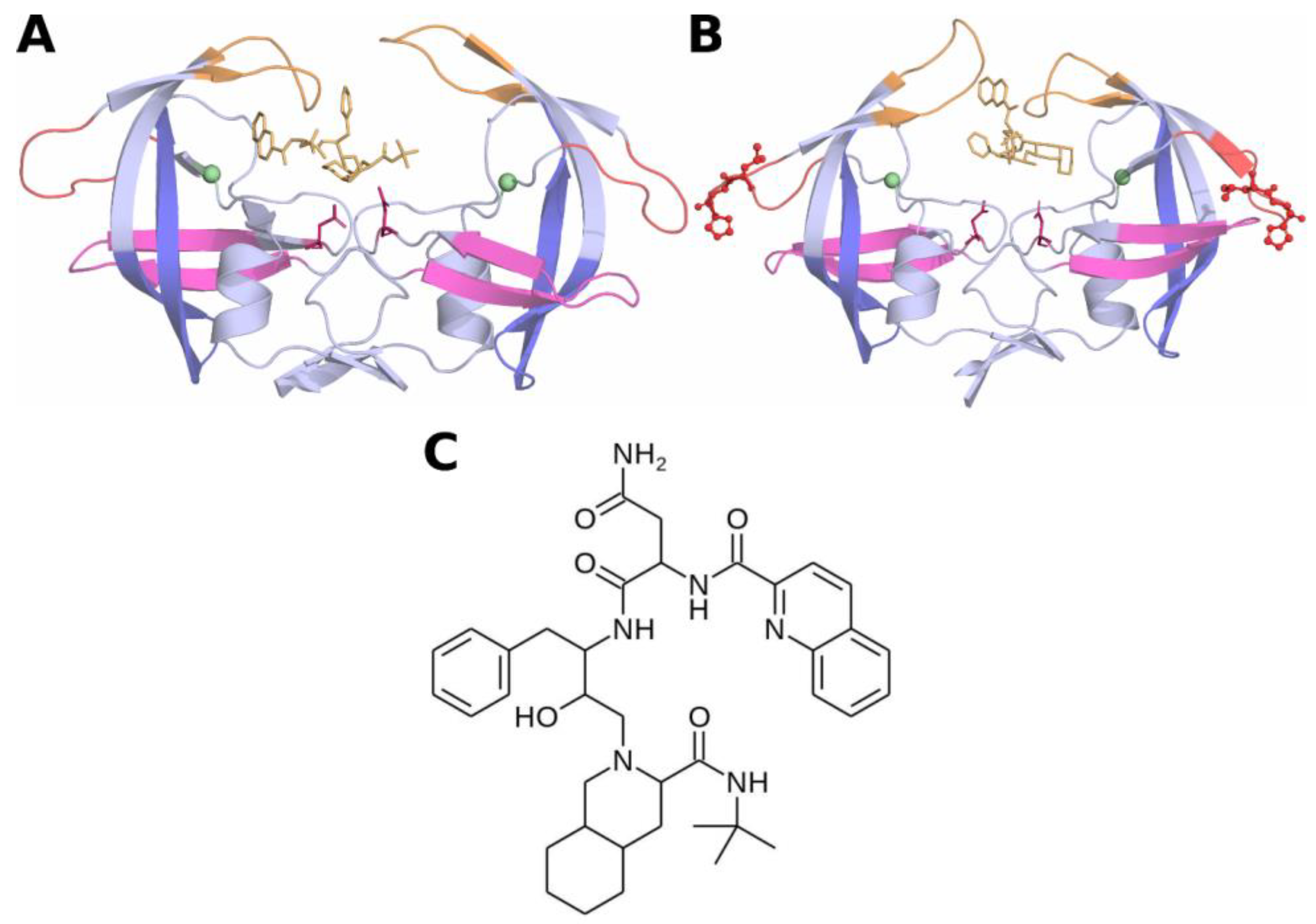
2.1. Initial Coordinates of HIV-Protease Subtype C and the L38HL Variant
2.2. MD Simulation Protocol
2.3. Trajectory Analysis
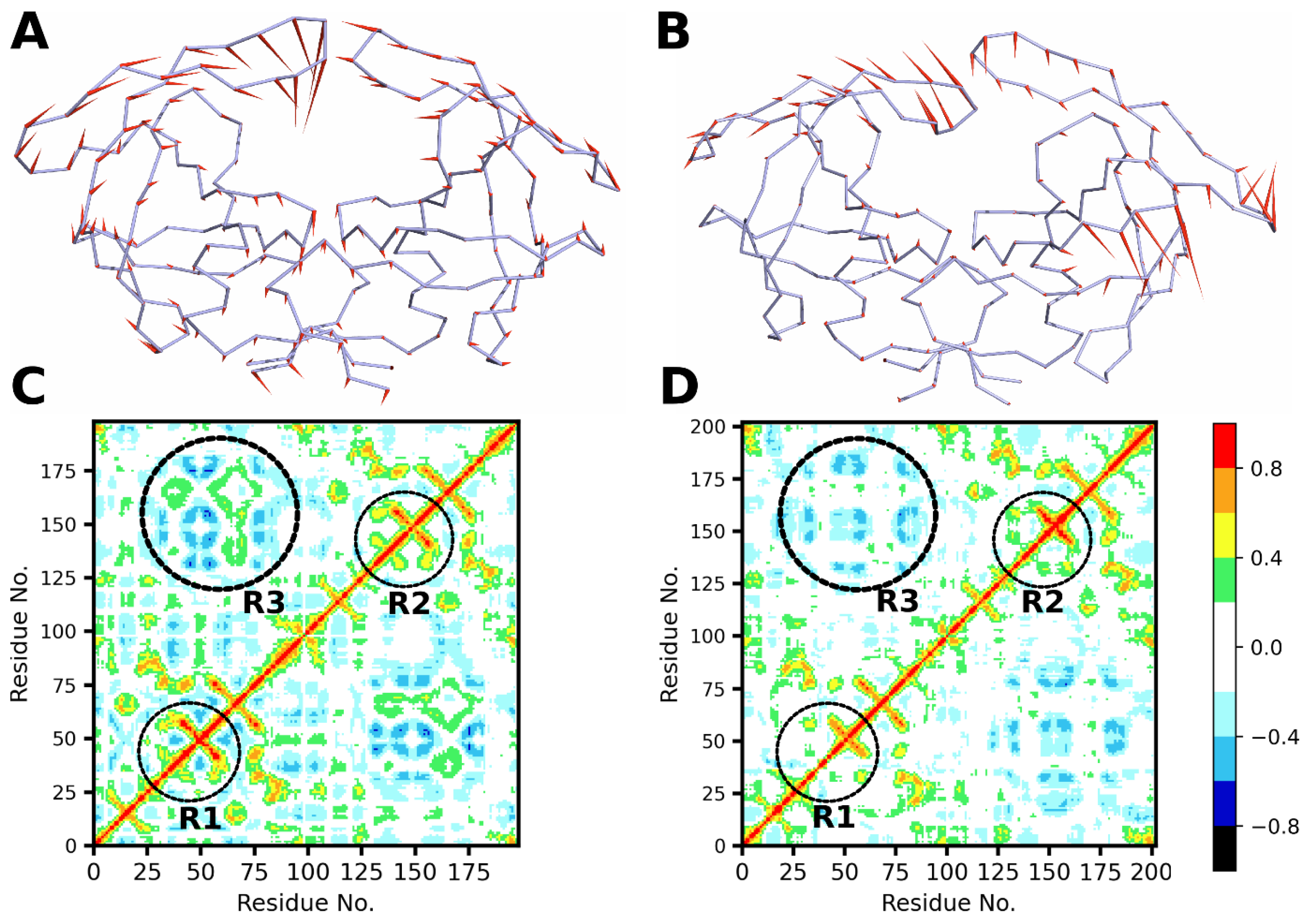
2.4. Dynamic Cross-Correlation
2.5. Principal Component Analysis
2.6. Binding Free Energy Calculations Using MM/GBSA
3. Results and Discussion
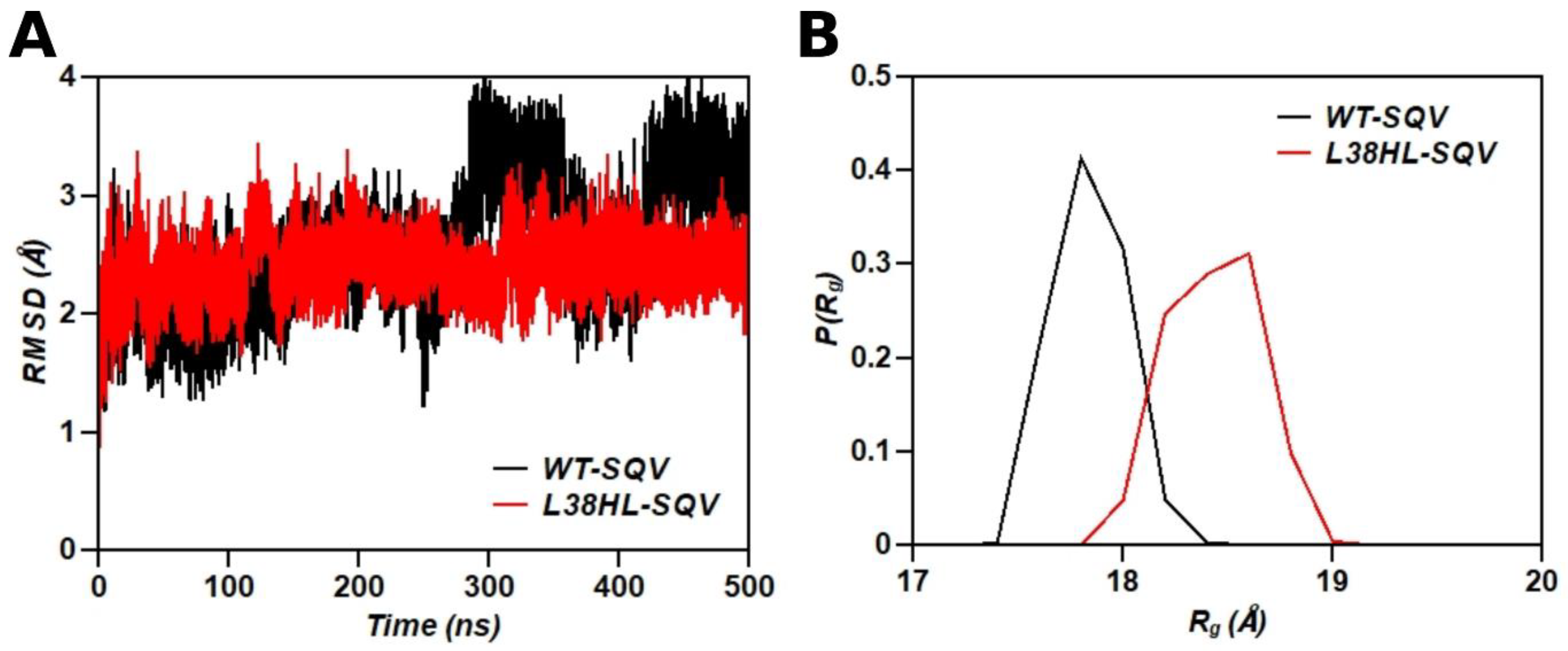
3.1. Global Structural Stability
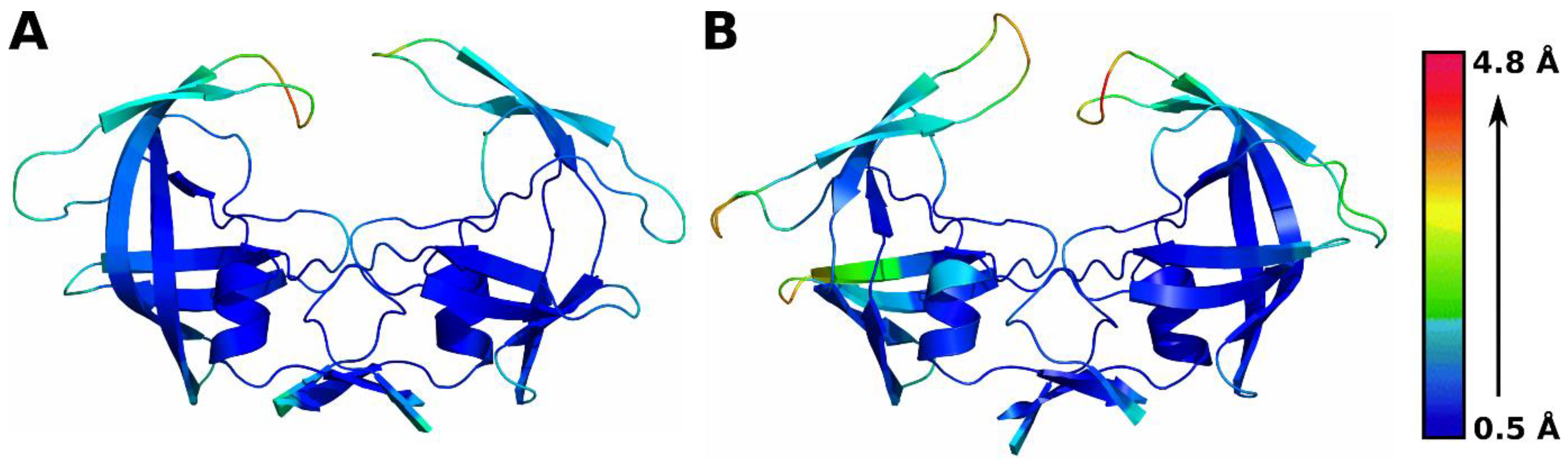
3.2. Residue-Level Fluctuations in Protein Conformation
3.3. Local Conformational Changes
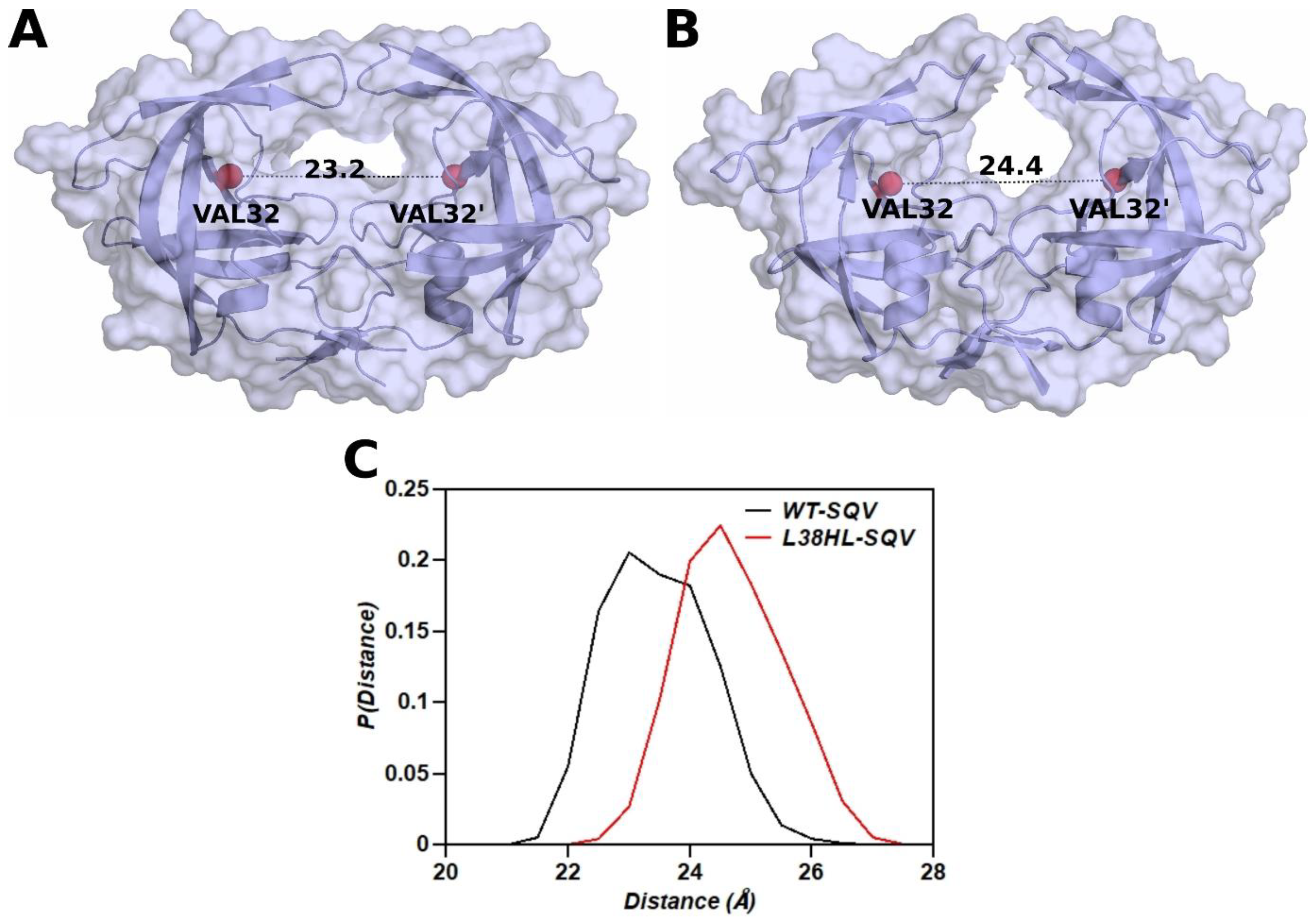
3.4. Distance between the Binding Site Residues
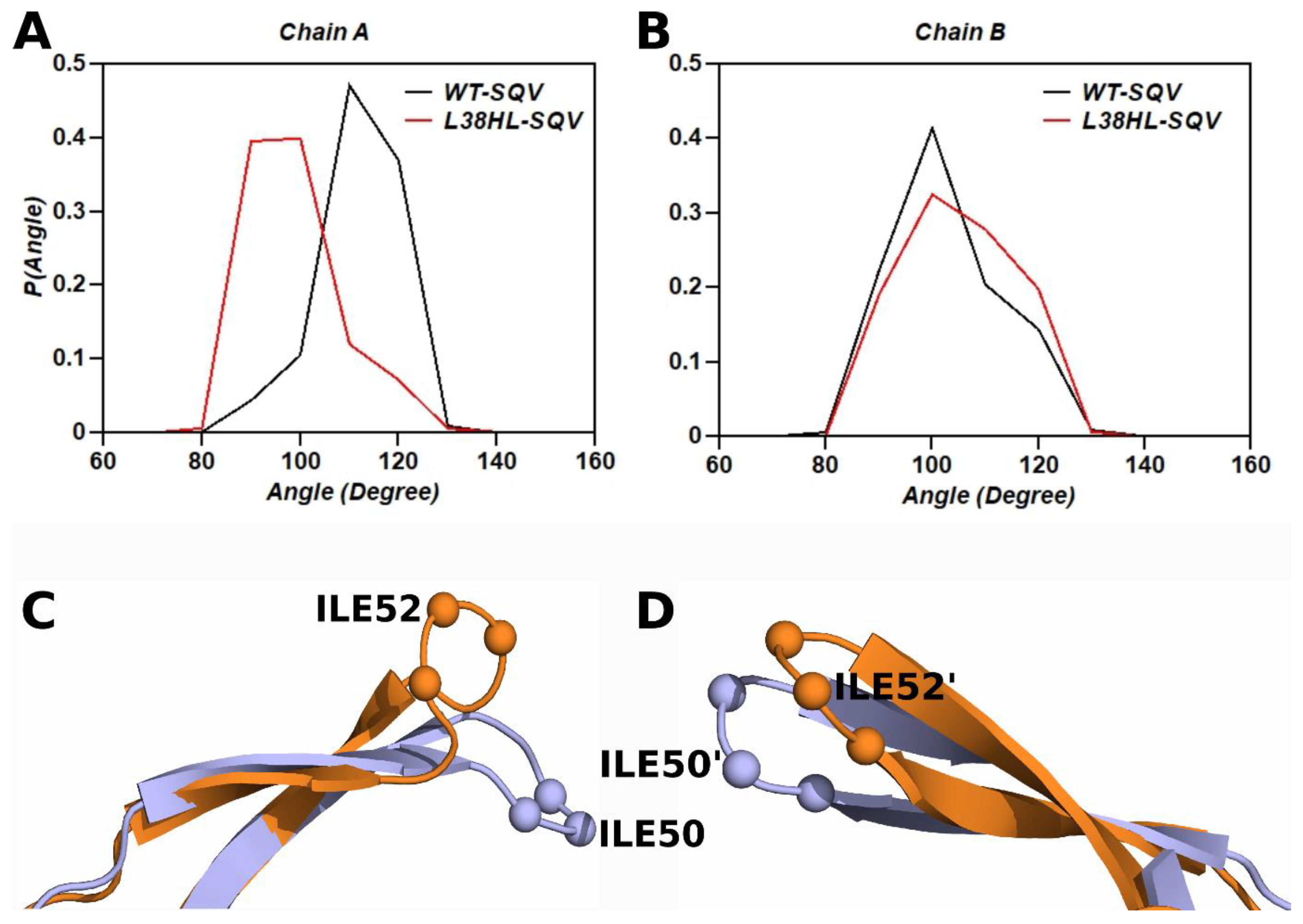
3.5. Analysis of Flap Tri Cα Angles
3.6. Analysis of Hinge Tri Cα Angle
3.7. Principal Component Analysis of Wild-Type and L38HL Bound to SQV
3.8. Dynamic Cross-Correlation Analysis
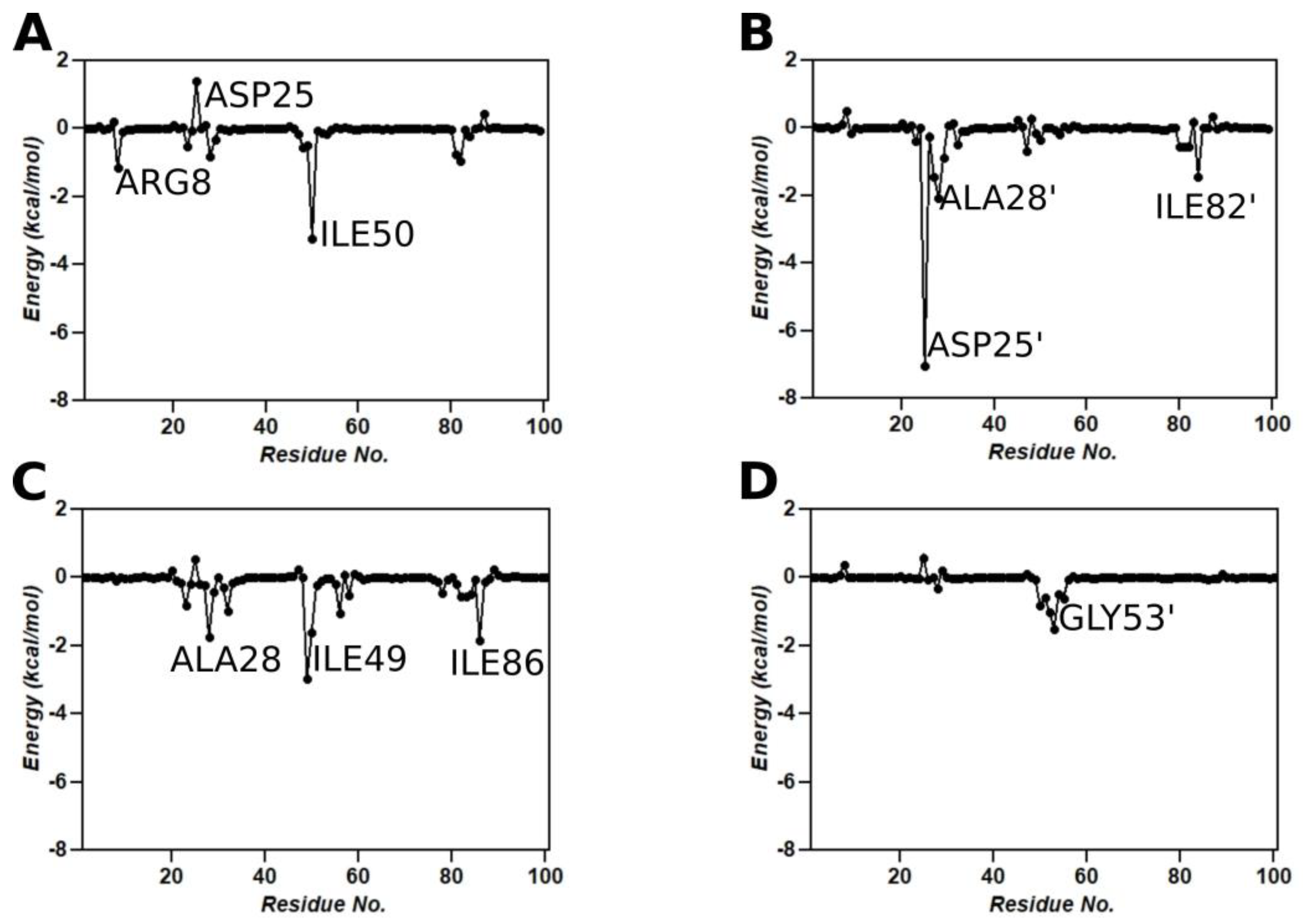
3.9. Binding Free Energy Calculations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elangovan, R.; Jenks, M.; Yun, J.; Dickson-Tetteh, L.; Kirtley, S.; Hemelaar, J. Global and Regional Estimates for Subtype-Specific Therapeutic and Prophylactic HIV-1 Vaccines: A Modeling Study. Front. Microbiol. 2021, 12, 690647. [Google Scholar] [CrossRef] [PubMed]
- Bbosa, N.; Kaleebu, P.; Ssemwanga, D. HIV subtype diversity worldwide. Curr. Opin. HIV AIDS 2019, 14, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Ciccozzi, M.; Parolin, C.; Borsetti, A. Molecular Epidemiology of HIV-1 in African Countries: A Comprehensive Overview. Pathogens 2020, 9, 1072. [Google Scholar] [CrossRef] [PubMed]
- Naicker, P.; Sayed, Y. Non-B HIV-1 subtypes in sub-Saharan Africa: Impact of subtype on protease inhibitor efficacy. Biol. Chem. 2014, 395, 1151–1161. [Google Scholar] [CrossRef]
- Häggblom, A.; Svedhem, V.; Singh, K.; Sönnerborg, A.; Neogi, U. Virological failure in patients with HIV-1 subtype C receiving antiretroviral therapy: An analysis of a prospective national cohort in Sweden. Lancet HIV 2016, 3, e166–e174. [Google Scholar] [CrossRef] [Green Version]
- Velázquez-Campoy, A.; Vega, S.; Fleming, E.; Bacha, U.; Sayed, Y.; Dirr, H.W.; Freire, E. Protease inhibition in African subtypes of HIV-1. AIDS Rev. 2003, 5, 165–171. [Google Scholar]
- Mosebi, S.; Morris, L.; Dirr, H.W.; Sayed, Y. Active-site mutations in the South african human immunodeficiency virus type 1 subtype C protease have a significant impact on clinical inhibitor binding: Kinetic and thermodynamic study. J. Virol. 2008, 82, 11476–11479. [Google Scholar] [CrossRef] [Green Version]
- Eche, S.; Kumar, A.; Sonela, N.; Gordon, M.L. Acquired HIV-1 Protease Conformational Flexibility Associated with Lopinavir Failure May Shape the Outcome of Darunavir Therapy after Antiretroviral Therapy Switch. Biomolecules 2021, 11, 489. [Google Scholar] [CrossRef]
- Shafer, R.W.; Eisen, J.A.; Merigan, T.C.; Katzenstein, D.A. Sequence and drug susceptibility of subtype C reverse transcriptase from human immunodeficiency virus type 1 seroconverters in Zimbabwe. J. Virol. 1997, 71, 5441–5448. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, Y.; Bello, G.; Castillo Mewa, J.; Martínez, A.A.; González, C.; García-Morales, C.; Avila-Ríos, S.; Reyes-Terán, G.; Pascale, J.M. Molecular epidemiology of HIV-1 in Panama: Origin of non-B subtypes in samples collected from 2007 to 2013. PLoS ONE 2014, 9, e85153. [Google Scholar] [CrossRef]
- Foster, G.M.; Ambrose, J.C.; Hué, S.; Delpech, V.C.; Fearnhill, E.; Abecasis, A.B.; Leign Brown, A.J.; Geretti, A.M. Novel HIV-1 recombinants spreading across multiple risk groups in the United Kingdom: The identification and phylogeography of Circulating Recombinant Form (CRF) 50_A1D. PLoS ONE 2014, 9, e83337. [Google Scholar] [CrossRef] [Green Version]
- Naicker, P.; Achilonu, I.; Fanucchi, S.; Fernandes, M.; Ibrahim, M.A.A.; Dirr, H.W.; Soliman, M.E.S.; Sayed, Y. Structural insights into the South African HIV-1 subtype C protease: Impact of hinge region dynamics and flap flexibility in drug resistance. J. Biomol. Struct. Dyn. 2013, 31, 1370–1380. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Kruger, H.G.; Govender, T.; Maguire, G.E.M.; Sayed, Y.; Ibrahim, M.A.A.; Naicker, P.; Soliman, M.E.S. Comparison of the Molecular Dynamics and Calculated Binding Free Energies for Nine FDA-Approved HIV-1 PR Drugs Against Subtype B and C-SA HIV PR. Chem. Biol. Drug. Des. 2013, 81, 208–218. [Google Scholar] [CrossRef]
- Ragland, D.A.; Nalivaika, E.A.; Nalam, M.N.L.; Prachanronarong, K.L.; Cao, H.; Bandaranayake, R.M.; Cai, Y.; Kurt-Yilmaz, N.; Schiffer, C.A. Drug Resistance Conferred by Mutations Outside the Active Site through Alterations in the Dynamic and Structural Ensemble of HIV-1 Protease. J. Am. Chem. Soc. 2014, 136, 11956–11963. [Google Scholar] [CrossRef] [Green Version]
- Obasa, A.E.; Ambikan, A.T.; Gupta, S.; Neogi, U.; Jacobs, G.B. Increased acquired protease inhibitor drug resistance mutations in minor HIV-1 quasispecies from infected patients suspected of failing on national second-line therapy in South Africa. BMC Infect. Dis. 2021, 21, 214. [Google Scholar] [CrossRef]
- Obasa, A.E.; Mikasi, S.G.; Brado, D.; Cloete, R.; Singh, K.; Neogi, U.; Jacobs, G.B. Drug Resistance Mutations Against Protease, Reverse Transcriptase and Integrase Inhibitors in People Living With HIV-1 Receiving Boosted Protease Inhibitors in South Africa. Front. Microbiol. 2020, 11, 438. [Google Scholar] [CrossRef] [Green Version]
- Ledwaba, J.; Sayed, Y.; Pillay, V.; Morris, L.; Hunt, G. Low Frequency of Protease Inhibitor Resistance Mutations and Insertions in HIV-1 Subtype C Protease Inhibitor-Naïve Sequences. AIDS Res. Hum. Retrovir. 2019, 35, 673–678. [Google Scholar] [CrossRef]
- Jacob, K.S.; Ganguly, S.; Kumar, P.; Poddar, R.; Kumar, A. Homology model, molecular dynamics simulation and novel pyrazole analogs design of Candida albicans CYP450 lanosterol 14 α-demethylase, a target enzyme for antifungal therapy. J. Biomol. Struct. Dyn. 2017, 35, 1446–1463. [Google Scholar] [CrossRef]
- Collier, T.A.; Piggot, T.J.; Allison, J.R. Molecular Dynamics Simulation of Proteins. Methods Mol. Biol. 2020, 2073, 311–327. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.; Sharma, R.; Kumar, A. Docking techniques in pharmacology: How much promising? Comput. Biol. Chem. 2018, 76, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L. Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J. Am. Chem. Soc. 1981, 103, 2. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Makatini, M.M.; Petzold, K.; Sriharsha, S.N.; Ndlovu, N.; Soliman, M.E.S.; Honarparvar, B.; Parboosing, R.; Naidoo, A.; Arvidsson, P.I.; Sayed, Y.; et al. Synthesis and structural studies of pentacycloundecane-based HIV-1 PR inhibitors: A hybrid 2D NMR and docking/QM/MM/MD approach. Eur. J. Med. Chem. 2011, 46, 3976–3985. [Google Scholar] [CrossRef]
- Makatini, M.M.; Petzold, K.; Sriharsha, S.N.; Soliman, M.E.S.; Honarparvar, B.; Arvidsson, P.I.; Sayed, Y.; Govender, P.; Maguire, G.E.M.; Kruger, H.G.; et al. Pentacycloundecane-based inhibitors of wild-type C-South African HIV-protease. Bioorg. Med. Chem. Lett. 2011, 21, 2274–2277. [Google Scholar] [CrossRef]
- Kasahara, K.; Fukuda, I.; Nakamura, H. A Novel Approach of Dynamic Cross Correlation Analysis on Molecular Dynamics Simulations and Its Application to Ets1 Dimer–DNA Complex. PLoS ONE 2014, 9, e112419. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, V.; Jagannathan, S.; Shaikh, A.R.; Rajagopalan, R. Dynamic and Structural Changes in the Minimally Restructuring EcoRI Bound to a Minimally Mutated DNA Chain. J. Biomol. Struct. Dyn. 2012, 29, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Tsui, V.; Case, D.A. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 2001, 56, 275–291. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Sherry, D.; Worth, R.; Ismail, Z.S.; Sayed, Y. Cantilever-centric mechanism of cooperative non-active site mutations in HIV protease: Implications for flap dynamics. J. Mol. Graph. Model 2021, 106, 107931. [Google Scholar] [CrossRef]
- Wang, R.-G.; Zhang, H.-X.; Zheng, Q.-C. Revealing the binding and drug resistance mechanism of amprenavir, indinavir, ritonavir, and nelfinavir complexed with HIV-1 protease due to double mutations G48T/L89M by molecular dynamics simulations and free energy analyses. Phys. Chem. Chem. Phys. 2020, 22, 4464–4480. [Google Scholar] [CrossRef]
- Weber, I.T.; Wang, Y.-F.; Harrison, R.W. HIV Protease: Historical Perspective and Current Research. Viruses 2021, 13, 839. [Google Scholar] [CrossRef]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS 2015, 7, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.X.; Liu, W.T.; Li, H.Y.; Wang, W.; Sun, H.B.; Zhang, L.L.; Wu, S.L. Decoding molecular mechanism underlying binding of drugs to HIV-1 protease with molecular dynamics simulations and MM-GBSA calculations. SAR QSAR Environ. Res. 2021, 32, 889–915. [Google Scholar] [CrossRef]
- Maseko, S.B.; Padayachee, E.; Govender, T.; Sayed, Y.; Kruger, G.; Maguire, G.E.M.; Lin, J. I36T↑T mutation in South African subtype C (C-SA) HIV-1 protease significantly alters protease-drug interactions. Biol. Chem. 2017, 398, 1109–1117. [Google Scholar] [CrossRef]
- Salminen, M.O.; Johansson, B.; Sönnerborg, A.; Ayehunie, S.; Gotte, D.; Leinikki, P.; Burke, D.S.; McCutchan, F.E. Full-length sequence of an ethiopian human immunodeficiency virus type 1 (HIV-1) isolate of genetic subtype C. AIDS Res. Hum. Retrovir. 1996, 12, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- McCormack, G.P.; Glynn, J.R.; Crampin, A.C.; Sibande, F.; Mulawa, D.; Bliss, L.; Broadbent, P.; Abarca, K.; Pönnighaus, J.M.; Fine, P.E.M.; et al. Early evolution of the human immunodeficiency virus type 1 subtype C epidemic in rural Malawi. J. Virol. 2002, 76, 12890–12899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lihana, R.W.; Ssemwanga, D.; Abimiku, A.; Ndembi, N. Update on HIV-1 diversity in Africa: A decade in review. AIDS Rev. 2012, 14, 83–100. [Google Scholar] [PubMed]
- Kozísek, M.; Sasková, K.G.; Rezácová, P.; Brynda, J.; van Maarseveen, N.M.; De Jong, D.; Boucher, C.A.; Kagan, R.M.; Nijhuis, M.; Konvalinka, J. Ninety-nine is not enough: Molecular characterization of inhibitor-resistant human immunodeficiency virus type 1 protease mutants with insertions in the flap region. J. Virol. 2008, 82, 5869–5878. [Google Scholar] [CrossRef] [Green Version]
- Vora, S.; Marcelin, A.-G.; Günthard, H.F.; Flandre, P.; Hirsch, H.H.; Masquelier, B.; Zinkernagel, A.; Peytavin, G.; Calvez, V.; Perrin, L.; et al. Clinical validation of atazanavir/ritonavir genotypic resistance score in protease inhibitor-experienced patients. AIDS 2006, 20, 35–40. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, J.; Chen, Z.; Wang, G.; Shao, Q.; Shi, J.; Zhu, W. Structural insights into HIV-1 protease flap opening processes and key intermediates. RSC Adv. 2017, 7, 45121–45128. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild-type (kcal/mol) | L38HL (kcal/mol) | |
|---|---|---|
| −57.22 ± 4.04 | −58.10 ± 5.30 | |
| −13.14 ± 12.66 | 35.11 ± 11.95 | |
| 29.31 ± 10.59 | −15.10 ± 11.33 | |
| −7.89 ± 0.45 | −7.72 ± 0.56 | |
| 16.17 ± 11.62 | 20.01 ± 11.64 | |
| −65.11 ± 8.35 | −65.82 ± 2.93 | |
| −48.94 ± 4.637 | −45.81 ± 5.67 |
| Acceptor | Donor | Average Distance (Å) | Angle (°) | Occupancy | |
|---|---|---|---|---|---|
| wild-type | ASP124@OD1 | SQV@N5 | 2.74 | 156.17 | 0.628 |
| SQV@O3 | ARG8@NH1 | 2.81 | 160.26 | 0.51 | |
| ASP124@OD1 | SQV@N6 | 2.85 | 157.83 | 0.47 | |
| SQV@O2 | ASP128@N | 2.86 | 162.61 | 0.25 | |
| ASP124@OD2 | SQV@N5 | 2.75 | 157.34 | 0.12 | |
| ASP124@OD1 | SQV@O4 | 2.75 | 162.47 | 0.10 | |
| GLY126@O | SQV@N1 | 2.88 | 159.20 | 0.10 | |
| L38HL | ASP25@OD2 | SQV@O4 | 2.63 | 164.89 | 0.45 |
| ASP25@OD1 | SQV@O4 | 2.63 | 164.42 | 0.32 | |
| SQV@O3 | GLY53@N | 2.86 | 157.20 | 0.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venkatachalam, S.; Murlidharan, N.; Krishnan, S.R.; Ramakrishnan, C.; Setshedi, M.; Pandian, R.; Barh, D.; Tiwari, S.; Azevedo, V.; Sayed, Y.; et al. Understanding Drug Resistance of Wild-Type and L38HL Insertion Mutant of HIV-1 C Protease to Saquinavir. Genes 2023, 14, 533. https://doi.org/10.3390/genes14020533
Venkatachalam S, Murlidharan N, Krishnan SR, Ramakrishnan C, Setshedi M, Pandian R, Barh D, Tiwari S, Azevedo V, Sayed Y, et al. Understanding Drug Resistance of Wild-Type and L38HL Insertion Mutant of HIV-1 C Protease to Saquinavir. Genes. 2023; 14(2):533. https://doi.org/10.3390/genes14020533
Chicago/Turabian StyleVenkatachalam, Sankaran, Nisha Murlidharan, Sowmya R. Krishnan, C. Ramakrishnan, Mpho Setshedi, Ramesh Pandian, Debmalya Barh, Sandeep Tiwari, Vasco Azevedo, Yasien Sayed, and et al. 2023. "Understanding Drug Resistance of Wild-Type and L38HL Insertion Mutant of HIV-1 C Protease to Saquinavir" Genes 14, no. 2: 533. https://doi.org/10.3390/genes14020533