
Status Epilepticus in Chromosomal Disorders Associated with Epilepsy: A Systematic Review

 , , , , and
, , , , and
Abstract
:1. Introduction
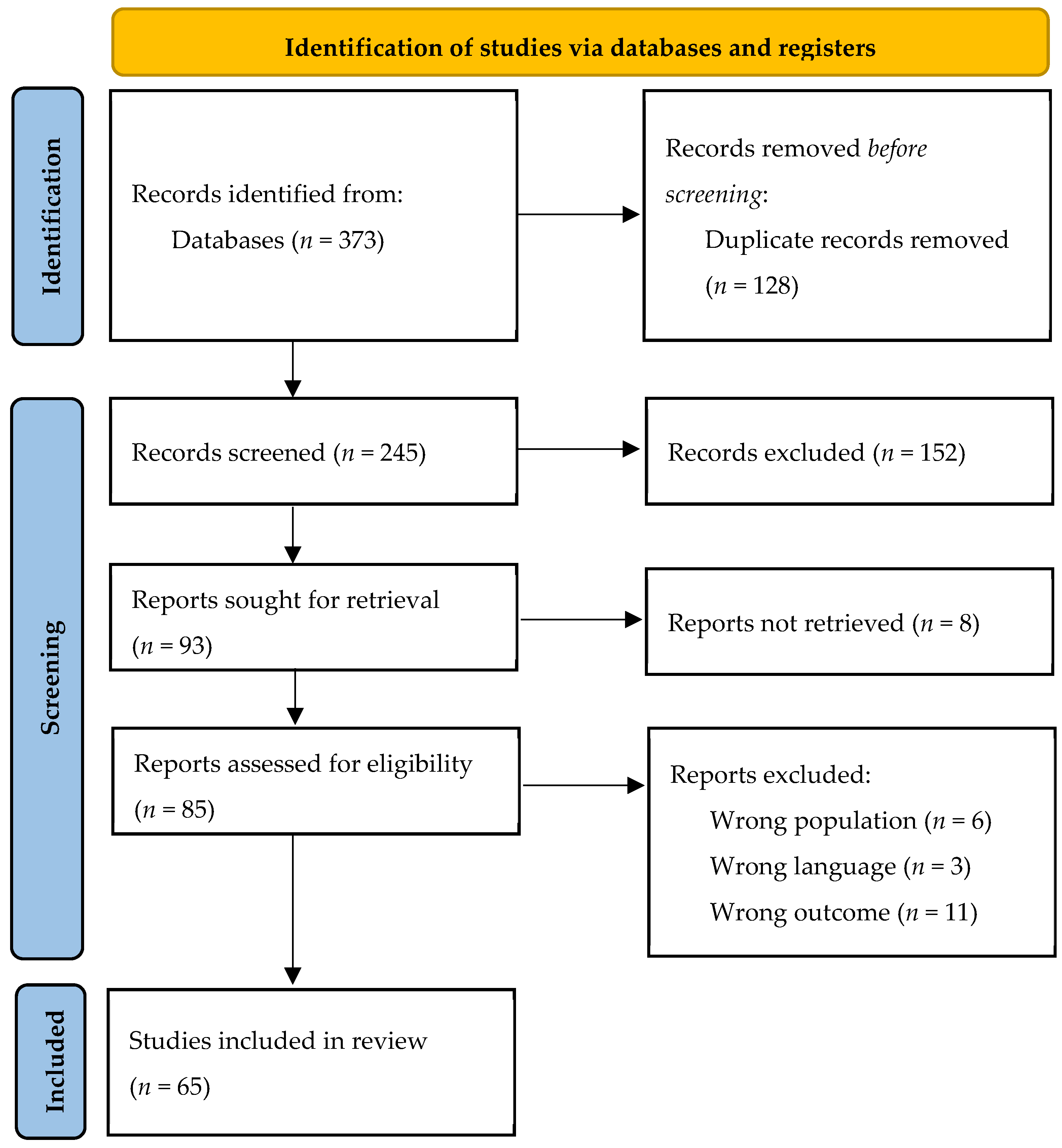
2. Materials and Methods
2.1. Protocol
- Inclusion criteria:
- (1)
- Date: published before October 2022.
- (2)
- Population:
- Infants, children, adolescents, and adults;
- Patients with a diagnosis of syndromes due to chromosomal abnormalities frequently seen in people with epilepsy or with distinct seizure and EEG features, as listed by the ILAE, https://www.epilepsydiagnosis.org/aetiology/chromosomal-abnormalities-overview.html (accessed on 1 October 2022);
- Patients who experienced at least one episode of SE during their lifetime.
- (3)
- Language: English.
- (4)
- Study Design: RCT, cohort studies, cross-sectional studies, retrospective studies, case reports.
- (5)
- Outcomes: reporting at least one between:
- SE clinical features (semeiology, symptoms/signs, duration before diagnosis);
- SE treatment;
- SE outcome (response to treatment and recovery).
- Exclusion criteria:
- (1)
- Population: other than humans;
- (2)
- Language: other than English;
- (3)
- Study design: descriptive studies, reviews, protocols;
- (4)
- Missing outcomes of interest.
2.2. Statistical Analysis
3. Results
3.1. Angelman Syndrome
3.1.1. Epilepsy History
3.1.2. Clinical Features of Status Epilepticus in Angelman Syndrome
3.1.3. Treatment of Status Epilepticus in Angelman Syndrome
3.1.4. Outcome of Status Epilepticus in Angelman Syndrome
3.2. Ring 20 Syndrome
3.2.1. Epilepsy History
3.2.2. Features of Status Epilepticus in Ring 20 Syndrome
3.2.3. Treatment of Status Epilepticus in Ring 20 Syndrome
3.2.4. Outcome of Status Epilepticus in Ring 20 Syndrome
3.3. Wolf–Hirschhorn Syndrome
3.4. Down Syndrome
3.5. Ring 14 Syndrome
3.6. Other CDAE
4. Discussion
4.1. Status Epilepticus in Angelman Syndrome
4.2. Status Epilepticus in Ring 20 Syndrome
4.3. Status Epilepticus in Wolf–Hirschhorn Syndrome
4.4. Status Epilepticus in Down Syndrome
4.5. Status Epilepticus in Ring 14 Syndrome
4.6. Other CDAE
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trinka, E.; Cock, H.; Hesdorffer, D.; Rossetti, A.O.; Scheffer, I.E.; Shinnar, S.; Shorvon, S.; Lowenstein, D.H. A definition and classification of status epilepticus—Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015, 56, 1515–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wylie, T.; Sandhu, D.S.; Murr, N. Status Epilepticus. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK430686/ (accessed on 20 December 2022).
- Sankar, R.; Shin, D.H.; Liu, H.; Mazarati, A.; De Vasconcelos, A.P.; Wasterlain, C.G. Patterns of Status Epilepticus-Induced Neuronal Injury during Development and Long-Term Consequences. J. Neurosci. 1998, 18, 8382–8393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, R.F.M. The outcomes of childhood convulsive status epilepticus. Epilepsy Behav. 2019, 101 Pt B, 106286. [Google Scholar] [CrossRef]
- Smith, D.M.; McGinnis, E.L.; Walleigh, D.J.; Abend, N.S. Management of Status Epilepticus in Children. J. Clin. Med. 2016, 5, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EpilepsyDiagnosis.org. Available online: https://www.epilepsydiagnosis.org/ (accessed on 21 December 2022).
- Sorge, G.; Sorge, A. Epilepsy and chromosomal abnormalities. Ital. J. Pediatr. 2010, 36, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; McKinlay Gardner, R.J.; Crossland, K.M.; Scheffer, I.E.; Berkovic, S.F. Chromosomal Abnormalities and Epilepsy: A Review for Clinicians and Gene Hunters. Epilepsia 2002, 43, 127–140. [Google Scholar] [CrossRef] [Green Version]
- Riservati, I.U.-T.I.D. Orphanet: Malattie. Available online: https://www.orpha.net/consor/cgi-bin/Disease.php?lng=IT (accessed on 21 December 2022).
- Valente, K.D.; Koiffmann, C.P.; Fridman, C.; Varella, M.; Kok, F.; Andrade, J.Q.; Grossmann, R.M.; Marques-Dias, M.J. Epilepsy in Patients with Angelman Syndrome Caused by Deletion of the Chromosome 15q11-13. Arch. Neurol. 2006, 63, 122. [Google Scholar] [CrossRef]
- Sugimoto, T.; Yasuhara, A.; Ohta, T.; Nishida, N.; Saitoh, S.; Hamabe, J.-I.; Niikawa, N. Angelman Syndrome in Three Siblings: Characteristic Epileptic Seizures and EEG Abnormalities. Epilepsia 1992, 33, 1078–1082. [Google Scholar] [CrossRef]
- Matsumoto, A.; Kumagai, T.; Miura, K.; Miyazaki, S.; Hayakawa, C.; Yamanaka, T. Epilepsy in Angelman Syndrome Associated with Chromosome 15q Deletion. Epilepsia 1992, 33, 1083–1090. [Google Scholar] [CrossRef]
- Viani, F.; Romeo, A.; Viri, M.; Mastrangelo, M.; Lalatta, F.; Selicorni, A.; Gobbi, G.; Lanzi, G.; Bettio, D.; Briscioli, V.; et al. Seizure and EEG patterns in Angelman’s syndrome. J. Child Neurol. 1995, 10, 467–471. [Google Scholar] [CrossRef]
- Rubin, D.I.; Patterson, M.C.; Westmoreland, B.F.; Klass, D.W. Angelman’s syndrome: Clinical and electroencephalographic findings. Electroencephalogr. Clin. Neurophysiol. 1997, 102, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Laan, L.A.E.M.; Renier, W.O.; Arts, W.F.M.; Buntinx, I.M.; Burgt, I.J.A.M.V.; Stroink, H.; Beuten, J.; Zwinderman, K.H.; Dijk, J.G.; Brouwer, O.F. Evolution of Epilepsy and EEG Findings in Angelman Syndrome. Epilepsia 1997, 38, 195–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galván-Manso, M.; Campistol, J.; Conill, J.; Sanmartí, F.-X. Analysis of the characteristics of epilepsy in 37 patients with the molecular diagnosis of Angelman syndrome. Epileptic Disord. 2005, 7, 19–25. [Google Scholar]
- Espay, A.J.; Andrade, D.M.; Wennberg, R.A.; Lang, A.E. Atypical absences and recurrent absence status in an adult with Angelman syndrome due to the UBE3A mutation. Epileptic Disord. 2005, 7, 227–230. [Google Scholar]
- Uemura, N.; Matsumoto, A.; Nakamura, M.; Watanabe, K.; Negoro, T.; Kumagai, T.; Miura, K.; Ohki, T.; Mizuno, S.; Okumura, A.; et al. Evolution of seizures and electroencephalographical findings in 23 cases of deletion type Angelman syndrome. Brain Dev. 2005, 27, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, Y.; Kobayashi, K.; Yoshinaga, H.; Ogino, T.; Ohmori, I.; Ogawa, K.; Oka, E. Relationship between severity of epilepsy and developmental outcome in Angelman syndromeq. Brain Dev. 2005, 27, 6. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, R.H.; Cersósimo, R.O.; Espeche, A.; Arroyo, H.A.; Fejerman, N. Myoclonic Status in Nonprogressive Encephalopathies: Study of 29 Cases. Epilepsia 2007, 48, 107–113. [Google Scholar] [CrossRef]
- Yang, X.Y.; Zou, L.P.; Song, F.; Zhang, L.P.; Zheng, H.; Wu, H.S.; Xiao, J. Clinical manifestation and EEG characteristics of Angelman syndrome. Zhonghua Er Ke Za Zhi 2010, 48, 783–786. [Google Scholar]
- Weber, P. Levetiracetam in Nonconvulsive Status Epilepticus in a Child With Angelman Syndrome. J. Child Neurol. 2010, 25, 393–396. [Google Scholar] [CrossRef]
- Valente, K.D.; Varela, M.C.; Koiffmann, C.P.; Andrade, J.Q.; Grossmann, R.; Kok, F.; Marques-Dias, M.J. Angelman syndrome caused by deletion: A genotype–phenotype correlation determined by breakpoint. Epilepsy Res. 2013, 105, 234–239. [Google Scholar] [CrossRef]
- Nicita, F.; Garone, G.; Papetti, L.; Consoli, F.; Magliozzi, M.; De Luca, A.; Spalice, A. Myoclonic status and central fever in Angelman syndrome due to paternal uniparental disomy. J. Neurogenet. 2015, 29, 178–182. [Google Scholar] [CrossRef]
- Grocott, O.R.; Herrington, K.S.; Pfeifer, H.H.; Thiele, E.A.; Thibert, R.L. Low glycemic index treatment for seizure control in Angelman syndrome: A case series from the Center for Dietary Therapy of Epilepsy at the Massachusetts General Hospital. Epilepsy Behav. 2017, 68, 45–50. [Google Scholar] [CrossRef]
- Worden, L.; Grocott, O.; Tourjee, A.; Chan, F.; Thibert, R. Diazepam for outpatient treatment of nonconvulsive status epilepticus in pediatric patients with Angelman syndrome. Epilepsy Behav. 2018, 82, 74–80. [Google Scholar] [CrossRef]
- Pollack, S.F.; Grocott, O.R.; Parkin, K.A.; Larson, A.M.; Thibert, R.L. Myoclonus in Angelman syndrome. Epilepsy Behav. 2018, 82, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Heus, K.G.C.B.B.; Mous, S.E.; Hooven-Radstaake, M.T.; van Iperen-Kolk, B.M.; Navis, C.; Rietman, A.B.; Hoopen, L.W.T.; Brooks, A.S.; Elgersma, Y.; Moll, H.A.; et al. An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am. J. Med. Genet. Part A 2020, 182, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melikishvili, G.; Bienvenu, T.; Tabatadze, N.; Gachechiladze, T.; Kurua, E.; Gverdtsiteli, S.; Melikishvili, M.; Dulac, O. Novel UBE3A pathogenic variant in a large Georgian family produces non-convulsive status epilepticus responsive to ketogenic diet. Seizure 2022, 94, 70–73. [Google Scholar] [CrossRef]
- Freire de Moura, M.; Flores-Guevara, R.; Gueguen, B.; Biraben, A.; Renault, F. Long-term EEG in patients with the ring chromosome 20 epilepsy syndrome. Epilepsia 2016, 57, e94–e96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gago-Veiga, A.B.; Toledano, R.; García-Morales, I.; Pérez-Jiménez, M.A.; Bernar, J.; Gil-Nagel, A. Specificity of electroclinical features in the diagnosis of ring chromosome 20. Epilepsy Behav. 2018, 80, 215–220. [Google Scholar] [CrossRef]
- Inoue, Y.; Fujiwara, T.; Matsuda, K.; Kubota, H.; Tanaka, M.; Yagi, K.; Yamamori, K.; Takahashi, Y. Ring chromosome 20 and nonconvulsive status epilepticus. A new epileptic syndrome. Brain 1997, 120 Pt 6, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Inagaki, M.; Sasaki, M.; Sugai, K.; Ohta, S.; Hashimoto, T. Characteristic EEG findings in ring 20 syndrome as a diagnostic clue. Electroencephalogr. Clin. Neurophysiol. 1998, 107, 258–262. [Google Scholar] [CrossRef]
- Petit, J.; Roubertie, A.; Inoue, Y.; Genton, P. Non-convulsive status in the ring chromosome 20 syndrome: A video illustration of 3 cases. Epileptic Disord. 1999, 1, 237–241. [Google Scholar] [PubMed]
- Augustijn, P.B.; Parra, J.; Wouters, C.H.; Joosten, P.; Lindhout, D.; van Emde Boas, W. Ring chromosome 20 epilepsy syndrome in children: Electroclinical features. Neurology 2001, 57, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- da Gomes, M.M.; Lucca, I.; Bezerra, S.A.M.; Llerena, J.J.; Moreira, D.M. Epilepsy and ring chromosome 20: Case report. Arq. Neuropsiquiatr. 2002, 60, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Delgado, M.; Salas, J.; Hernando, I.; Calleja, S.; Hernandez, C. Ring chromosome 20: A distinctive syndrome identifiable by electroclinical diagnosis. Neurologia 2004, 19, 215–219. [Google Scholar]
- Locharernkul, C.; Ebner, A.; Promchainant, C. Ring chromosome 20 with nonconvulsive status epilepticus: Electroclinical correlation of a rare epileptic syndrome. Clin. EEG Neurosci. 2005, 36, 151–160. [Google Scholar] [CrossRef]
- Alpman, A.; Serdaroglu, G.; Cogulu, O.; Tekgul, H.; Gokben, S.; Ozkinay, F. Ring chromosome 20 syndrome with intractable epilepsy. Dev. Med. Child Neurol. 2005, 47, 343–346. [Google Scholar] [CrossRef]
- de Falco, F.; Olivieri, P.; Concolino, D.; Battaglia, F.; Verardi, R.; Grande, G.; Stabile, M. Electroclinical evolution in ring chromosome 20 epilepsy syndrome: A case with severe phenotypic features followed for 25 years. Seizure 2006, 15, 449–453. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Van Dyke, D.L.; Thorland, E.C.; Chhabra, H.S.; Michels, V.V.; Keefe, J.G.; Lega, M.A.; Feely, M.A.; Uphoff, T.S.; Jalal, S.M. Mosaic ring 20 with no detectable deletion by FISH analysis: Characteristic seizure disorder and literature review. Am. J. Med. Genet. A 2006, 140, 1696–1706. [Google Scholar] [CrossRef]
- Elghezal, H.; Hannachi, H.; Mougou, S.; Kammoun, H.; Triki, C.; Saad, A. Ring chromosome 20 syndrome without deletions of the subtelomeric and CHRNA4—KCNQ2 genes loci. Eur. J. Med. Genet. 2007, 50, 441–445. [Google Scholar] [CrossRef]
- Jacobs, J.; Bernard, G.; Andermann, E.; Dubeau, F.; Andermann, F. Refractory and lethal status epilepticus in a patient with ring chromosome 20 syndrome. Epileptic Disord. 2008, 10, 254–259. [Google Scholar] [CrossRef]
- Elens, I.; Vanrykel, K.; De Waele, L.; Jansen, K.; Segeren, M.; Van Paesschen, W.; Ceulemans, B.; Boel, M.; Frijns, J.-P.; Buyse, G.; et al. Ring chromosome 20 syndrome: Electroclinical description of six patients and review of the literature. Epilepsy Behav. 2012, 23, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, A.; Menon, R.N.; Hariharan, S.; Radhakrishnan, K. The evolving electroclinical syndrome of “epilepsy with ring chromosome 20”. Seizure 2012, 21, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Wechapinan, T.; Sri-Udomkajorn, S.; Suwannachote, S. Rare epileptic syndrome of ring chromosome 20 with epileptic encephalopathy: A case report. J. Med. Assoc. Thail. 2014, 97 (Suppl. S6), S239–S242. [Google Scholar]
- Onder, H.; Tezer, F.I. Significant Improvements of EEG and Clinical Findings with Oral Lacosamide in a Patient with Ring Chromosome 20. Clin. EEG Neurosci. 2016, 47, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Vignoli, A.; Bisulli, F.; Darra, F.; Mastrangelo, M.; Barba, C.; Giordano, L.; Turner, K.; Zambrelli, E.; Chiesa, V.; Bova, S.; et al. Epilepsy in ring chromosome 20 syndrome. Epilepsy Res. 2016, 128, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Oguni, H.; Nagata, S. Refractory and severe status epilepticus in a patient with ring chromosome 20 syndrome. Brain Dev. 2016, 38, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-F.; Chi, C.-S. Intractable epilepsy, growth failure, hypothyroidism, and cataract: Rare clinical manifestations in a patient with ring chromosome 20 syndrome. Neurol. Asia 2020, 25, 63–66. [Google Scholar]
- Yamagishi, H.; Goto, M.; Osaka, H.; Kuwajima, M.; Muramatsu, K.; Yamagata, T. Praxis-induced reflex seizures in two Japanese cases with ring chromosome 20 syndrome. Epileptic Disord. 2020, 22, 214–218. [Google Scholar] [CrossRef]
- Kishore, V.K.; Viswanathan, L.G.; Asranna, A.; Kenchiah, R.; Chowdary, M.; Sinha, S. Intravenous methylprednisolone is a potential add on therapy for Ring chromosome 20 syndrome. Seizure 2022, 96, 118–120. [Google Scholar] [CrossRef]
- Borković, M.; Čuturilo, G.; Cerovac, N. Ring chromosome 20: A further contribution to the delineation of epileptic phenotype. Vojnosanit. Pregl. 2022, 79, 196–200. [Google Scholar] [CrossRef]
- Kagitani-Shimono, K.; Imai, K.; Otani, K.; Kamio, N.; Okinaga, T.; Toribe, Y.; Suzuki, Y.; Ozono, K. Epilepsy in Wolf-Hirschhorn syndrome (4p-). Epilepsia 2005, 46, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Valente, K.D.; Freitas, A.; Fiore, L.A.; Kim, C.A. A study of EEG and epilepsy profile in Wolf-Hirschhorn syndrome and considerations regarding its correlation with other chromosomal disorders. Brain Dev. 2003, 25, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, O.; Irie, N.; Kawai, I. Epileptic Seizures in the 4p- Syndrome: Report of Two Cases. Psychiatry Clin. Neurosci. 1991, 45, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Mitic, V.; Cuturilo, G.; Novakovic, I.; Dimitrijevic, N.; Damnjanovic, T.; Dimitrijević, A.; Dobricic, V.; Kostic, V.; Radlovic, N. Epilepsy in a child with Wolf-Hirschhorn syndrome. Srp. Arh. Celok. Lek. 2011, 139, 795–799. [Google Scholar] [CrossRef]
- Battaglia, A.; Filippi, T.; South, S.T.; Carey, J.C. Spectrum of epilepsy and electroencephalogram patterns in Wolf–Hirschhorn syndrome: Experience with 87 patients. Dev. Med. Child Neurol. 2009, 51, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Wakui, K.; Kosho, T.; Okamoto, N.; Mizuno, S.; Itomi, K.; Hattori, S.; Nishio, K.; Samura, O.; Kobayashi, Y.; et al. Microarray and FISH-based genotype-phenotype analysis of 22 Japanese patients with Wolf-Hirschhorn syndrome. Am. J. Med. Genet. A 2014, 164, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Itakura, A.; Saito, Y.; Nishimura, Y.; Okazaki, T.; Ohno, K.; Sejima, H.; Yamamoto, T.; Maegaki, Y. Successful treatment of migrating partial seizures in Wolf-Hirschhorn syndrome with bromide. Brain Dev. 2016, 38, 658–662. [Google Scholar] [CrossRef]
- Vignoli, A.; Zambrelli, E.; Chiesa, V.; Savini, M.; La Briola, F.; Gardella, E.; Canevini, M.P. Epilepsy in adult patients with Down syndrome: A clinical-video EEG study. Epileptic Disord. 2011, 13, 125–132. [Google Scholar] [CrossRef]
- Takasugi, H.; Maemoto, T.; Kitazawa, K.; Honda, A. A case of Down syndrome with moyamoya syndrome presenting extensive multiple cerebral infarction during measles infection. No To Hattatsu 2000, 32, 39–43. [Google Scholar]
- Giovannini, S.; Frattini, D.; Scarano, A.; Fusco, C.; Bertani, G.; Della Giustina, E.; Martinelli, P.; Orteschi, D.; Zollino, M.; Neri, G.; et al. Partial epilepsy complicated by convulsive and nonconvulsive episodes of status epilepticus in a patient with ring chromosome 14 syndrome. Epileptic Disord. 2010, 12, 222–227. [Google Scholar] [CrossRef]
- Giovannini, S.; Marangio, L.; Fusco, C.; Scarano, A.; Frattini, D.; Della Giustina, E.; Zollino, M.; Neri, G.; Gobbi, G. Epilepsy in ring 14 syndrome: A clinical and EEG study of 22 patients. Epilepsia 2013, 54, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- Whitney, R.; Nair, A.; McCready, E.; Keller, A.E.; Adil, I.S.; Aziz, A.S.; Borys, O.; Siu, K.; Shah, C.; Meaney, B.F.; et al. The spectrum of epilepsy in children with 15q13.3 microdeletion syndrome. Seizure 2021, 92, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Kevelam, S.H.G.; Jansen, F.E.; Van Binsbergen, E.; Braun, K.P.; Verbeek, N.E.; Lindhout, D.; Poot, M.; Brilstra, E.H. Copy number variations in patients with electrical status epilepticus in sleep. J. Child Neurol. 2012, 27, 178–182. [Google Scholar] [CrossRef]
- Kanazawa, O.; Naruto, T.; Irie, N.; Kawai, I.; Takagi, R. A case of 18 q-syndrome associated with status epilepticus. No To Hattatsu 1989, 21, 470–474. [Google Scholar]
- Kanabar, G.; Boyd, S.; Schugal, A.; Bhate, S. Multiple causes of apnea in 1p36 deletion syndrome include seizures. Seizure 2012, 21, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Friedman, D.; Thaler, A.; Thaler, J.; Rai, S.; Cook, E.; Schanen, C.; Devinsky, O. Mortality in isodicentric chromosome 15 syndrome: The role of SUDEP. Epilepsy Behav. 2016, 61, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Brault, J.; Walsh, L.; Vance, G.H.; Weaver, D.D. Klinefelter’s Syndrome with Maternal Uniparental Disomy X, Interstitial Xp22.31 Deletion, X-linked Ichthyosis, and Severe Central Nervous System Regression. J. Pediatr. Genet. 2021, 10, 222–229. [Google Scholar] [CrossRef]
- Ricard-Mousnier, B.; N’Guyen, S.; Dubas, F.; Pouplard, F.; Guichet, A. Ring chromosome 17 epilepsy may resemble that of ring chromosome 20 syndrome. Epileptic Disord. 2007, 9, 327–331. [Google Scholar] [CrossRef]
- de Palma, L.; De Carlo, D.; Lenzini, E.; Boniver, C.; Tarantino, V.; Bacci, B.; Vecchi, M. Ring 17 syndrome: First clinical report without intellectual disability. Epileptic Disord. 2015, 17, 84–87. [Google Scholar] [CrossRef]
- Coppola, A.; Morrogh, D.; Farrell, F.; Balestrini, S.; Hernandez-Hernandez, L.; Krithika, S.; Sander, J.; Waters, J.J.; Sisodiya, S.M. Ring Chromosome 17 Not Involving the Miller-Dieker Region: A Case with Drug-Resistant Epilepsy. Mol. Syndromol. 2017, 9, 38–44. [Google Scholar] [CrossRef]
- Pelc, K.; Boyd, S.G.; Cheron, G.; Dan, B. Epilepsy in Angelman syndrome. Seizure 2008, 17, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Clynen, E.; Swijsen, A.; Raijmakers, M.; Hoogland, G.; Rigo, J.-M. Neuropeptides as Targets for the Development of Anticonvulsant Drugs. Mol. Neurobiol. 2014, 50, 626–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Besten, I.; de Jong, R.F.; Geerts-Haages, A.; Bruggenwirth, H.T.; Koopmans, M.; Brooks, A.; Elgersma, Y.; Festen, D.A.M.; Valstar, M.J. Clinical aspects of a large group of adults with Angelman syndrome. Am. J. Med. Genet. Part A 2021, 185, 168–181. [Google Scholar] [CrossRef]
- Peron, A.; Catusi, I.; Recalcati, M.P.; Calzari, L.; Larizza, L.; Vignoli, A.; Canevini, M.P. Ring Chromosome 20 Syndrome: Genetics, Clinical Characteristics, and Overlapping Phenotypes. Front. Neurol. 2020, 11, 613035. Available online: https://www.frontiersin.org/articles/10.3389/fneur.2020.613035 (accessed on 19 November 2022). [CrossRef] [PubMed]
- Vignoli, A.; Canevini, M.P.; Darra, F.; La Selva, L.; Fiorini, E.; Piazzini, A.; Lazzarotto, F.; Zucca, C.; Bernardina, B.D. Ring chromosome 20 syndrome: A link between epilepsy onset and neuropsychological impairment in three children. Epilepsia 2009, 50, 2420–2427. [Google Scholar] [CrossRef] [PubMed]
- Zollino, M.; Ponzi, E.; Gobbi, G.; Neri, G. The ring 14 syndrome. Eur. J. Med. Genet. 2012, 55, 374–380. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Chromosomal Disorders and Epilepsy |
|---|
| 15q13.3 Microdeletion Syndrome (Del15q13.3) |
| 18q- Syndrome (Del18q) |
| InvDup (15) or IDIC (15) |
| Del 1p36 |
| Angelman Syndrome (AS) |
| Down Syndrome (DS—Trisomy 21) |
| Klinefelter Syndrome (XXY) |
| Miller Dieker Syndrome (Del 17p) |
| Pallister Killian Syndrome (Tetrasomy 12p) |
| Ring 14 (R14) Syndrome |
| Ring 20 (R20) Syndrome |
| Trisomy 12p |
| Wolf-Hirschhorn Syndrome (WHS—Del 4p) |
| Authors | Population (Number, Sex, Age) | Patients with SE (Number, Sex, Age) | Previous Epilepsy | SE Semeiology | SE Recurrence | SE Treatment |
|---|---|---|---|---|---|---|
| Sugimoto T. et al., 1992 [11] | N:3, M:2; 1–6 yo | N:1; M; 6 yo | Yes | NCSE | - | CZP |
| Matsumoto A. et al., 1992 [12] | N:8; Na; Na | N: 2; Na; 8–10 yo | Yes | NCSE | Yes | 1: DZP; DZP and TRH (1 mg/day) 2: DZP; |
| Viani F. et al., 1995 [13] | N:18; M:12, F:6; 8 m–27 yo | N: 9; Na; < 6 yo | - | MSE | - | DZP, VPA, CLB |
| Rubin D.I. et al., 1997 [14] | N:3; M:3; 2 y 3 m | N: 1; Na; Na | Yes | NCSE (ESES) | Yes | - |
| Laan L.A. et al., 1997 [15] | N: 36; M: 20, F:16; 1–32 yo | N: 7; Na; Na, | Yes (n:5) No (n: 2) | NCSE, CSE, MSE | Yes | - |
| Galván-Manso M. et al., 2005 [16] | N:37; F:19, M:18; 3–23 yo | N: 15; Na; Na | - | NCSE | - | - |
| Espay A.J. et al., 2005 [17] | N:1; M; 29 yo | N: 1; Na; Na | Yes | NCSE | Yes | - |
| Uemura N. et al., 2005 [18] | N:23; M:10, F:13; 3 m–37 yo | N: 11; Na; 1–15 yo | Yes (n: 10), No (n: 1) | NCSE, CSE | Yes | - |
| Ohtsuka Y. et al., 2005 [19] | N:11; M:6, F:5; 3–15 yo | N: 10; M: 5, F: 5; 8 m–8 yo | - | NCSE | Yes | 1: iv DZP, VPA; 3: iv DZP, CZP, CLB, VPA; 4: iv DZP, VPA; 5: iv DZP, VPA, ESM, ACTH; 6: iv DZP, CZP, VPA, ESM, ACTH; 7: iv DZP, CZP, VPA; 8: VPA; 9: VPA; 10: iv DZP, VPA; 11: CZP, VPA |
| Valente K.D. et al., 2006 [10] | N:19; Na; Na | N: 16; Na; Na | Yes | NCSE, MSE | Yes | - |
| Caraballo R.H. et al., 2007 [20] | N:15; Na; Na | N:17; Na; <1 yo | - | MSE | - | - |
| Yang X.Y. et al., 2010 [21] | N:14; M:4, F:10; 8 m–3 yo | N: 10; Na; Na | Yes (n: 9), No (n:1) | NCSE | Yes | - |
| Weber P., 2010 [22] | N:1; M:1; 7 yo | N: 1; M:1; 7 yo | Yes | CSE, NCSE | Yes | LEV |
| Valente K.D. et al., 2012 [23] | N:16; Na; Na | N:5; Na; Na | - | - | - | - |
| Nicita F. et al., 2015 [24] | N:1; M:1; Na | N:1; Na; Na | No | MSE | Yes | MDZ (two boluses), VPA, VGB, LEV was finally effective |
| Grocott O.R. et al., 2017 [25] | N:23; M:15, F:8; 2–31 yo; | N:3; Na; Na | - | NCSE | Yes | - |
| Worden L. et al., 2018 [26] | N:104; Na; <18 yo | N:13; M:9, F:4; 15 m–12 yo | Yes (n: 10), No (n: 3) | NCSE | Yes | Oral DZP |
| Pollack S.F. et al., 2018 [27] | N:185; M:102, F:83; 1–44 yo | N: 1; Na; Na | MSE | - | - | |
| Bindels-de Heus K.G.C.B. et al., 2020 [28] | N:100; M:50, F:50; <18 y | N: 24; Na; Na | NCSE, CSE | Yes | - | |
| Melikishvili G et al., 2022 [29] | N:2; Na; Na | N:2; Na; Na | NCSE | Yes | 1: DZP, VPA, B6; KD 4:1 radio;2: KD |
| Positive Findings | Negative Findings |
|---|---|
| Motor phenomena: clumsiness, drooling of saliva, eye deviation, eye dropping Minor motor components: blinking, trembling, and clusters of jerky movements (especially in the distal regions of the limbs) Behavioral changes: agitation, emotional liability Sleep: somnolence Seizures: increased seizure frequency | Motor phenomena: psychomotor arrest (staring), arrest of stereotyped movements (i.e.: hand flapping) Behavioral changes: Minor readiness to laugh, loss of interest in food Seizures: lower frequency of motor seizures in those regularly experiencing them Sleep: poor quality Consciousness: decreased alertness and responsiveness, poor communication, loss of acquired milestones |
| Authors | Population (Number, Sex, Age) | Patients with SE (Number, Sex, Age) | Previous Epilepsy | SE Semeiology | SE Recurrence | SE Treatment |
|---|---|---|---|---|---|---|
| Inoue Y. et al., 1997 [32] | N:6; F:4, M:2; 13–48 yo | N:6; F:4, M:2; 3–14 yo | No (N: 5); Yes (N: 1) | NCSE | Yes (daily) | 1: iv DZP; 2: iv lidocaine; 3-4-5-6, |
| Kobayashi K. et al., 1998 [33] | N:2; F:1, M:1; 22 yo | N:2; F:1, M:1; 22 | Yes | CSE | Yes | 1: PB; 2: Na |
| Petit J. et al., 1999 [34] | N:3; M:1, F:2; 22–43 yo; | N:3; M:1, F:2; Na | Yes | NCSE | Yes | - |
| Augustijn P. et al., 2001 [35] | N:4; F:3, M:1; 8–13 yo; | N:4; F:3; M:1; 8–14 yo | Yes | NCSE | Yes | - |
| Gomes M. et al., 2002 [36] | N:1; M:1; 12 yo | N:1; M:1, 11.5 | Yes | NCSE | Yes | - |
| Gonzalez-Delgado M. et al., 2004 [37] | N:1; Na; 18 yo | N:1; Na; Na | Yes | NCSE | - | CZP |
| Locharernkul C. et al., 2005 [38] | N:2; F:2; 25–37 yo | N:2; F:2; 21–27 yo | Yes | NCSE | - | 1: iv PHT, iv VPA; 2: iv VPA |
| Alpman A. et al., 2005 [39] | N:1; M:1; 14 yo | N:1; M:1; 11 yo | Yes | NCSE | Yes | - |
| de Falco F. et al., 2006 [40] | N:1; F:1; 46 yo | N:1 F;1; 39 | Yes | CSE + NCSE | Yes | - |
| Zou Y.S. et al., 2006 [41] | N:1; F:1; 26 yo | N:1; F:1; Na | Yes | NCSE | Yes | - |
| Elghezal H. et al., 2007 [42] | N:1; F:1; 12 yo | N:1; F:1; Na | Yes | NCSE | Yes | - |
| Jacobs J. et al., 2009 [43] | N:1; M:1; 13 yo | N:1; M:1; Na | Yes | NCSE | Yes | - |
| Elens I. et al., 2012 [44] | N:6; M:4, F:2; 4–53 yo | N:6; M:4, F:2; 4–54 yo | Yes | NCSE | - | - |
| Radhakrishnan A. et al., 2012 [45] | N:3; M:3; 10–20 yo | N:2; M:2; 10 | Yes | NCSE | - | - |
| Wechapinan T. et al., 2014 [46] | N:1; F:1; 11 yo | N:1; F:1; 11 yo | Yes | NCSE | - | iv MDZ, LTG, LEV, CBZ, ZNS |
| Onder H. et al., 2015 [47] | N:1; F:1; 25 yo | N:1; F:1, Na | Yes | NCSE | - | LCS |
| Vignoli A. et al., 2016 [48] | N:25; F:17, M:8; 8–59 yo | N:22; Na; Na | Yes | NCSE | Yes | - |
| Hirano Y. et al., 2016 [49] | N:1; F:1; 17 yo | N:1, F:1, 13 yo | Yes | CSE | Yes | MDZ, PHT, PB, sodium thiopental |
| de Moura F. et al., 2016 [30] | N:24; F:10, M:14; 17–57 yo | N:24; F:10, M:14; Na | - | - | - | - |
| Gago-Veiga A.B. et al., 2018 [31] | N:6; F:5, M:1; 11–30 yo | N:6; F:5, M:1; 2–15 yo | Yes | NCSE (N:6); CSE (N:1) | Yes | - |
| Lee H. et al., 2020 [50] | N:1; F:1; 17 yo | N:1; F:1; 8 yo | Yes | NCSE | - | - |
| Yamagishi H. et al., 2020 [51] | N:2; F:1, M:1, 6–11 yo | N:2; F:1, M:1, Na | Yes | NCSE | - | LCS |
| Kishore V.K. et al., 2022 [52] | N:1; Na; Na; | N:1; Na; Na | Yes | NCSE | - | Iv mPRED |
| Borkovic M. et al., 2022 [53] | N:4; F:3, M:1; 9–18 yo | N:2; F:2; Na | - | NCSE + CSE | - | - |
| Positive Findings | Negative Findings |
|---|---|
| Consciousness: confusion Motor phenomena: wandering Minor motor components: automatisms, jerks, myoclonias in the distal regions of the limbs Seizures: mild clonic or tonic seizures over a prevalent confusional state Behavioral changes: agitation, fear, emotional liability, screaming Speech: Sudden post-aphasic speech | Consciousness: impaired vigilance Behavioral changes: Loss of emotional facial expressions, unresponsiveness, or mental slowing; behavioral arrest Speech: Diminished spontaneous speech, aphasia, mutism. |
| Syndrome | Authors | Patients with the Syndrome (Number, Sex, Age) | Patients with SE (Number, Sex, Age) | Previous Epilepsy | SE Semeiology | SE Recurrence | SE Treatment |
|---|---|---|---|---|---|---|---|
| WHS | Kanazawa O. et al., 1991 [56] | N:2; M:1, F:1; 2–3 yo | N:1; F:1; 2 yo | No | CSE, Hemiclonic SE | Yes | DZP, PHT, ZNS |
| WHS | Kagitani K. et al, 2005 [54] | N: 11; F:8; M:3; 2–25 yo | N:11; F:8; M:3; 2–3 yo | Yes | CSE, Hemiclonic SE, NCSE | Yes | DZP (N: 8); LD (N:1); TS (N:1); TP (N:1); Bromide (N:4) |
| WHS | Mitić V. et al., 2011 [57] | N:1; F:1; 9 m | N:1; F:1; 9 m | No | Hemiclonic SE | Yes | PB |
| WHS | Caraballo R.H. et al., 2007 [20] | N:2; Na; Na | N:2; Na; Na | Na | MSE | Yes | - |
| WHS | Battaglia A. et al., 2009 [58] | N:87; F:54; M:33; 1–25.6 yo | N:36; Na; <3y yo | Yes | CSE | - | - |
| WHS | Valente K.D. et al., 2003 [55] | N:1; F:1; 2 yo | N:1; F:1; 2 yo | Yes | NCSE, MSE | Yes | PB |
| WHS | Shimizu K. et al., 2014 [59] | N: 22; F: 18, M: 4; Na | N: 14; F:11; M:3; Na | Yes | - | - | Bromide (N:4) |
| WHS | Itakura A. et al., 2016 [60] | N:1; F:1; 3 m | N:1; F:1; 3 m | No | Hemiclonic SE, CSE | - | MDZ; PB |
| DS | Vignoli A. et al., 2011 [61] | N:22; F:11, M:11; 28–64 yo | N:3; M:2, F:1; Na | Yes | MSE | Yes | VPA |
| DS | Takasugi H. et al., 2000 [62] | N:1; Na; Na | N: 1; Na; Na | No | - | - | - |
| R14 | Giovannini S. et al., 2010 [63] | N:1; M:1; 9 yo | N:1; M:1; 15 m | No | CSE; NCSE | - | BZD, PHT |
| R14 | Giovannini S. et al., 2013 [64] | N:22; F:9, M:13; 2–22 yo | N:13; F:3, M:10; Na | Yes (N:5); No (N:3) | CSE (N:9); NCSE (N:3); Na (N:3) | - | BZD, PHT |
| Del15q13.3 | Whitney R. et al., 2021 [65] | N:13; F:9, M:4; 3–15 yo | N:3; F:2, M:1; 3–8.5 yo | - | NCSE (N:2, ESES (N:1)); CSE (N:1); | - | - |
| Del15q13.3 | Kevelam S.H. et al., 2012 [66] | N:13; Na; Na | N:1; Na; 7 yo | No | NCSE (ESES) | - | - |
| Del18q | Kanazawa O. et al., 1989 [67] | N:1; M:1; 15 yo | N:1; M:1; 15 yo | Yes | Hemiclonic SE | - | - |
| Del1p36 | Kanabar G. et al., 2012 [68] | N:4; M:3, F:1; 2–16 yo | N:4; M:3, F:1; 2–16 yo | Yes | - | - | - |
| InvDup15 | Friedman D. et al., 2016 [69] | N:19; F:8, M:11; 2.5–39 yo | N:7; F:3, M:4; 10–26 yo | Yes | - | - | - |
| XXY | Brault J. et al., 2021 [70] | N:1; M:1,7 m | N:1; M:1; 7 m | No | - | - | BZD, PB, PHT, LEV |
| R17 | Ricard-Mousnier B. et al., 2007 [71] | N:1; M:1; 4 yo | N:1; M:1; 4 yo | Yes | NCSE | - | - |
| R17 | de Palma L. et al., 2015 [72] | N:1, F:1, 17 yo | N:1; F:1; 11 yo | Yes | NCSE | - | - |
| R17 | Coppola A. et al., 2017 [73] | N:1, F:1, 31 yo | N: 1; F: 1; 28 yo | Yes | NCSE | - | BDZ |
| Semiology | Number of Patients |
|---|---|
| Generalized tonic–clonic | 4 [54] |
| Tonic | 2 [54] |
| Myoclonic | 2 [58] |
| Focal motor, Hemiclonic | 7 [54], 4 [54,56,57,60] |
| Generalized or focal motor SE | 36 [55,58] |
| Syndrome | SE Semeiology | Patients with SE Reported (Number) | Age Range of SE | SE Recurrence | Ictal EEG | SE Treatment |
|---|---|---|---|---|---|---|
| AS | NCSE, MSE | 150 | 8 m–15 yo | Yes | Diffuse high voltage 1.5–3 Hz SW | Remove triggers; BZD; VPA; LEV |
| R20 | NCS | 93 | 2–54 yo | Yes | Generalized or predominantly frontal slow waves and spikes or SW | BZD, LCS |
| WHS | CSE, Hemiclonic | 67 | 3 m–3 yo | Yes | - | BZD, Bromide, PB |
| DS | MSE | 4 | - | - | - | VPA |
| R14 | CSE | 14 | - | - | - | BZD, PHT |
| Del15q13.3 | ESES | 4 | 3–8.5 yo | - | SW during sleep | - |
| Del18q | Hemiclonic | 1 | 15 yo | - | - | - |
| Del1p36 | - | 4 | 2–16 yo | - | - | - |
| InvDup15 | - | 7 | 10–26 yo | - | - | - |
| XXY | - | 1 | 7 m | - | - | - |
| Ring 17 | NCSE | 3 | 4–28 yo | - | Sub-continuous diffuse slow SW, slow background activity. | BZD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergonzini, L.; Pruccoli, J.; Pettenuzzo, I.; Pugliano, R.; Soliani, L.; Fetta, A.; Cordelli, D.M. Status Epilepticus in Chromosomal Disorders Associated with Epilepsy: A Systematic Review. Genes 2023, 14, 299. https://doi.org/10.3390/genes14020299
Bergonzini L, Pruccoli J, Pettenuzzo I, Pugliano R, Soliani L, Fetta A, Cordelli DM. Status Epilepticus in Chromosomal Disorders Associated with Epilepsy: A Systematic Review. Genes. 2023; 14(2):299. https://doi.org/10.3390/genes14020299
Chicago/Turabian StyleBergonzini, Luca, Jacopo Pruccoli, Ilaria Pettenuzzo, Rosa Pugliano, Luca Soliani, Anna Fetta, and Duccio Maria Cordelli. 2023. "Status Epilepticus in Chromosomal Disorders Associated with Epilepsy: A Systematic Review" Genes 14, no. 2: 299. https://doi.org/10.3390/genes14020299