
Improved Outcomes in Patients with 22q11.2 Deletion Syndrome and Diagnosis of Interrupted Aortic Arch Prior to Birth Hospital Discharge, a Retrospective Study
 , , , , ,
, , , , , 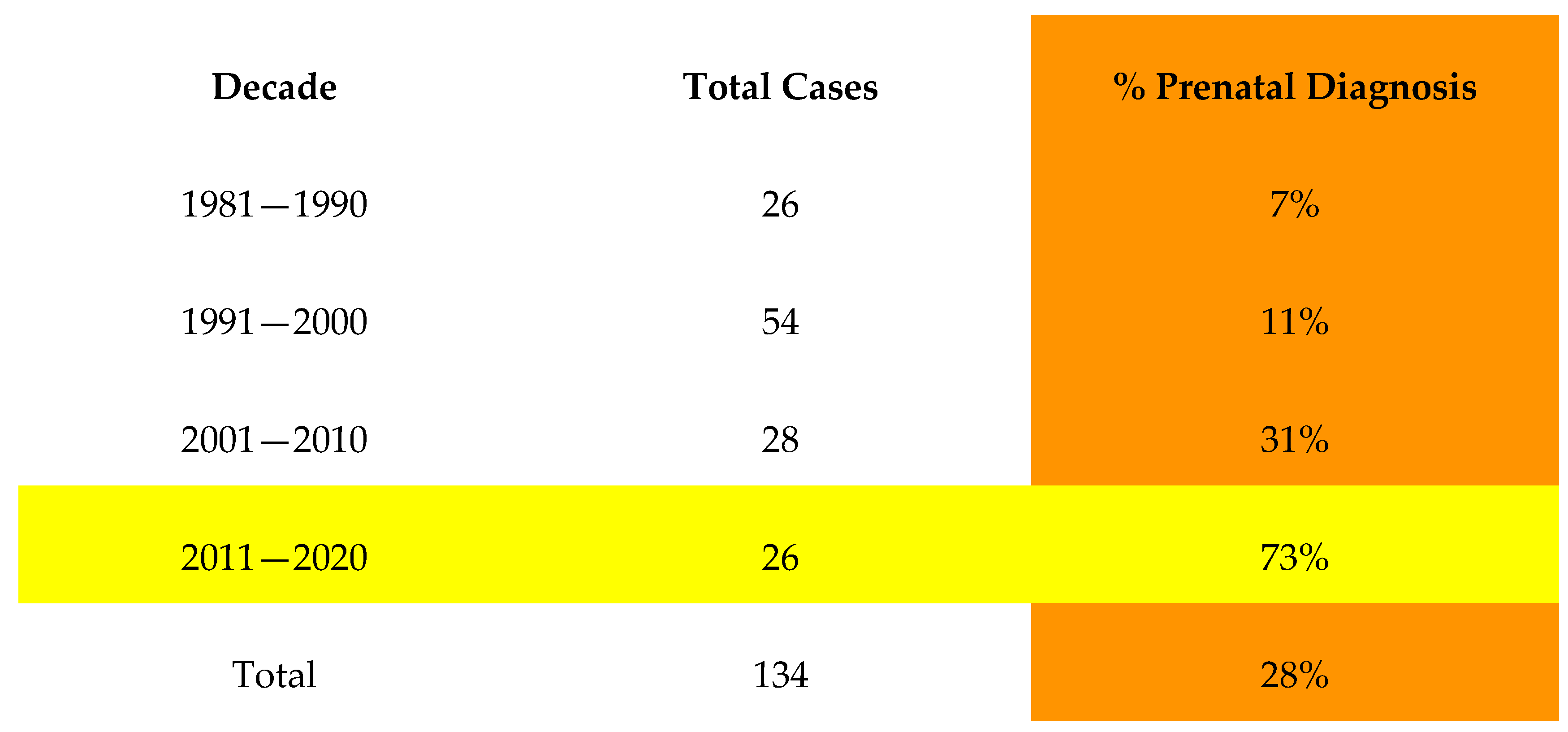{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
1.1. Anatomy of IAA
1.2. Prevalence and Implication of 22q11.2DS in IAA
1.3. Prenatal Diagnosis of IAA
1.4. Deletion Size
2. Materials and Methods
3. Results
3.1. Cohort
3.2. Deletion Size
3.3. Prenatal Diagnosis
3.4. Postnatal Detection after Birth Hospital Discharge
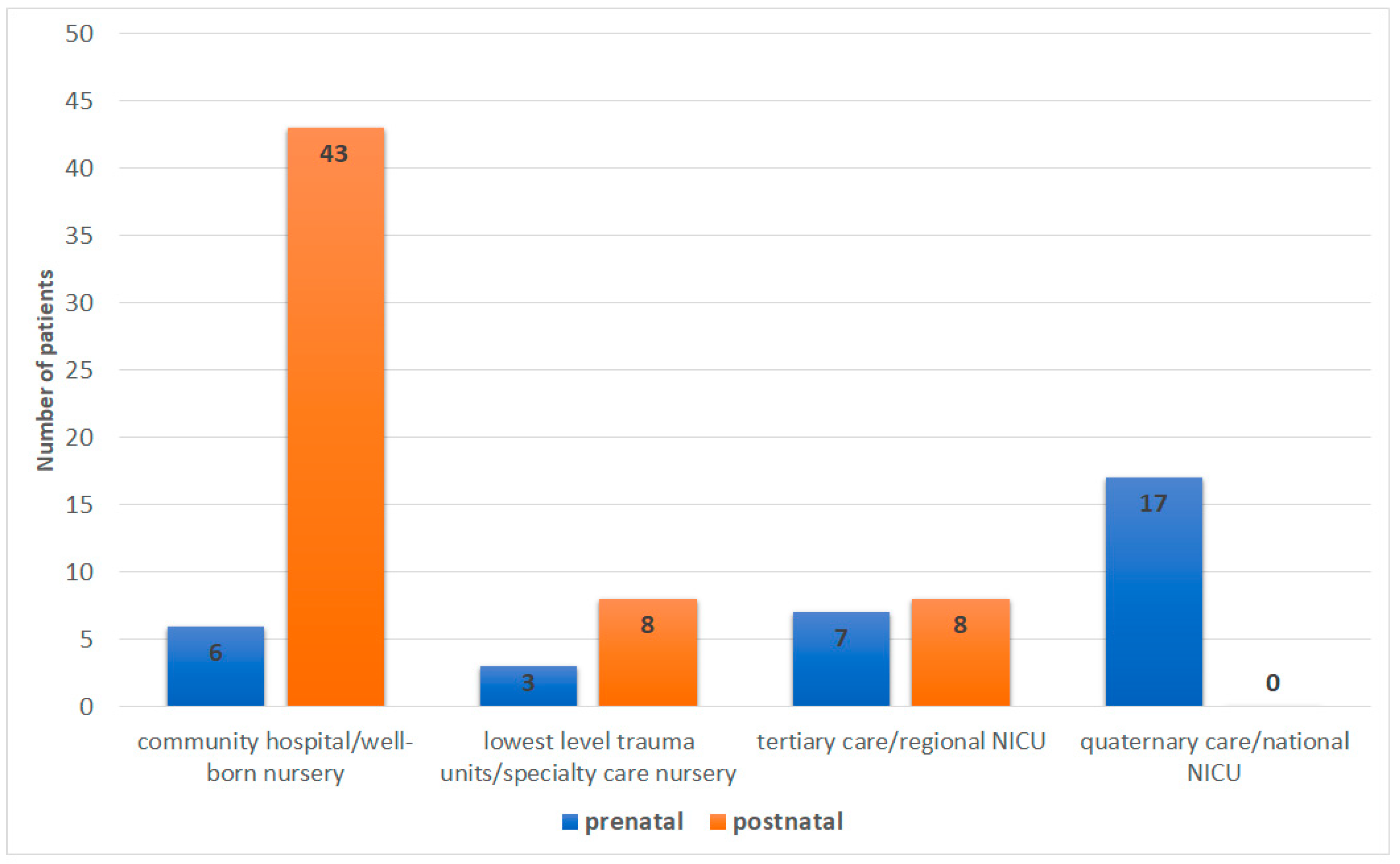
3.5. Level of Care of Birth Hospital
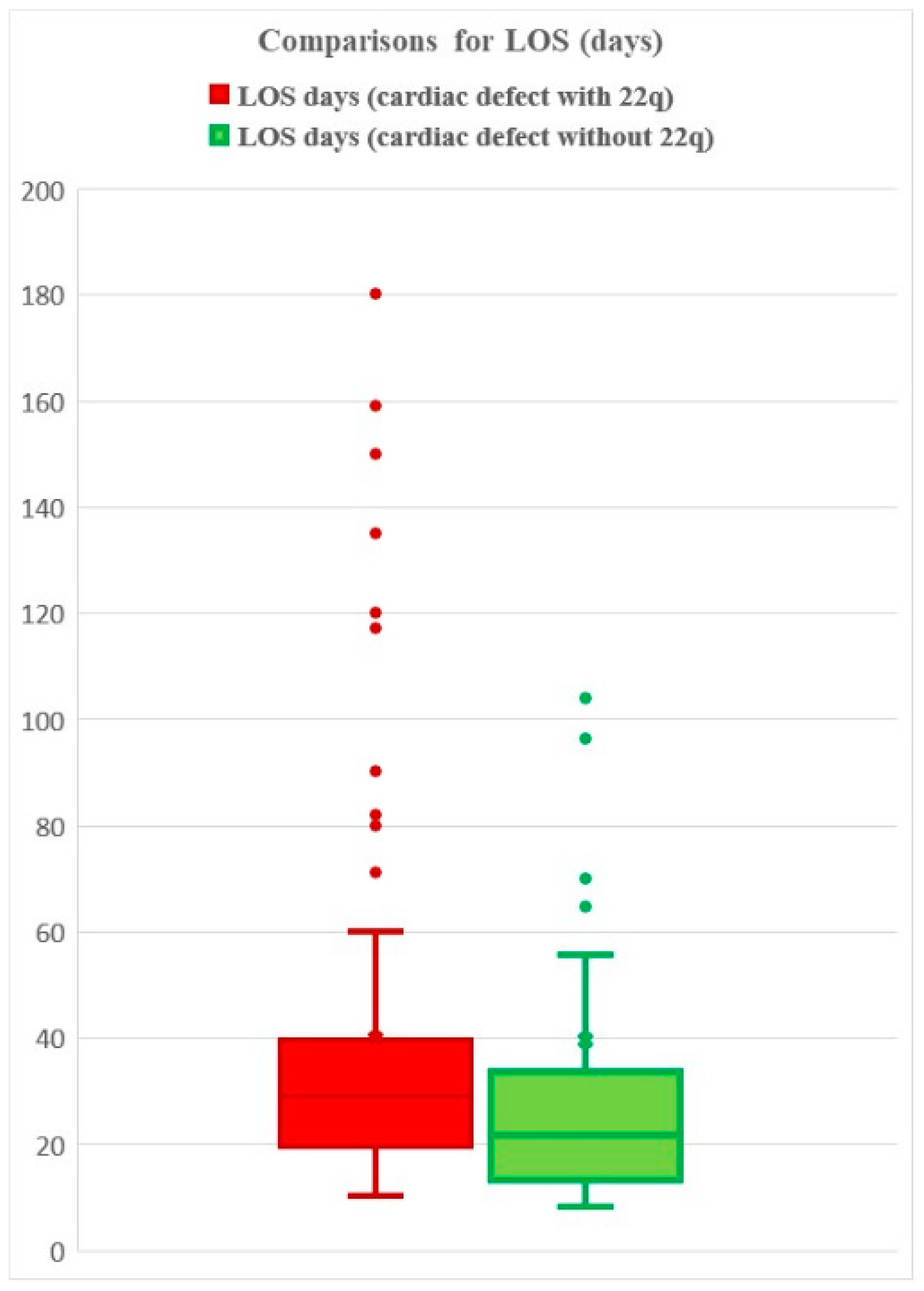
3.6. Length of Hospital Stay
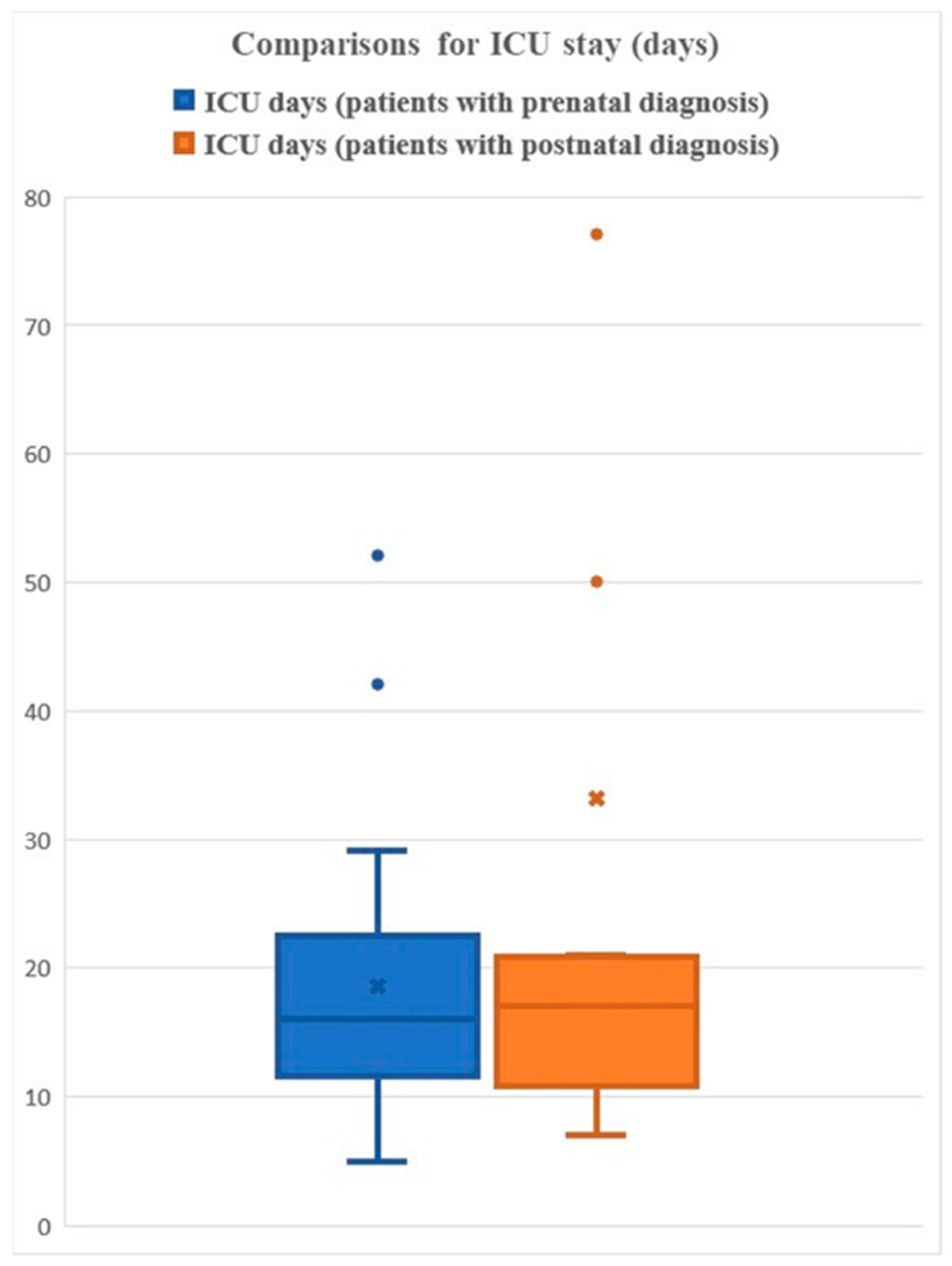
3.7. Length of ICU Stay
3.8. Duration of Intubation
3.9. Duration of Inotrope Usage
3.10. Shock
3.11. Mortality
3.12. Comparison to Non-Deleted Patients with IAA
3.12.1. ICU
3.12.2. LOS
3.13. Parental Deletion Status
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perloff, J.K.; Marelli, A.J. Coarctation of the Aorta and Interrupted Aortic Arch. In Clinical Recognition of Congenital Heart Disease; Elsevier: Amsterdam, The Netherlands, 2012; pp. 101–128. [Google Scholar]
- Takashina, T.; Ishikura, Y.; Yamane, K.; Yorifuji, S.; Iwasaki, T.; Yoshida, Y.; Takeshita, I.; Oka, K. The congenital cardiovascular anomalies of the interruption of the aorta-Steidel’s complex. Am. Heart J. 1972, 83, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, G.; Schreiber, R.; Meisner, H.; Lorenz, H.P.; Sebening, F.; Bühlmeyer, K. Interrupted aortic arch: Natural history and operative results. Pediatr. Cardiol. 1986, 7, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.M.; Sheppard, S.E.; Crowley, T.B.; McGinn, D.E.; Bailey, A.; McGinn, M.J.; Unolt, M.; Homans, J.F.; Chen, E.Y.; Salmons, H.I.; et al. What is new with 22q? An update from the 22q and You Center at the Children’s Hospital of Philadelphia. Am. J. Med. Genet. Part A 2018, 176, 2058–2069. [Google Scholar] [CrossRef]
- Goldmuntz, E.; Clark, B.J.; Mitchell, L.E.; Jawad, A.F.; Cuneo, B.F.; Reed, L.; McDonald-McGinn, D.; Chien, P.; Feuer, J.; Zackai, E.H.; et al. Frequency of 22q11 deletions in patients with conotruncal defects. J. Am. Coll. Cardiol. 1998, 32, 492–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyvandi, S.; Lupo, P.J.; Garbarini, J.; Woyciechowski, S.; Edman, S.; Emanuel, B.S.; Mitchell, L.E.; Goldmuntz, E. 22q11.2 Deletions in Patients with Conotruncal Defects: Data from 1,610 Consecutive Cases. Pediatr. Cardiol. 2013, 34, 1687–1694. [Google Scholar] [CrossRef]
- Volpe, P.; Marasini, M.; Caruso, G.; Gentile, M. Prenatal diagnosis of interruption of the aortic arch and its association with deletion of chromosome 22q11. Ultrasound Obstet. Gynecol. 2002, 20, 327–331. [Google Scholar] [CrossRef]
- Lindsay, E.A.; Goldberg, R.; Jurecic, V.; Morrow, B.; Carlson, C.; Kucherlapati, R.S.; Shprintzen, R.J.; Baldini, A. Velo-cardio-facial syndrome: Frequency and extent of 22q1l deletions. Am. J. Med. Genet. 1995, 57, 514–522. [Google Scholar] [CrossRef]
- Takahashi, K.; Kuwahara, T.; Nagatsu, M. Interruption of the aortic arch at the isthmus with DiGeorge syndrome and 22q11.2 deletion. Cardiol. Young 1999, 9, 516–518. [Google Scholar] [CrossRef]
- O’Byrne, M.L.; Yang, W.; Mercer-Rosa, L.; Parnell, A.S.; Oster, M.E.; Levenbrown, Y.; Tanel, R.E.; Goldmuntz, E. 22q11.2 Deletion syndrome is associated with increased perioperative events and more complicated postoperative course in infants undergoing infant operative correction of truncus arteriosus communis or interrupted aortic arch. J Thorac Cardiovasc Surg. 2014, 148(4), 1597–1605. [Google Scholar] [CrossRef] [Green Version]
- Vogel, M.; Vernon, M.M.; McElhinney, D.B.; Brown, D.W.; Colan, S.D.; Tworetzky, W. Fetal Diagnosis of Interrupted Aortic Arch. Am. J. Cardiol. 2010, 105, 727–734. [Google Scholar] [CrossRef]
- Axt-Fliedner, R.; Kawecki, A.; Enzensberger, C.; Wienhard, J.; Degenhardt, J.; Schranz, D.; Vogel, M. Fetal and neonatal diagnosis of interrupted aortic arch: Associations and outcomes. Fetal Diagn. Ther. 2011, 30, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Committee on Practice Bulletins—Obstetrics and the American Institute of Ultrasound in Medicine. Practice Bulletin No. 175: Ultrasound in Pregnancy. Obstet. Gynecol. 2016, 128, e241–e256. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, D.A.; Spinner, N.B.; Budarf, M.L.; McDonald-McGinn, D.M.; Zackai, E.H.; Goldberg, R.B.; Shprintzen, R.J.; Saal, H.M.; Zonana, J.; Jones, M.C.; et al. Deletions and microdeletions of 22q11.2 in velo-cardio-facial syndrome. Am. J. Med. Genet. 1992, 44, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Scambler, P.J.; Carey, A.H.; Wyse, R.K.; Roach, S.; Dumanski, J.P.; Nordenskjold, M.; Williamson, R. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics 1991, 10, 201–206. [Google Scholar] [CrossRef]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 Deletion Syndrome. Nat. Rev. Dis. Prim. 2015, 1, 15071. [Google Scholar] [CrossRef] [Green Version]
- Trauma Centers. American College of Surgeons. Available online: https://www.facs.org/hospital-and-facilities/ (accessed on 21 July 2022).
- Mcdonald-Mcginn, D.M.; Tonnesen, M.K.; Laufer-Cahana, A.; Finucane, B.; Driscoll, D.A.; Emanuel, B.S.; Zackai, E.H. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: Cast a wide FISHing net! Genet. Med. 2001, 3, 23–29. [Google Scholar] [CrossRef] [Green Version]
- ROBERTS, W.C.; MORROW, A.G.; BRAUNWALD, E. Complete interruption of the aortic arch. Circulation 1962, 26, 39–59. [Google Scholar] [CrossRef] [Green Version]
- Higgins, C.B.; French, J.W.; Silverman, J.F.; Wexler, L. Interruption of the aortic arch: Preoperative and postoperative clinical, hemodynamic and angiographic features. Am. J. Cardiol. 1977, 39, 563–571. [Google Scholar] [CrossRef]
- Jonas, R.A. Management of Interrupted Aortic Arch. Semin. Thorac. Cardiovasc. Surg. 2015, 27, 177–188. [Google Scholar] [CrossRef]
- Ailes, E.C.; Gilboa, S.M.; Honein, M.A.; Oster, M.E. Estimated Number of Infants Detected and Missed by Critical Congenital Heart Defect Screening. Pediatrics 2015, 135, 1000–1008. [Google Scholar] [CrossRef]
- Blagojevic, C.; Heung, T.; Theriault, M.; Tomita-Mitchell, A.; Chakraborty, P.; Kernohan, K.; Bulman, D.E.; Bassett, A.S. Estimate of the contemporary live-birth prevalence of recurrent 22q11.2 deletions: A cross-sectional analysis from population-based newborn screening. CMAJ Open 2021, 9, E802–E809. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, B.; Prettenhofe, P.; Weis, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ron, H.A.; Crowley, T.B.; Liu, Y.; Unolt, M.; Schindewolf, E.; Moldenhauer, J.; Rychik, J.; Goldmuntz, E.; Emanuel, B.S.; Ryba, D.; et al. Improved Outcomes in Patients with 22q11.2 Deletion Syndrome and Diagnosis of Interrupted Aortic Arch Prior to Birth Hospital Discharge, a Retrospective Study. Genes 2023, 14, 62. https://doi.org/10.3390/genes14010062
Ron HA, Crowley TB, Liu Y, Unolt M, Schindewolf E, Moldenhauer J, Rychik J, Goldmuntz E, Emanuel BS, Ryba D, et al. Improved Outcomes in Patients with 22q11.2 Deletion Syndrome and Diagnosis of Interrupted Aortic Arch Prior to Birth Hospital Discharge, a Retrospective Study. Genes. 2023; 14(1):62. https://doi.org/10.3390/genes14010062
Chicago/Turabian StyleRon, Hayley A., Terrence Blaine Crowley, Yichuan Liu, Marta Unolt, Erica Schindewolf, Julie Moldenhauer, Jack Rychik, Elizabeth Goldmuntz, Beverly S. Emanuel, Douglas Ryba, and et al. 2023. "Improved Outcomes in Patients with 22q11.2 Deletion Syndrome and Diagnosis of Interrupted Aortic Arch Prior to Birth Hospital Discharge, a Retrospective Study" Genes 14, no. 1: 62. https://doi.org/10.3390/genes14010062