
The Power of Clinical Diagnosis for Deciphering Complex Genetic Mechanisms in Rare Diseases


Abstract
:1. Introduction
2. Unravelling Complex Genetic Mechanisms of RDs Based on Clinical Diagnosis
2.1. Non-Coding Variants in Patients with Established Clinical Phenotypes
2.1.1. Single-Nucleotide Variants
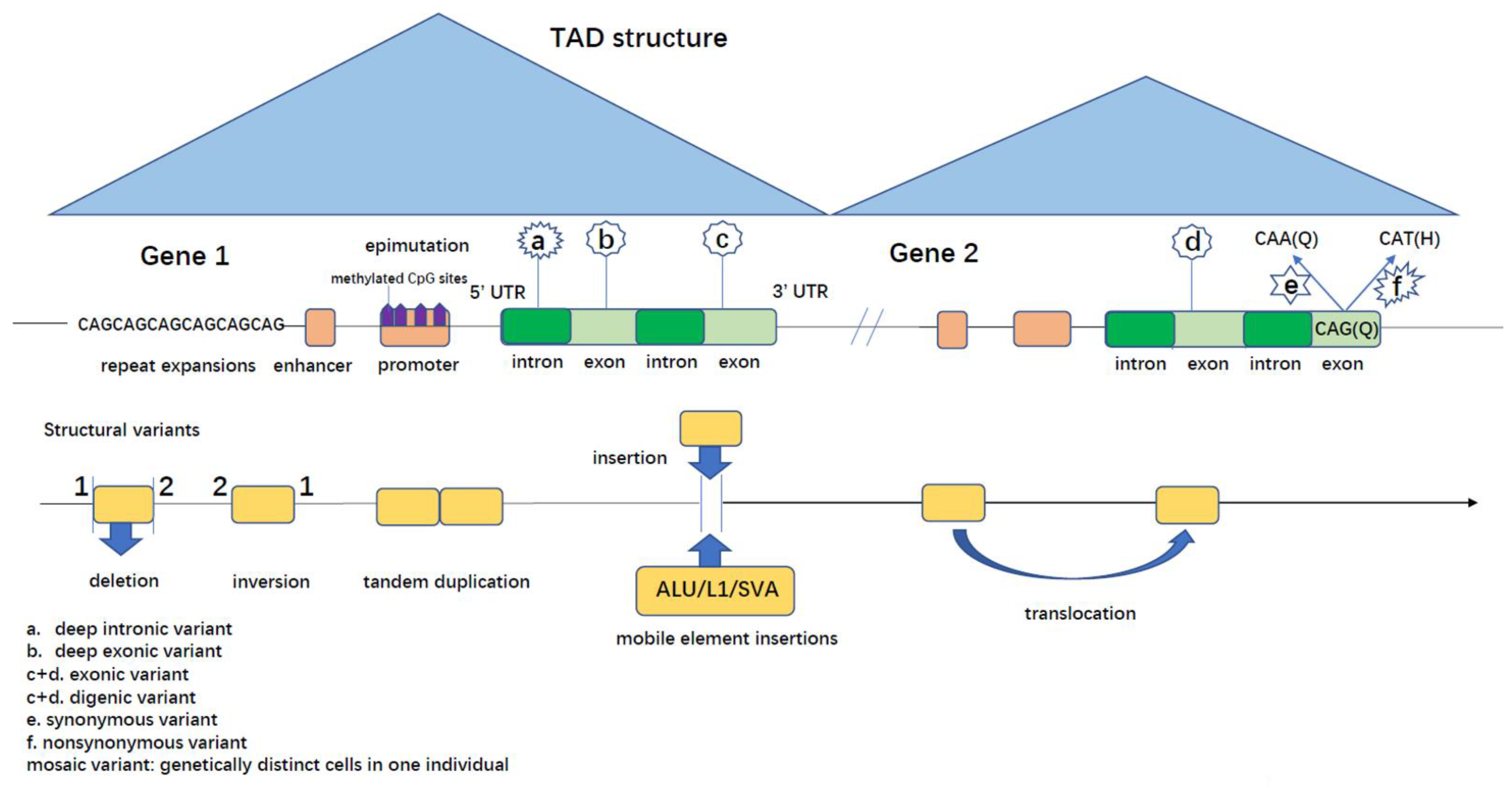
- Deep Intronic Variants
- b.
- 5′ and 3′ Untranslated Regions (UTRs) Variants
- c.
- Regulatory Elements
2.1.2. Complex Variants
- Mobile Element Insertions (MEIs)
- b.
- Repeat Expansions (REs)
- c.
- Genomic Rearrangements (GRs)
- d.
- Epigenetic Changes
2.2. Coding Variants in Patients with Established Clinical Phenotypes
2.2.1. Single-Nucleotide Variants
- Deep Exonic Variants
- b.
- Synonymous Variants
2.2.2. Complex Variants
- MEIs
- b.
- GRs
- c.
- Mosaic Variants
- d.
- Oligogenic Inheritance
3. Functional Confirmation of the Role of Complex Genetic Mechanisms in RDs
4. Clinical Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Wright, C.F.; FitzPatrick, D.R.; Firth, H.V. Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 2018, 19, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, J.; Sedlmayr, M.; Schaefer, J.; Storf, H. Diagnosis of Rare Diseases: A scoping review of clinical decision support systems. Orphanet J. Rare Dis. 2020, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, S.D.; Avramović, V.; Maroilley, T.; Lehman, A.; Arbour, L.; Tarailo-Graovac, M. Rare disorders have many faces: In silico characterization of rare disorder spectrum. Orphanet J. Rare Dis. 2022, 17, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Groft, S.C.; Posada, M.; Taruscio, D. Progress, challenges and global approaches to rare diseases. Acta Paediatr. 2021, 110, 2711–2716. [Google Scholar] [CrossRef] [PubMed]
- Maroilley, T.; Tarailo-Graovac, M. Uncovering Missing Heritability in Rare Diseases. Genes 2019, 10, 275. [Google Scholar] [CrossRef] [Green Version]
- Ellingford, J.M.; Ahn, J.W.; Bagnall, R.D.; Baralle, D.; Barton, S.; Campbell, C.; Downes, K.; Ellard, S.; Duff-Farrier, C.; FitzPatrick, D.R.; et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 2022, 14, 1–19. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.; Tarailo-Graovac, M.; Richmond, P.A.; Drögemöller, B.I.; Pouladi, M.A.; Leen, R.; Brand-Arzamendi, K.; Dobritzsch, D.; Dolzhenko, E.; Eberle, M.A.; et al. Glutaminase Deficiency Caused by Short Tandem Repeat Expansion in GLS. N. Engl. J. Med. 2019, 380, 1433–1441. [Google Scholar] [CrossRef]
- Ishiura, H.; Doi, K.; Mitsui, J.; Yoshimura, J.; Matsukawa, M.K.; Fujiyama, A.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; Suzuki, Y.; et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat. Genet. 2018, 50, 581–590. [Google Scholar] [CrossRef]
- Smedley, D.; Smith, K.R.; Martin, A.; Thomas, E.A.; McDonagh, E.M.; Cipriani, V.; Ellingford, J.M.; Arno, G.; Tucci, A.; Vandrovcova, J.; et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care—Preliminary Report. N. Engl. J. Med. 2021, 385, 1868–1880. [Google Scholar] [CrossRef]
- Martinez-Delgado, B.; Barrero, M.J. Epigenomic Approaches for the Diagnosis of Rare Diseases. Epigenomes 2022, 6, 21. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Drögemöller, B.I.; Wasserman, W.W.; Ross, C.J.D.; Ouweland, A.M.W.V.D.; Darin, N.; Kollberg, G.; van Karnebeek, C.D.M.; Blomqvist, M. Identification of a large intronic transposal insertion in SLC17A5 causing sialic acid storage disease. Orphanet J. Rare Dis. 2017, 12, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caorsi, R.; Penco, F.; Grossi, A.; Insalaco, A.; Omenetti, A.; Alessio, M.; Conti, G.; Marchetti, F.; Picco, P.; Tommasini, A.; et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: A multicentre national study. Ann. Rheum. Dis. 2017, 76, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Van Kuilenburg, A.B.; Tarailo-Graovac, M.; Meijer, J.; Drogemoller, B.; Vockley, J.; Maurer, D.; Dobritzsch, D.; Ross, C.J.; Wasserman, W.; Meinsma, R.; et al. Genome sequencing reveals a novel genetic mechanism underlying dihydropyrimidine dehydrogenase deficiency: A novel missense variant c.1700G>A and a large intragenic inversion in DPYD spanning intron 8 to intron 12. Hum. Mutat. 2018, 39, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Eichler, E.E. Genetic Variation, Comparative Genomics, and the Diagnosis of Disease. N. Engl. J Med. 2019, 381, 64–74. [Google Scholar] [CrossRef]
- Miller, D.E.; Sulovari, A.; Wang, T.; Loucks, H.; Hoekzema, K.; Munson, K.M.; Lewis, A.P.; Fuerte, E.P.A.; Paschal, C.R.; Walsh, T.; et al. Targeted long-read sequencing identifies missing disease-causing variation. Am. J. Hum. Genet. 2021, 108, 1436–1449. [Google Scholar] [CrossRef] [PubMed]
- Marwaha, S.; Knowles, J.W.; Ashley, E.A. A guide for the diagnosis of rare and undiagnosed disease: Beyond the exome. Genome Med. 2022, 14, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Davis-Turak, J.; Courtney, S.M.; Hazard, E.S.; Glen, W.B., Jr.; da Silveira, W.A.; Wesselman, T.; Harbin, L.P.; Wolf, B.J.; Chung, D.; Hardiman, G. Genomics pipelines and data integration: Challenges and opportunities in the research setting. Expert Rev. Mol. Diagn. 2017, 17, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, M.; Gobet, N.; Cruz-Dávalos, D.I.; Mounier, N.; Dessimoz, C.; Sedlazeck, F.J. Structural variant calling: The long and the short of it. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wooderchak-Donahue, W.L.; McDonald, J.; Farrell, A.; Akay, G.; Velinder, M.; Johnson, P.; VanSant-Webb, C.; Margraf, R.; Briggs, E.; Whitehead, K.J.; et al. Genome sequencing reveals a deep intronic splicing ACVRL1 mutation hotspot in Hereditary Haemorrhagic Telangiectasia. J. Med. Genet. 2018, 55, 824–830. [Google Scholar] [CrossRef]
- Maroilley, T.; Wright, N.A.M.; Diao, C.; MacLaren, L.; Pfeffer, G.; Sarna, J.R.; Au, P.Y.B.; Tarailo-Graovac, M. Case Report: Biallelic Loss of Function ATM due to Pathogenic Synonymous and Novel Deep Intronic Variant c.1803-270T > G Identified by Genome Sequencing in a Child With Ataxia–Telangiectasia. Front. Genet. 2022, 13, 815210. [Google Scholar] [CrossRef] [PubMed]
- Whiffin, N.; Karczewski, K.J.; Zhang, X.; Chothani, S.; Smith, M.J.; Evans, D.G.; Ware, J.S. Characterising the loss-of-function impact of 5’ untranslated region variants in 15,708 individuals. Nat. Commun. 2020, 11, 2523. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; Quaife, N.M.; Ramos-Hernández, L.; Danecek, P.; Ferla, M.P.; Samocha, K.E.; Kaplanis, J.; Gardner, E.J.; Eberhardt, R.Y.; Chao, K.R.; et al. Non-coding region variants upstream of MEF2C cause severe developmental disorder through three distinct loss-of-function mechanisms. Am. J. Hum. Genet. 2021, 108, 1083–1094. [Google Scholar] [CrossRef]
- Hornig, N.C.; de Beaufort, C.; Denzer, F.; Cools, M.; Wabitsch, M.; Ukat, M.; Kulle, A.E.; Schweikert, H.-U.; Werner, R.; Hiort, O.; et al. A Recurrent Germline Mutation in the 5’UTR of the Androgen Receptor Causes Complete Androgen Insensitivity by Activating Aberrant uORF Translation. PLoS ONE 2016, 11, e0154158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnappauf, O.; Zhou, Q.; Moura, N.S.; Ombrello, A.K.; Michael, D.G.; Deuitch, N.; Barron, K.; Stone, D.L.; Hoffmann, P.; Hershfield, M.; et al. Deficiency of Adenosine Deaminase 2 (DADA2): Hidden Variants, Reduced Penetrance, and Unusual Inheritance. J. Clin. Immunol. 2020, 40, 917–926. [Google Scholar] [CrossRef]
- Bhatia, S.; Bengani, H.; Fish, M.; Brown, A.; Divizia, M.T.; de Marco, R.; Damante, G.; Grainger, R.; van Heyningen, V.; Kleinjan, D.A. Disruption of Autoregulatory Feedback by a Mutation in a Remote, Ultraconserved PAX6 Enhancer Causes Aniridia. Am. J. Hum. Genet. 2013, 93, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]
- LaCroix, A.J.; Stabley, D.; Sahraoui, R.; Adam, M.P.; Mehaffey, M.; Kernan, K.; Myers, C.T.; Fagerstrom, C.; Anadiotis, G.; Akkari, Y.M.; et al. GGC Repeat Expansion and Exon 1 Methylation of XYLT1 Is a Common Pathogenic Variant in Baratela-Scott Syndrome. Am. J. Hum. Genet. 2019, 104, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Corbett, M.A.; Kroes, T.; Veneziano, L.; Bennett, M.F.; Florian, R.; Schneider, A.L.; Coppola, A.; Licchetta, L.; Franceschetti, S.; Suppa, A.; et al. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Florian, R.T.; Kraft, F.; Leitão, E.; Kaya, S.; Klebe, S.; Magnin, E.; van Rootselaar, A.-F.; Buratti, J.; Kühnel, T.; Schröder, C.; et al. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with Familial Adult Myoclonic Epilepsy type 3. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Yeetong, P.; Pongpanich, M.; Srichomthong, C.; Assawapitaksakul, A.; Shotelersuk, V.; Tantirukdham, N.; Chunharas, C.; Suphapeetiporn, K.; Shotelersuk, V. TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain 2019, 142, 3360–3366. [Google Scholar] [CrossRef] [PubMed]
- Garland, J.; Stephen, J.; Class, B.; Gruber, A.; Ciccone, C.; Poliak, A.; Hayes, C.P.; Singhal, V.; Slota, C.; Perreault, J.; et al. Identification of an Alu element-mediated deletion in the promoter region of GNE in siblings with GNE myopathy. Mol. Genet. Genom. Med. 2017, 5, 410–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schanze, D.; Neubauer, D.; Cormier-Daire, V.; Delrue, M.A.; Dieux-Coeslier, A.; Hasegawa, T.; Zenker, M. Deletions in the 3’ part of the NFIX gene including a recurrent Alu-mediated deletion of exon 6 and 7 account for previously unexplained cases of Marshall-Smith syndrome. Hum. Mutat. 2014, 35, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Todorov, T.; Balakrishnan, P.; Savov, A.; Socha, P.; Schmidt, H.H.J. Intragenic Deletions in ATP7B as an Unusual Molecular Genetics Mechanism of Wilson’s Disease Pathogenesis. PLoS ONE 2016, 11, e0168372. [Google Scholar] [CrossRef] [Green Version]
- De Bruijn, S.E.; Fiorentino, A.; Ottaviani, D.; Fanucchi, S.; Melo, U.S.; Corral-Serrano, J.C.; Mulders, T.; Georgiou, M.; Rivolta, C.; Pontikos, N.; et al. Structural Variants Create New Topological-Associated Domains and Ectopic Retinal Enhancer-Gene Contact in Dominant Retinitis Pigmentosa. Am. J. Hum. Genet. 2020, 107, 802–814. [Google Scholar] [CrossRef]
- Guéant, J.L.; Chery, C.; Oussalah, A.; Nadaf, J.; Coelho, D.; Josse, T.; Rosenblatt, D.S. APRDX1 mutant allele causes a MMACHC secondary epimutation in cblC patients. Nat. Commun. 2018, 9, 67. [Google Scholar] [CrossRef] [Green Version]
- Dionnet, E.; Defour, A.; Da Silva, N.; Salvi, A.; Lévy, N.; Krahn, M.; Bartoli, M.; Puppo, F.; Gorokhova, S. Splicing impact of deep exonic missense variants in CAPN3 explored systematically by minigene functional assay. Hum. Mutat. 2020, 41, 1797–1810. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Mishima, E.; Shima, H.; Akiyama, Y.; Suzuki, C.; Suzuki, T.; Kobayashi, T.; Suzuki, Y.; Nakayama, T.; Takeshima, Y.; et al. Exonic Mutations in the SLC12A3 Gene Cause Exon Skipping and Premature Termination in Gitelman Syndrome. J. Am. Soc. Nephrol. 2014, 26, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, Y.; Pan, Y.; Wang, J.; Yu, W.; Wang, X. Unraveling synonymous and deep intronic variants causing aberrant splicing in two genetically undiagnosed epilepsy families. BMC Med. Genom. 2021, 14, 1–9. [Google Scholar] [CrossRef]
- Ferri, L.; Dionisi-Vici, C.; Taurisano, R.; Vaz, F.M.; Guerrini, R.; Morrone, A. When silence is noise: Infantile-onset Barth syndrome caused by a synonymous substitution affecting TAZ gene transcription. Clin. Genet. 2016, 90, 461–465. [Google Scholar] [CrossRef]
- Tavares, E.; Tang, C.Y.; Vig, A.; Li, S.; Billingsley, G.; Sung, W.; Vincent, A.; Thiruvahindrapuram, B.; Héon, E. Retrotransposon insertion as a novel mutational event in Bardet-Biedl syndrome. Mol. Genet. Genom. Med. 2018, 7, e00521. [Google Scholar] [CrossRef] [Green Version]
- Hacıhamdioğlu, B.; Özgürhan, G.; Pereira, C.; Tepeli, E.; Acar, G.; Cömert, S. A Case of the Perinatal Form Hypophosphatasia Caused by a Novel Large Duplication of the ALPL Gene and Report of One Year Follow-up with Enzyme Replacement Therapy. J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Burin-des-Roziers, C.; Rothschild, P.R.; Layet, V.; Chen, J.M.; Ghiotti, T.; Leroux, C.; Valleix, S. Deletions Overlapping VCAN Exon 8 Are New Molecular Defects for Wagner Disease. Hum. Mutat. 2017, 38, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Yang, F.; Wang, J.; Yang, F.; Liang, M.; Yang, H. Exon skipping caused by a complex structural variation in SH2D1A resulted in X-linked lymphoproliferative syndrome type 1. Mol. Genet. Genom. Med. 2022, 10, e1873. [Google Scholar] [CrossRef] [PubMed]
- Sukalo, M.; Schäflein, E.; Schanze, I.; Everman, D.B.; Rezaei, N.; Argente, J.; Lorda-Sanchez, I.; Deshpande, C.; Takahashi, T.; Kleger, A.; et al. Expanding the mutational spectrum in Johanson-Blizzard syndrome: Identification of whole exon deletions and duplications in the UBR1 gene by multiplex ligation-dependent probe amplification analysis. Mol. Genet. Genom. Med. 2017, 5, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Ribierre, T.; Deleuze, C.; Bacq, A.; Baldassari, S.; Marsan, E.; Chipaux, M.; Muraca, G.; Roussel, D.; Navarro, V.; LeGuern, E.; et al. Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia–associated epilepsy. J. Clin. Investig. 2018, 128, 2452–2458. [Google Scholar] [CrossRef] [Green Version]
- Matthews, A.; Tarailo-Graovac, M.; Price, E.; Blydt-Hansen, I.; Ghani, A.; Drögemöller, B.; Robinson, W.; Ross, C.; Wasserman, W.; Siden, H.; et al. A de novo mosaic mutation in SPAST with two novel alternative alleles and chromosomal copy number variant in a boy with spastic paraplegia and autism spectrum disorder. Eur. J. Med Genet. 2017, 60, 548–552. [Google Scholar] [CrossRef]
- Joyce, C.M.; Houghton, J.A.; O’Halloran, D.J.; O’Shea, P.; O’Connell, S.M. Inheritance of a paternal ABCC8 variant and maternal loss of heterozygosity at 11p15 retrospectively unmasks the etiology in a case of Congenital hyperinsulinism. Clin. Case Rep. 2020, 8, 1217–1222. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Savary, C.; Dubourg, C.; Carré, W.; Mouden, C.; Hamdi-Rozé, H.; Guyodo, H.; Le Douce, J.; Genin, E.; Campion, D.; et al. Integrated clinical and omics approach to rare diseases: Novel genes and oligogenic inheritance in holoprosencephaly. Brain 2018, 142, 35–49. [Google Scholar] [CrossRef]
- König, E.; Volpato, C.B.; Motta, B.M.; Blankenburg, H.; Picard, A.; Pramstaller, P.; Casella, M.; Rauhe, W.; Pompilio, G.; Meraviglia, V.; et al. Exploring digenic inheritance in arrhythmogenic cardiomyopathy. BMC Med. Genet. 2017, 18, 145. [Google Scholar] [CrossRef]
- Bennett, M.F.; Hildebrand, M.S.; Kayumi, S.; Corbett, M.A.; Gupta, S.; Ye, Z.; Krivanek, M.; Burgess, R.; Henry, O.J.; Damiano, J.A.; et al. Evidence for a Dual-Pathway, 2-Hit Genetic Model for Focal Cortical Dysplasia and Epilepsy. Neurol. Genet. 2022, 8, e0652. [Google Scholar] [CrossRef]
- Vaz-Drago, R.; Custódio, N.; Carmo-Fonseca, M. Deep intronic mutations and human disease. Hum. Genet. 2017, 136, 1093–1111. [Google Scholar] [CrossRef] [PubMed]
- De Moor, C.H.; Meijer, H.; Lissenden, S. Mechanisms of translational control by the 3′ UTR in development and differentiation. Semin. Cell Dev. Biol. 2005, 16, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Araujo, P.R.; Yoon, K.; Ko, D.; Smith, A.D.; Qiao, M.; Suresh, U.; Burns, S.C.; Penalva, L.O.F. Before It Gets Started: Regulating Translation at the 5′ UTR. Comp. Funct. Genom. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Gerstein, M.B.; Kundaje, A.; Hariharan, M.; Landt, S.G.; Yan, K.-K.; Cheng, C.; Mu, X.J.; Khurana, E.; Rozowsky, J.; Alexander, R.; et al. Architecture of the human regulatory network derived from ENCODE data. Nature 2012, 489, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solyom, S.; Kazazian, H.H. Mobile elements in the human genome: Implications for disease. Genome Med. 2012, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Depienne, C.; Mandel, J.L. 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am. J. Hum. Genet. 2021, 108, 764–785. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, C.R., 2nd; Scharer, G.H.; Shaikh, T.H. Clinical impact of copy number variation analysis using high-resolution microarray technologies: Advantages, limitations and concerns. Genome Med. 2012, 4, 80. [Google Scholar] [CrossRef] [Green Version]
- Maroilley, T.; Li, X.; Oldach, M.; Jean, F.; Stasiuk, S.J.; Tarailo-Graovac, M. Deciphering complex genome rearrangements in C. elegans using short-read whole genome sequencing. Sci. Rep. 2021, 11, 18258. [Google Scholar] [CrossRef]
- McArthur, E.; Capra, J.A. Topologically associating domain boundaries that are stable across diverse cell types are evolutionarily constrained and enriched for heritability. Am. J. Hum. Genet. 2021, 108, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Jadhav, B.; Rodriguez, O.L.; Patel, N.; Martin-Trujillo, A.; Jain, M.; Metsu, S.; Olsen, H.; Paten, B.; Ritz, B.; et al. A Survey of Rare Epigenetic Variation in 23,116 Human Genomes Identifies Disease-Relevant Epivariations and CGG Expansions. Am. J. Hum. Genet. 2020, 107, 654–669. [Google Scholar] [CrossRef]
- Buiting, K.; Groß, S.; Lich, C.; Gillessen-Kaesbach, G.; El-Maarri, O.; Horsthemke, B. Epimutations in Prader-Willi and Angelman Syndromes: A Molecular Study of 136 Patients with an Imprinting Defect. Am. J. Hum. Genet. 2003, 72, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savisaar, R.; Hurst, L.D. Estimating the prevalence of functional exonic splice regulatory information. Hum. Genet. 2017, 136, 1059–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromberg, Y.; Kahn, P.C.; Rost, B. Neutral and weakly nonneutral sequence variants may define individuality. Proc. Natl. Acad. Sci. USA 2013, 110, 14255–14260. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Bromberg, Y. Predicting Functional Effects of Synonymous Variants: A Systematic Review and Perspectives. Front. Genet. 2019, 10, 914. [Google Scholar] [CrossRef] [Green Version]
- Biesecker, L.G.; Spinner, N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013, 14, 307–320. [Google Scholar] [CrossRef]
- Miller, C.R.; Lee, K.; Pfau, R.B.; Reshmi, S.C.; Corsmeier, D.J.; Hashimoto, S.; Dave-Wala, A.; Jayaraman, V.; Koboldt, D.C.; Matthews, T.; et al. Disease-associated mosaic variation in clinical exome sequencing: A two-year pediatric tertiary care experience. Mol. Case Stud. 2020, 6, a005231. [Google Scholar] [CrossRef]
- Stosser, M.B.; Lindy, A.S.; Butler, E.; Retterer, K.; Piccirillo-Stosser, C.M.; Richard, G.; McKnight, D.A. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet. Med. 2018, 20, 403–410. [Google Scholar] [CrossRef]
- Heinzen, E.L. Somatic variants in epilepsy—Advancing gene discovery and disease mechanisms. Curr. Opin. Genet. Dev. 2020, 65, 1–7. [Google Scholar] [CrossRef]
- Dremsek, P.; Schwarz, T.; Weil, B.; Malashka, A.; Laccone, F.; Neesen, J. Optical Genome Mapping in Routine Human Genetic Diagnostics—Its Advantages and Limitations. Genes 2021, 12, 1958. [Google Scholar] [CrossRef] [PubMed]
- Deltas, C. Digenic inheritance and genetic modifiers. Clin. Genet. 2018, 93, 429–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajiwara, K.; Berson, E.L.; Dryja, T.P. Digenic Retinitis Pigmentosa Due to Mutations at the Unlinked Peripherin/RDS and ROM1 Loci. Science 1994, 264, 1604–1608. [Google Scholar] [CrossRef] [Green Version]
- Kerner, G.; Bouaziz, M.; Cobat, A.; Bigio, B.; Timberlake, A.T.; Bustamante, J.; Lifton, R.P.; Casanova, J.-L.; Abel, L. A genome-wide case-only test for the detection of digenic inheritance in human exomes. Proc. Natl. Acad. Sci. USA 2020, 117, 19367–19375. [Google Scholar] [CrossRef]
- Gazzo, A.M.; Daneels, D.; Cilia, E.; Bonduelle, M.-L.; Abramowicz, M.; Van Dooren, S.; Smits, G.; Lenaerts, T. DIDA: A curated and annotated digenic diseases database. Nucleic Acids Res. 2015, 44, D900–D907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodenburg, R.J. The functional genomics laboratory: Functional validation of genetic variants. J. Inherit. Metab. Dis. 2018, 41, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Matlin, A.J.; Clark, F.; Smith, C.W.J. Understanding alternative splicing: Towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005, 6, 386–398. [Google Scholar] [CrossRef]
- Savatt, J.M.; Myers, S.M. Genetic Testing in Neurodevelopmental Disorders. Front. Pediatr. 2021, 9, 526779. [Google Scholar] [CrossRef]
- Hiraide, T.; Shimizu, K.; Miyamoto, S.; Aoto, K.; Nakashima, M.; Yamaguchi, T.; Kosho, T.; Ogata, T.; Saitsu, H. Genome sequencing and RNA sequencing of urinary cells reveal an intronic FBN1 variant causing aberrant splicing. J. Hum. Genet. 2022, 67, 387–392. [Google Scholar] [CrossRef]
- Walker, S.; Lamoureux, S.; Khan, T.; Joynt, A.C.M.; Bradley, M.; Branson, H.M.; Carter, M.T.; Hayeems, R.Z.; Jagiello, L.; Marshall, C.R.; et al. Genome sequencing for detection of pathogenic deep intronic variation: A clinical case report illustrating opportunities and challenges. Am. J. Med. Genet. Part A 2021, 185, 3129–3135. [Google Scholar] [CrossRef]
- Hiraide, T.; Nakashima, M.; Ikeda, T.; Tanaka, D.; Osaka, H.; Saitsu, H. Identification of a deep intronic POLR3A variant causing inclusion of a pseudoexon derived from an Alu element in Pol III-related leukodystrophy. J. Hum. Genet. 2020, 65, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Youk, J.; Kwon, H.W.; Kim, R.; Ju, Y.S. Dissecting single-cell genomes through the clonal organoid technique. Exp. Mol. Med. 2021, 53, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Melo, U.S.; Schöpflin, R.; Acuna-Hidalgo, R.; Mensah, M.A.; Fischer-Zirnsak, B.; Holtgrewe, M.; Klever, M.-K.; Türkmen, S.; Heinrich, V.; Pluym, I.D.; et al. Hi-C Identifies Complex Genomic Rearrangements and TAD-Shuffling in Developmental Diseases. Am. J. Hum. Genet. 2020, 106, 872–884. [Google Scholar] [CrossRef]
- King, E.A.; Davis, J.W.; Degner, J.F. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 2019, 15, e1008489. [Google Scholar] [CrossRef]
- Marek-Yagel, D.; Eliyahu, A.; Veber, A.; Shalva, N.; Philosoph, A.M.; Barel, O.; Javasky, E.; Pode-Shakked, B.; Loewenthal, N.; Anikster, Y.; et al. Deep intronic variant in the ARSB gene as the genetic cause for Maroteaux–Lamy syndrome (MPS VI). Am. J. Med. Genet. Part A 2021, 185, 3804–3809. [Google Scholar] [CrossRef]
- Cavalieri, S.; Pozzi, E.; Gatti, R.A.; Brusco, A. Deep-intronic ATM mutation detected by genomic resequencing and corrected in vitro by antisense morpholino oligonucleotide (AMO). Eur. J. Hum. Genet. 2012, 21, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Oura, S.; Noda, T.; Morimura, N.; Hitoshi, S.; Nishimasu, H.; Nagai, Y.; Nureki, O.; Ikawa, M. Precise CAG repeat contraction in a Huntington’s Disease mouse model is enabled by gene editing with SpCas9-NG. Commun. Biol. 2021, 4, 1–13. [Google Scholar] [CrossRef]
- Lappalainen, T.; MacArthur, D.G. From variant to function in human disease genetics. Science 2021, 373, 1464–1468. [Google Scholar] [CrossRef]


{kind=link}
| Author | Publication Year | Clinical Diagnosis | Inheritance | Causative Gene | Previous Genetic Tests | Previous Incomplete Genetic Findings | Further Analysis to Make a Definite Molecular Diagnosis | Further Genetic Findings | PMID |
|---|---|---|---|---|---|---|---|---|---|
| Wooderchak-Donahue, WL. et al. [20] | 2018 | hereditary hemorrhagic telangiectasia (HHT) (HHT1; OMIM: #187300/HHT2; OMIM: #600376/Juvenile polyposis/HTT syndrome; OMIM: #175050) | AD | ENG, ACVRL1, SMAD4 | ES | negative results | SR-GS, SR-GS panel sequencing | deep intronic variants, one translocation in ACVRL1 | 30244195 |
| Maroilley, T. et al. [21] | 2022 | ataxia–telangiectasia (AT; OMIM: #208900) | AR | ATM | clinical ataxia gene panel | Heterozygous synonymous variant | SR-GS | deep intronic variant | 35145552 |
| Whiffin, N. et al. [22] | 2020 | neurofibromatosis, type 2 (NF2; OMIM: #101000) | AD | NF2 | targeted sequencing | negative results | analyzing targeted sequencing data | 5′ untranslated region variants | 32461616 |
| Wright, C. F. et al. [23] | 2021 | neurodevelopmental disorder with hypotonia, stereotypic hand movements, and impaired language (NEDHSIL; OMIM: #613443) | AD | MEF2C | ES | negative results | ES data analysis | 5′ UTR variants | 34022131 |
| Hornig, NC. et al. [24] | 2016 | androgen insensitivity syndrome (AIS; OMIM #300068) | XLR | AR | Sanger sequencing | negative results | SR-GS of AR genomic locus | 5′UTR variant | 27110943 |
| Schnappauf, O. et al. [25] | 2020 | vasculitis, autoinflammation, immunodeficiency, and hematologic defects syndrome (VAIHS; OMIM: #615688) | AR | ADA2 | ES/Sanger sequencing of ADA2, chromosomal microarray, ES | heterozygosity for the known pathogenic variant in one family/negative results in the other family | SR-GS/MLPA in combination with long-read PCR sequencing | 5′UTR variant/a homozygous 800bp duplication | 32638197 |
| Bhatia, S. et al. [26] | 2013 | aniridia (AN1; OMIM: #106210) | AD | PAX6 | ES, array-CGH, MLPA testing | negative results | screening a selection of eye-related cis-regulatory elements | cis-element (SIMO enhancer) variant | 24290376 |
| Tarailo-Graovac, M. et al. [11] | 2017 | sialic acid storage disease (SASDs) [infantile sialic acid storage disease (ISSD; OMIM: #269920)/Salla disease (SD; OMIM: #604369)] | AR | SLC17A5 | Sanger sequencing | no pathogenic variant | ES, RNA and genomic DNA analysis | homozygous 6040 bp intronic transposal insertion in intron 9 of SLC17A5 | 28187749 |
| Kim, J. et al. [27] | 2019 | ceroid lipofuscinosis, neuronal, 7 (CLN7; OMIM: #610951) | AR | MFSD8 | genetic panel testing (including deletion–duplication analysis)for known Batten’s disease genes | single known missense variant | SR-GS | an insertion of an SVA (SINE–VNTR–Alu) retrotransposon | 31597037 |
| van Kuilenburg, ABP. et al. [7] | 2019 | global developmental delay, progressive ataxia, and elevated glutamine (GDPAG; OMIM #618412) | AR | GLS | ES | missense in patient 1 and a duplication variant in patient 3 | SR-GS | GCA trinucleotide expansion | 30970188 |
| LaCroix, A. J. et al. [28] | 2019 | Baratela-Scott syndrome (BSS; OMIM: #615777) | AR | XYLT1 | ES, clinical chromosome microarray, Sanger sequencing | single or no causative variants in XYLT1 | Southern Blot and SR-GS analysis | GGC repeat expansion | 30554721 |
| Ishiura, H. et al. [8] | 2018 | epilepsy, familial adult myoclonic (FAME) | AD | SAMD12, TNRC6A, RAPGEF2 | analysis of the exons of 38 genes located in the candidate region, including copy-number analysis | negative results | single-molecule, real-time sequencing of BAC clones and nanopore sequencing | expansions of TTTCA and TTTTA repeats | 29507423 |
| Corbett, M. A. et al. [29] | 2019 | epilepsy, familial adult myoclonic, 2 (FAME2; OMIM: #607876) | AD | STARD7 | NA | NA | SR-GS | ATTTC repeat expansions | 31664034 |
| Florian, R. T. et al. [30] | 2019 | epilepsy, familial adult myoclonic, 3 (FAME3; OMIM: #613608) | AD | MARCH6 | ES | negative results | SR-GS and repeat-primed PCR | intronic TTTTA/TTTCA expansions | 31664039 |
| Yeetong, P. et al. [31] | 2019 | benign adult familial myoclonic epilepsy type 4 (FAME4; OMIM: #615127) | AD | YEATS2 | targeted resequencing of the 10-Mbp critical region, array CGH, ES and SR-GS | negative results | single-molecule real-time sequencing | TTTCA repeat insertions | 31539032 |
| van Kuilenburg, ABP. et al. [13] | 2018 | dihydropyrimidine dehydrogenase deficiency (DPD deficiency; OMIM: #274270) | AR | DPYD | Sanger sequencing | heterozygous missense variant | SR-GS | large intragenic inversion | 29691939 |
| Garland, J. et al. [32] | 2017 | Nonaka myopathy (NM; OMIM: #605820) | AR | GNE | Sanger sequencing | heterozygous mutation | copy number variant analysis of GNE | deletion in the promoter region | 28717665 |
| Schanze D, et al. [33] | 2014 | Marshall-Smith syndrome (MRSHSS; OMIM: #602535) | AD | NFIX | conventional sequencing of NFIX | causes for part of the patients including frameshift and splice-site mutations | MLPA | a recurrent large deletion | 24924640 |
| Todorov T, et al. [34] | 2016 | Wilson disease (WD; OMIM: #277900) | AR | ATP7B | extensive sequence analysis of promoter, coding region and associated intron-exon boundaries | negative results | selective amplification and MLPA | intragenic deletions | 27992490 |
| de Bruijn, S. E. et al. [35] | 2020 | retinitis pigmentosa (RP; OMIM: #268000) | AD | genomic region spanning YPEL2 to LINC01476 | SR-GS | SVs | Hi-C | topological-associated domains | 33022222 |
| Gueant, JL. et al. [36] | 2018 | methylmalonic aciduria and homocystinuria, cobalamin C type (MAHCC; OMIM: #277400) | AR | MMACHC | Sanger sequencing | single heterozygous mutations | methylation analysis (Sanger sequencing of bisulfite-converted DNA) | heterozygous promoter hypermethylation | 29302025 |
| Dionnet, E. et al. [37] | 2020 | muscular dystrophy, limb-girdle, autosomal recessive 1 (LGMDR1; OMIM: #253600) | AR | CAPN3 | present machine learning-based computational tools | negative predictions | minigene assay | deep exonic missense variants | 32668095 |
| Takeuchi, Y. et al. [38] | 2015 | Gitelman syndrome (GTLMNS; OMIM: #263800) | AR | SLC12A3 | previously reported or are accessible from the PubMed database | missense variants | minigene assay | exonic variants affecting mRNA splicing | 25060058 |
| Li, Q. et al. [39] | 2021 | seizures, benign neonatal, 1 (BFNS1; OMIM: #121200) | AD | KCNQ2 | ES | negative results | ES reanalysis | synonymous variant | 34107977 |
| Ferri, L. et al. [40] | 2016 | Barth syndrome (BTHS; OMIM: #302060) | XLR | TAZ | NA | NA | sequencing of the TAZ gene | new synonymous variant | 26853223 |
| Miller, DE. et al. [15] | 2021 | strongly suspected clinical diagnoses such as Hermansky-Pudlaksyndrome (HPS1; OMIM: # 203300), glycogen storagedisease III (GSD3; OMIM: #232400) etc. | AR, X-linked | ALMS1,NPHP4,VARS2 etc. | chromosomal microarray, karyotype, clinical ES, or research SR-GS | single variant missed in a recessive condition or no variants found in an X-linked condition | T-LRS | deletions, mobile element insertions, inversions, repeat expansions, and intronic variants predicted to affect splicing | 34216551 |
| Tavares, E. et al. [41] | 2019 | Bardet-Biedl syndrome 1 (BBS1; OMIM: #209900) | AR | BBS1 | SR-GS on 19 BBS genes | missense allele | SR-GS | Novel ~1.7-kb retrotransposon insertion | 30484961 |
| Hacıhamdioğlu, B. et al. [42] | 2019 | Hypophosphatasia (HPP) (HPPC; OMIM: #241510/HPPI; OMIM: #241500/HPPA; OMIM: #146300) | AR | ALPL | ES | negative results | quantitative PCR | large duplication | 30468149 |
| Burin-des-Roziers, C. et al. [43] | 2016 | Wagner syndrome 1 (WGN1; OMIM: #143200) | AD | VCAN | Sanger sequencing | no nucleotide variations at exon 8 boundaries | targeted deep SR-GS, quantitative real-time PCR, and long-range PCR | heterozygous deletions | 27667122 |
| Wu, L.et al. [44] | 2022 | lymphoproliferative syndrome, X-linked, 1 (XLP1; OMIM: #308240) | XLR | SH2D1A | ES | negative results | extended ES analysis | complex structural variant including two deletions and one inversion | 35092357 |
| Sukalo M, et al. [45] | 2017 | Johanson-Blizzard syndrome (JBS; OMIM: #243800) | AR | UBR1 | Sanger sequencing | negative results or only a single variant | MLPA | exon deletions/duplications | 29178640 |
| Ribierre, T. et al. [46] | 2018 | focal cortical dysplasia type II (FCORD2; OMIM: #607341) | 2-hit genetic model | DEPDC5 | deep sequencing of a panel of mTORC1 genes | heterozygous variant in blood | Sanger sequencing | brain somatic variant | 29708508 |
| Matthews, A. M. et al. [47] | 2017 | spastic paraplegia-4 disorder (SPG4; OMIM: #182601) | AD | SPAST | chromosome microarray | copy number variant | ES, pyrosequencing | de novo mosaic bi-alternative variants | 28778789 |
| Joyce, C. M. et al. [48] | 2020 | hyperinsulinemic hypoglycemia, familial, 1 (HHF1; OMIM: # 256450) | 2-hit genetic model | ABCC8 | Sanger sequencing | paternally inherited ABCC8 nonsense variant | further analysis for microsatellite markers | somatic maternal loss of heterozygosity at 11p15 | 32695361 |
| Kim, A. et al. [49] | 2019 | holoprosencephaly 1 (HPE1; OMIM: #236100) | AD | 180 genes directly linked to the SHH signalling, cilium and Wnt/PCP pathways | targeted HPE gene-panel sequencing, CGH, MLPA | negative results | ES | oligogenic variants | 30508070 |
| Konig, E. et al. [50] | 2017 | arrhythmogenic cardiomyopathy (ACM) | digenic inheritance | PKP2 and TTN | diagnostic tests | PKP2 mutations | ES | TTN mutations | 29221435 |
| Bennett, MF. et al. [51] | 2022 | focal cortical dysplasia type II (FCORD2; OMIM: #607341) | 2-hit genetic model | mTOR and related pathway genes | ES | truncating variant in NPRL3 | ES | mosaic missense variant in brain-derived DNA in the WNT2 gene | 35097204 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, L.; Maroilley, T.; Tarailo-Graovac, M. The Power of Clinical Diagnosis for Deciphering Complex Genetic Mechanisms in Rare Diseases. Genes 2023, 14, 196. https://doi.org/10.3390/genes14010196
Shu L, Maroilley T, Tarailo-Graovac M. The Power of Clinical Diagnosis for Deciphering Complex Genetic Mechanisms in Rare Diseases. Genes. 2023; 14(1):196. https://doi.org/10.3390/genes14010196
Chicago/Turabian StyleShu, Li, Tatiana Maroilley, and Maja Tarailo-Graovac. 2023. "The Power of Clinical Diagnosis for Deciphering Complex Genetic Mechanisms in Rare Diseases" Genes 14, no. 1: 196. https://doi.org/10.3390/genes14010196