
Multi-Cell-Type Openness-Weighted Association Studies for Trait-Associated Genomic Segments Prioritization


Abstract
:1. Introduction
2. Materials and Methods
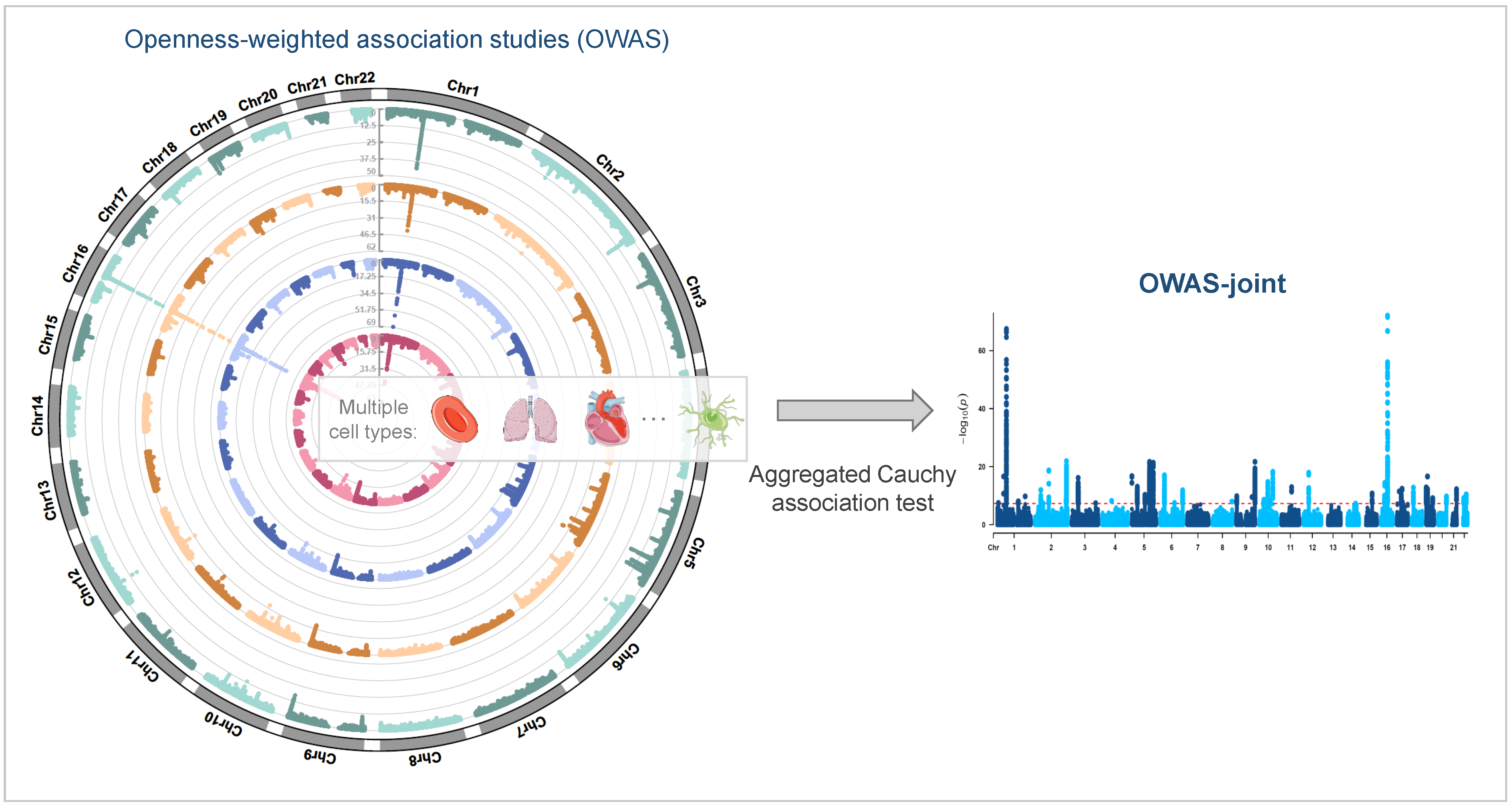
2.1. Jointly Modeling Multi-Cell-Type Openness Scores
2.2. Linkage Disequilibrium (LD) Shrinkage
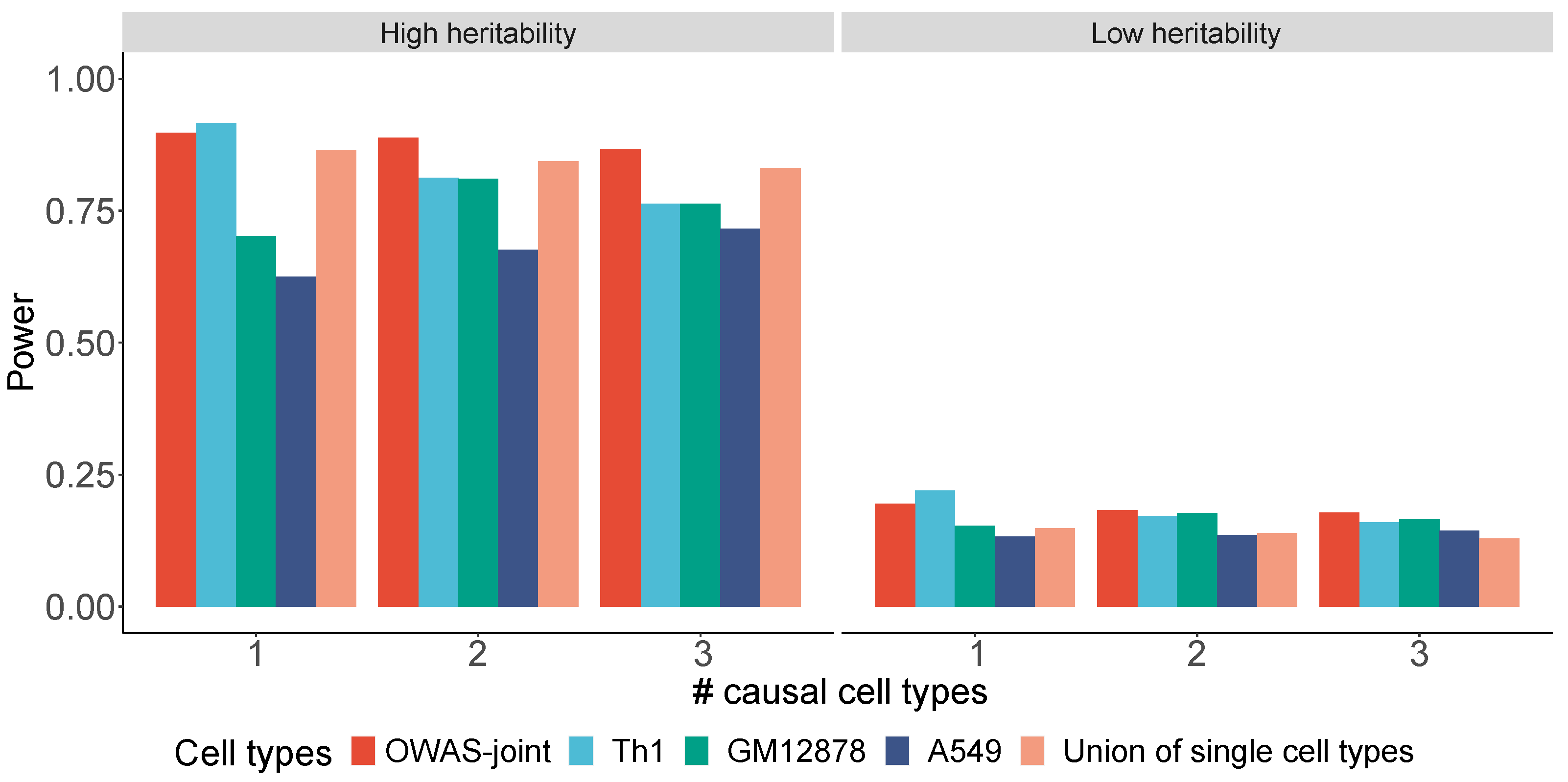
2.3. Simulation Settings
- Only the first cell type (Th1) is causal to the phenotype.
- . The cell types Th1 and GM12878 are causal, while A549 is not.
- . All three cell types are causal.
2.4. GWAS Datasets
2.4.1. GWAS Summary Statistics
2.4.2. Individual-Level Genotype Data
2.5. Predicted Openness of Personal Genomes
2.6. Pathway Enrichment Analysis
3. Results
3.1. Simulations
3.2. Real Data Applications
3.2.1. OWAS-Joint Identifies More Genetic Signals
3.2.2. OWAS-Joint Provides Novel Biological Interpretation
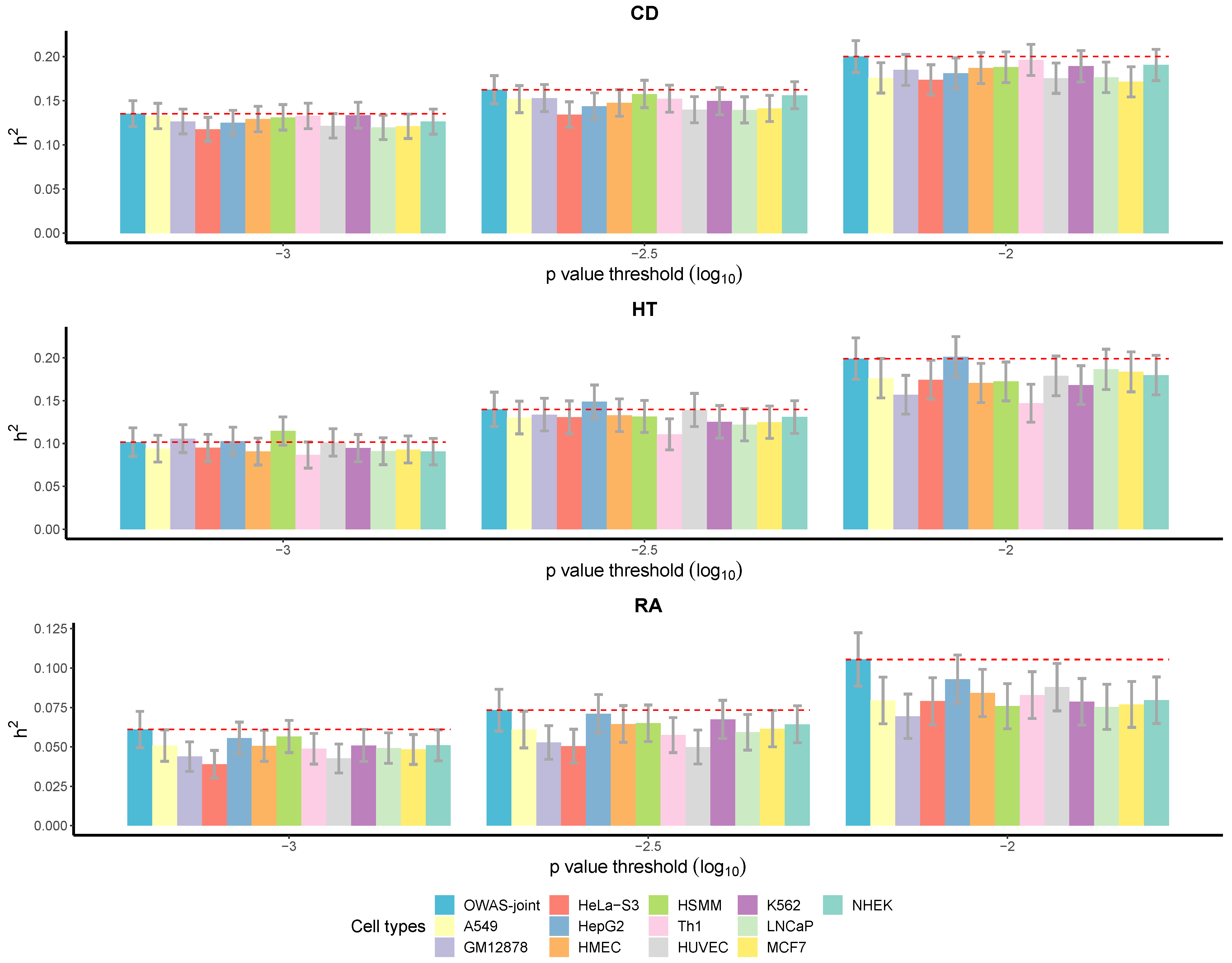
3.2.3. More Heritability Explained by OWAS-Joint Segments
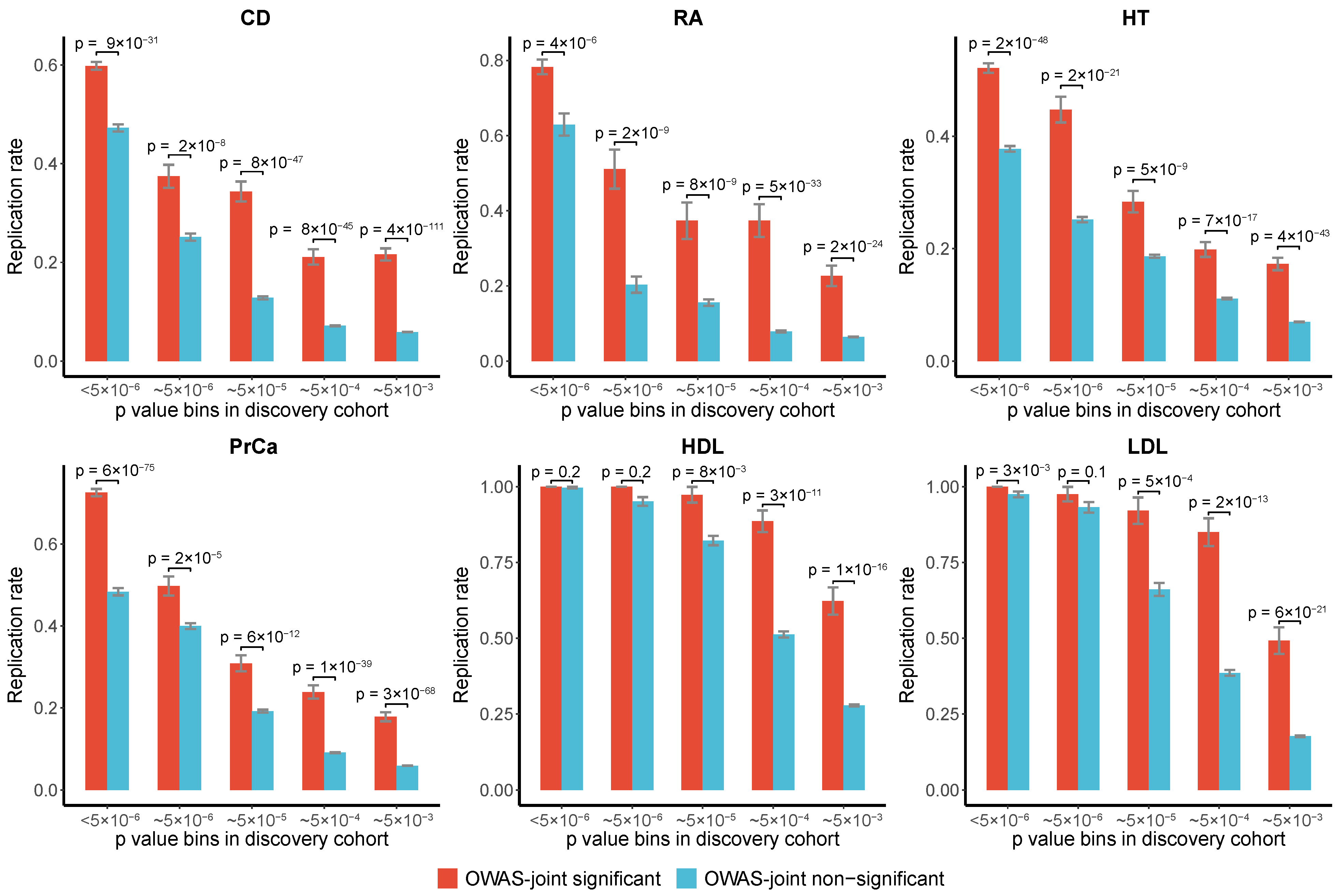
3.2.4. Replication Rates of OWAS-Joint Segments
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 1000G | 1000 Genomes Project |
| ACAT | Aggregated Cauchy association test |
| CD | Crohn’s disease |
| GWAS | Genome wide association studies |
| HDL | High-density lipoprotein |
| HT | Hypertension |
| LD | Linkage disequilibrium |
| LDL | Low-density lipoprotein |
| OWAS | Openness-weighted association studies |
| PrCa | Prostate cancer |
| RA | Rheumatoid arthritis |
| SD | Standard deviation |
| SNP | Single nucleotide polymorphism |
| UKBB | UK Biobank |
| UC | Ulcerative colitis |
| WTCCC | Wellcome Trust Case Control Consortium |
References
- Jostins, L.; Barrett, J.C. Genetic risk prediction in complex disease. Hum. Mol. Genet. 2011, 20, R182–R188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusev, A.; Lee, S.H.; Trynka, G.; Finucane, H.; Vilhjálmsson, B.J.; Xu, H.; Zang, C.; Ripke, S.; Bulik-Sullivan, B.; Stahl, E.; et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet. 2014, 95, 535–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano-Gamez, E.; Trynka, G. From GWAS to function: Using functional genomics to identify the mechanisms underlying complex diseases. Front. Genet. 2020, 11, 424. [Google Scholar] [CrossRef]
- Hou, L.; Zhao, H. A review of post-GWAS prioritization approaches. Front. Genet. 2013, 4, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, M.D.; Chen-Plotkin, A.S. The post-GWAS era: From association to function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Minnoye, L.; Marinov, G.K.; Krausgruber, T.; Pan, L.; Marand, A.P.; Secchia, S.; Greenleaf, W.J.; Furlong, E.E.; Zhao, K.; Schmitz, R.J.; et al. Chromatin accessibility profiling methods. Nat. Rev. Methods Prim. 2021, 1, 10. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G.; Kaya-Okur, H.S.; Ahmad, K. Efficient chromatin accessibility mapping in situ by nucleosome-tethered tagmentation. eLife 2020, 9, e63274. [Google Scholar] [CrossRef]
- Pique-Regi, R.; Degner, J.F.; Pai, A.A.; Gaffney, D.J.; Gilad, Y.; Pritchard, J.K. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 2011, 21, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, P.; Palidwor, G.A.; Perkins, T.J. BIDCHIPS: Bias decomposition and removal from ChIP-seq data clarifies true binding signal and its functional correlates. Epigenet. Chromatin 2015, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Gorkin, D.U.; Baker, M.; Strober, B.J.; Asoni, A.L.; McCallion, A.S.; Beer, M.A. A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet. 2015, 47, 955–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Hua, K.; Zhang, X.; Wong, W.H.; Jiang, R. DeepCAGE: Incorporating transcription factors in genome-wide prediction of chromatin accessibility. Genom. Proteom. Bioinform. 2022. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Shan, N.; Wang, G.; Yan, X.; Liu, J.S.; Hou, L. Openness weighted association studies: Leveraging personal genome information to prioritize non-coding variants. Bioinformatics 2021, 37, 4737–4743. [Google Scholar] [CrossRef] [PubMed]
- Calderon, D.; Bhaskar, A.; Knowles, D.A.; Golan, D.; Raj, T.; Fu, A.Q.; Pritchard, J.K. Inferring relevant cell types for complex traits by using single-cell gene expression. Am. J. Hum. Genet. 2017, 101, 686–699. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Xue, C.; Dai, S.; Chen, S.; Chen, P.; Sham, P.C.; Wang, H.; Li, M. DESE: Estimating driver tissues by selective expression of genes associated with complex diseases or traits. Genome Biol. 2019, 20, 233. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An expanded view of complex traits: From polygenic to omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Schaid, D.J.; Chen, W.; Larson, N.B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 2018, 19, 491–504. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Iotchkova, V.; Ritchie, G.R.; Geihs, M.; Morganella, S.; Min, J.L.; Walter, K.; Timpson, N.J.; Dunham, I.; Birney, E.; Soranzo, N. GARFIELD classifies disease-relevant genomic features through integration of functional annotations with association signals. Nat. Genet. 2019, 51, 343–353. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.; Li, Z.; Morrison, A.C.; Boerwinkle, E.; Lin, X. ACAT: A fast and powerful p value combination method for rare-variant analysis in sequencing studies. Am. J. Hum. Genet. 2019, 104, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Nasser, J.; Bergman, D.T.; Fulco, C.P.; Guckelberger, P.; Doughty, B.R.; Patwardhan, T.A.; Jones, T.R.; Nguyen, T.H.; Ulirsch, J.C.; Lekschas, F.; et al. Genome-wide enhancer maps link risk variants to disease genes. Nature 2021, 593, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xie, J. Cauchy combination test: A powerful test with analytic p-value calculation under arbitrary dependency structures. J. Am. Stat. Assoc. 2020, 115, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, J.; Strimmer, K. A Shrinkage Approach to Large-Scale Covariance Matrix Estimation and Implications for Functional Genomics. Stat. Appl. Genet. Mol. Biol. 2005, 4. [Google Scholar] [CrossRef] [Green Version]
- Opgen-Rhein, R.; Strimmer, K. Accurate ranking of differentially expressed genes by a distribution-free shrinkage approach. Stat. Appl. Genet. Mol. Biol. 2007, 6. [Google Scholar] [CrossRef]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genome-wide experiments. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storey, J.D.; Xiao, W.; Leek, J.T.; Tompkins, R.G.; Davis, R.W. Significance analysis of time course microarray experiments. Proc. Natl. Acad. Sci. USA 2005, 102, 12837–12842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.Z.; Van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A.; et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010, 42, 508–514. [Google Scholar] [CrossRef]
- Schumacher, F.R.; Al Olama, A.A.; Berndt, S.I.; Benlloch, S.; Ahmed, M.; Saunders, E.J.; Dadaev, T.; Leongamornlert, D.; Anokian, E.; Cieza-Borrella, C.; et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2018, 50, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef]
- Wainberg, M.; Sinnott-Armstrong, N.; Mancuso, N.; Barbeira, A.N.; Knowles, D.A.; Golan, D.; Ermel, R.; Ruusalepp, A.; Quertermous, T.; Hao, K.; et al. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 2019, 51, 592–599. [Google Scholar] [CrossRef]
- Sartor, R.B. Microbial influences in inflammatory bowel diseases. Gastroenterology 2008, 134, 577–594. [Google Scholar] [CrossRef]
- Spiller, R.; Campbell, E. Post-infectious irritable bowel syndrome. Curr. Opin. Gastroenterol. 2006, 22, 13–17. [Google Scholar] [CrossRef]
- Stagg, A.; Hart, A.; Knight, S.; Kamm, M. The dendritic cell: Its role in intestinal inflammation and relationship with gut bacteria. Gut 2003, 52, 1522–1529. [Google Scholar] [CrossRef]
- Gusev, A.; Mancuso, N.; Won, H.; Kousi, M.; Finucane, H.K.; Reshef, Y.; Song, L.; Safi, A.; McCarroll, S.; Neale, B.M.; et al. Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet. 2018, 50, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekar, A.; Bialas, A.R.; De Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Benoist, C.; Birney, E.; Bodenmiller, B.; Campbell, P.; Carninci, P.; Clatworthy, M.; et al. Science forum: The human cell atlas. eLife 2017, 6, e27041. [Google Scholar] [CrossRef] [PubMed]
- Verlaan, D.J.; Berlivet, S.; Hunninghake, G.M.; Madore, A.M.; Larivière, M.; Moussette, S.; Grundberg, E.; Kwan, T.; Ouimet, M.; Ge, B.; et al. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am. J. Hum. Genet. 2009, 85, 377–393. [Google Scholar] [CrossRef] [Green Version]
- Chae, C.S.; Kim, G.C.; Park, E.S.; Lee, C.G.; Verma, R.; Cho, H.L.; Jun, C.D.; Yoo, Y.J.; Im, S.H. NFAT1 regulates systemic autoimmunity through the modulation of a dendritic cell property. J. Immunol. 2017, 199, 3051–3062. [Google Scholar] [CrossRef]
- Yang, Y.; Musco, H.; Simpson-Yap, S.; Zhu, Z.; Wang, Y.; Lin, X.; Zhang, J.; Taylor, B.; Gratten, J.; Zhou, Y. Investigating the shared genetic architecture between multiple sclerosis and inflammatory bowel diseases. Nat. Commun. 2021, 12, 5641. [Google Scholar] [CrossRef]
- Zhang, W.J.; Koltun, W.A.; Tilberg, A.F.; Page, M.J.; Chorney, M.J. Absence of GNAI2 codon 179 oncogene mutations in inflammatory bowel disease. Inflamm. Bowel Dis. 2000, 6, 103–106. [Google Scholar] [CrossRef]
- Rudolph, U.; Finegold, M.J.; Rich, S.S.; Harriman, G.R.; Srinivasan, Y.; Brabet, P.; Boulay, G.; Bradley, A.; Birnbaumer, L. Ulcerative colitis and adenocarcinoma of the colon in Gαi2-deficient mice. Nat. Genet. 1995, 10, 143–150. [Google Scholar] [CrossRef]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.; Jansen, R.; De Geus, E.J.; Boomsma, D.I.; Wright, F.A.; et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Mundade, R.; Ozer, H.G.; Wei, H.; Prabhu, L.; Lu, T. Role of ChIP-seq in the discovery of transcription factor binding sites, differential gene regulation mechanism, epigenetic marks and beyond. Cell Cycle 2014, 13, 2847–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | # Identified Segments | # Identified Genes | ||||
|---|---|---|---|---|---|---|
| OWAS-Joint | Union (Bonferroni) | Single-Cell-Type OWAS | OWAS-Joint | Union (Bonferroni) | Single-Cell-Type OWAS | |
| CD | 2743 | 2595 | 1138 (48) | 382 | 374 | 293 (13) |
| RA | 1571 | 1558 | 659 (54) | 595 | 590 | 204 (16) |
| HT | 6598 | 6308 | 2452 (111) | 978 | 944 | 776 (18) |
| PrCa | 1711 | 1650 | 635 (32) | 301 | 293 | 213 (14) |
| HDL | 3070 | 2944 | 1347 (41) | 1293 | 1264 | 441 (23) |
| LDL | 2811 | 2734 | 1196 (50) | 1229 | 1219 | 399 (22) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.; Sun, H.; Liu, J.S.; Hou, L. Multi-Cell-Type Openness-Weighted Association Studies for Trait-Associated Genomic Segments Prioritization. Genes 2022, 13, 1220. https://doi.org/10.3390/genes13071220
Song S, Sun H, Liu JS, Hou L. Multi-Cell-Type Openness-Weighted Association Studies for Trait-Associated Genomic Segments Prioritization. Genes. 2022; 13(7):1220. https://doi.org/10.3390/genes13071220
Chicago/Turabian StyleSong, Shuang, Hongyi Sun, Jun S. Liu, and Lin Hou. 2022. "Multi-Cell-Type Openness-Weighted Association Studies for Trait-Associated Genomic Segments Prioritization" Genes 13, no. 7: 1220. https://doi.org/10.3390/genes13071220