
Overview of Transcriptomic Research on Type 2 Diabetes: Challenges and Perspectives

 , , ,
, , , 

Abstract
:1. Introduction
2. Approaches for Assessing Gene Expression in T2D
3. Transcriptome Studies in T2D
3.1. DEGs Involved in Lipid Metabolism
3.2. DEGs Belonging to Ubiquitin–Proteasome System
3.3. DEGs Involved in Immune Response
3.4. DEGs Participating in Cancer Signaling and Cell-Cycle Pathways
3.5. DEGs in T2D Complications
3.6. The Role of Ethnic Differences
3.7. Early Expression Changes
3.8. MicroRNA Expression Changes
4. Common Features of Gene Expression in Type 1 Diabetes and Gestational Diabetes Mellitus
5. Findings from Single-Cell Sequencing Studies
6. Comparative Analysis of Genomic and Transcriptomic Patterns
7. Discussion
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2009, 32 (Suppl. 1), S62–S67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of type 2 diabetes—Global burden of disease and forecasted trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Global Report on Diabetes; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Jing, X.; Chen, J.; Dong, Y.; Han, D.; Zhao, H.; Wang, X.; Gao, F.; Li, C.; Cui, Z.; Liu, Y.; et al. Related factors of quality of life of type 2 diabetes patients: A systematic review and meta-analysis. Health Qual. Life Outcomes 2018, 16, 189. [Google Scholar] [CrossRef] [PubMed]
- Dean, L.; McEntyre, J. Chapter 3, Genetic Factors in Type 2 Diabetes. In The Genetic Landscape of Diabetes; National Center for Biotechnology Information: Bethesda, MD, USA, 2004. [Google Scholar]
- Xue, A.; Wu, Y.; Zhu, Z.; Zhang, F.; Kemper, K.E.; Zheng, Z.; Yengo, L.; Lloyd-Jones, L.R.; Sidorenko, J.; Wu, Y.; et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018, 9, 2941. [Google Scholar] [CrossRef] [Green Version]
- DeForest, N.; Majithia, A.R. Genetics of type 2 diabetes: Implications from large-scale studies. Curr. Diab. Rep. 2022, 22, 227–235. [Google Scholar] [CrossRef]
- Banerjee, M.; Saxena, M. Genetic polymorphisms of cytokine genes in type 2 diabetes mellitus. World J. Diabetes 2014, 5, 493–504. [Google Scholar] [CrossRef]
- Sun, X.; Yu, W.; Hu, C. Genetics of type 2 diabetes: Insights into the pathogenesis and its clinical application. BioMed Res. Int. 2014, 2014, 926713. [Google Scholar] [CrossRef] [Green Version]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Cano-Gamez, E.; Trynka, G. From GWAS to function: Using functional genomics to identify the mechanisms underlying complex diseases. Front. Genet. 2020, 11, 424. [Google Scholar] [CrossRef]
- Liu, W.; Zhuang, Z.; Wang, W.; Huang, T.; Liu, Z. An improved genome-wide polygenic score model for predicting the risk of type 2 diabetes. Front. Genet. 2021, 12, 632385. [Google Scholar] [CrossRef]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Udler, M.S.; McCarthy, M.I.; Florez, J.C.; Mahajan, A. Genetic Risk Scores for Diabetes Diagnosis and Precision Medicine. Endocr. Rev. 2019, 40, 1500–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkinson, C.P.; Göring, H.H.H.; Arya, R.; Blangero, J.; Duggirala, R.; DeFronzo, R.A. Transcriptomics in type 2 diabetes: Bridging the gap between genotype and phenotype. Genom. Data 2016, 8, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaizer, E.C.; Glaser, C.L.; Chaussabel, D.; Banchereau, J.; Pascual, V.; White, P.C. Gene expression in peripheral blood mononuclear cells from children with diabetes. J. Clin. Endocrinol. Metab. 2007, 92, 3705–3711. [Google Scholar] [CrossRef] [Green Version]
- Grayson, B.L.; Wang, L.; Aune, T.M. Peripheral blood gene expression profiles in metabolic syndrome, coronary artery disease and type 2 diabetes. Genes. Immun. 2011, 12, 341–351. [Google Scholar] [CrossRef]
- Mao, J.; Ai, J.; Zhou, X.; Shenwu, M.; Ong, M., Jr.; Blue, M.; Washington, J.T.; Wang, X.; Deng, Y. Transcriptomic profiles of peripheral white blood cells in type II diabetes and racial differences in expression profiles. BMC Genom. 2011, S12. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Mao, J.; Ai, J.; Shenwu, M.; Shi, T.; Zhang, D.; Wang, X.; Wang, Y.; Deng, Y. Integrated lipidomics and transcriptomic analysis of peripheral blood reveals significantly enriched pathways in type 2 diabetes mellitus. BMC Med. Genom. 2013, 6 (Suppl. 1), S12. [Google Scholar] [CrossRef] [Green Version]
- Bugliani, M.; Liechti, R.; Cheon, H.; Suleiman, M.; Marselli, L.; Kirkpatrick, C.; Filipponi, F.; Boggi, U.; Xenarios, I.; Syed, F.; et al. Microarray analysis of isolated human islet transcriptome in type 2 diabetes and the role of the ubiquitin-proteasome system in pancreatic β cell dysfunction. Mol. Cell. Endocrinol. 2013, 367, 1–10. [Google Scholar] [CrossRef]
- Collares, C.V.; Evangelista, A.F.; Xavier, D.J.; Takahashi, P.; Almeida, R.; Macedo, C.; Manoel-Caetano, F.; Foss, M.C.; Foss-Freitas, M.C.; Rassi, D.M.; et al. Transcriptome meta-analysis of peripheral lymphomononuclear cells indicates that gestational diabetes is closer to type 1 diabetes than to type 2 diabetes mellitus. Mol. Biol. Rep. 2013, 40, 5351–5358. [Google Scholar] [CrossRef]
- Taneera, J.; Fadista, J.; Ahlqvist, E.; Zhang, M.; Wierup, N.; Renström, E.; Groop, L. Expression profiling of cell cycle genes in human pancreatic islets with and without type 2 diabetes. Mol. Cell. Endocrinol. 2013, 375, 35–42. [Google Scholar] [CrossRef]
- Corbi, S.C.T.; Bastos, A.S.; Nepomuceno, R.; Cirelli, T.; Dos Santos, R.A.; Takahashi, C.S.; Rocha, C.S.; Orrico, S.R.P.; Maurer-Morelli, C.V.; Scarel-Caminaga, R.M. Expression profile of genes potentially associated with adequate glycemic control in patients with type 2 diabetes mellitus. J. Diabetes Res. 2017, 2017, 2180819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matone, A.; Derlindati, E.; Marchetti, L.; Spigoni, V.; Dei Cas, A.; Montanini, B.; Ardigò, D.; Zavaroni, I.; Priami, C.; Bonadonna, R.C. Identification of an early transcriptomic signature of insulin resistance and related diseases in lymphomonocytes of healthy subjects. PLoS ONE 2017, 12, e0182559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villegas-Ruiz, V.; Hendlmeier, F.; Buentello-Volante, B.; Rodríguez-Loaiza, J.L.; Miranda-Duarte, A.; Zenteno, J.C. Genome-wide mRNA analysis reveals a TUBD1 isoform profile as a potential biomarker for diabetic retinopathy development. Exp. Eye Res. 2017, 155, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ha, X.; Li, W.; Xu, P.; Zhang, Z.; Wang, T.; Li, J.; Wang, Y.; Li, S.; Xie, J.; et al. Comparative gene expression profile and DNA methylation status in diabetic patients of Kazak and Han people. Medicine 2018, 97, e11982. [Google Scholar] [CrossRef]
- Saxena, A.; Tiwari, P.; Wahi, N.; Soni, A.; Bansiwal, R.C.; Kumar, A.; Sharma, B.; Punjabi, P.; Gupta, N.; Malik, B.; et al. Transcriptome profiling reveals association of peripheral adipose tissue pathology with type-2 diabetes in Asian Indians. Adipocyte 2019, 8, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Corbi, S.C.T.; de Vasconcellos, J.F.; Bastos, A.S.; Bussaneli, D.G.; da Silva, B.R.; Santos, R.A.; Takahashi, C.S.; de S. Rocha, C.; de Sá Carvalho, B.; Maurer-Morelli, C.V.; et al. Circulating lymphocytes and monocytes transcriptomic analysis of patients with type 2 diabetes mellitus, dyslipidemia and periodontitis. Sci. Rep. 2020, 10, 8145. [Google Scholar] [CrossRef]
- Veroneze, R.; Cruz Tfaile Corbi, S.; Roque da Silva, B.; de S. Rocha, C.; Maurer-Morelli, V.C.; Perez Orrico, S.R.; Cirelli, J.A.; Von Zuben, F.J.; Mantuaneli Scarel-Caminaga, R. Using association rule mining to jointly detect clinical features and differentially expressed genes related to chronic inflammatory diseases. PLoS ONE 2020, 15, e0240269. [Google Scholar] [CrossRef]
- Noreen, Z.; Loffredo, C.A.; Bhatti, A.; Simhadri, J.J.; Nunlee-Bland, G.; Nnanabu, T.; John, P.; Khan, J.S.; Ghosh, S. Transcriptional profiling and biological pathway(s) analysis of type 2 diabetes mellitus in a Pakistani population. Int. J. Environ. Res. Public Health 2020, 17, 5866. [Google Scholar] [CrossRef]
- Almanza-Aguilera, E.; Hernáez, Á.; Corella, D.; Sanllorente, A.; Ros, E.; Portolés, O.; Valussi, J.; Estruch, R.; Coltell, O.; Subirana, I.; et al. Cancer signaling transcriptome is upregulated in type 2 diabetes mellitus. J. Clin. Med. 2020, 10, 85. [Google Scholar] [CrossRef]
- Bury, J.J.; Chambers, A.; Heath, P.R.; Paul, G.; Ince, P.J.; Shaw, F.E.; Matthews, C.B.; Simpson, J.E.; Wharton, S.B. Type 2 diabetes mellitus-associated transcriptome alterations in cortical neurones and associated neurovascular unit cells in the ageing brain. Acta Neuropathol. Commun. 2021, 9, 5. [Google Scholar] [CrossRef]
- Scott, L.J.; Erdos, M.R.; Huyghe, J.R.; Welch, R.P.; Beck, A.T.; Wolford, B.N.; Chines, P.S.; Didion, J.P.; Narisu, N.; Stringham, H.M.; et al. The genetic regulatory signature of type 2 diabetes in human skeletal muscle. Nat. Commun. 2016, 7, 11764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, R.; Chen, M.; Zhang, C.; Chen, M.; Li, H. A comparison of gene expression profiles in patients with coronary artery disease, type 2 diabetes, and their coexisting conditions. Diagn. Pathol. 2017, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, X.; Shu, J.; Lee, C.T. Whole-genome expression analyses of type 2 diabetes in human skin reveal altered immune function and burden of infection. Oncotarget 2017, 8, 34601–34609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckman, J.A.; Doherty, S.P.; Feldman, Z.B.; Banks, E.S.; Moslehi, J.; Jaffe, I.Z.; Hamburg, N.M.; Sheng, Q.; Brown, J.D. Comparative transcriptomics of Ex Vivo, patient-derived endothelial cells reveals novel pathways associated with type 2 diabetes mellitus. JACC Basic Transl. Sci. 2019, 4, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Bao, X.; Li, P.; Lian, J.; Wu, Y.; An, T.; Zhang, J.; Yang, X.; Wang, T.; Zhu, J.; et al. Transcriptome sequencing analysis of peripheral blood of type 2 diabetes mellitus patients with thirst and fatigue. Front. Endocrinol. 2020, 11, 558344. [Google Scholar] [CrossRef]
- Lin, Q.; Zhou, W.; Wang, Y.; Huang, J.; Hui, X.; Zhou, Z.; Xiao, Y. Abnormal peripheral neutrophil transcriptome in newly diagnosed type 2 diabetes patients. J. Diabetes Res. 2020, 2020, 9519072. [Google Scholar] [CrossRef] [Green Version]
- Carruthers, N.J.; Strieder-Barboza, C.; Caruso, J.A.; Flesher, C.G.; Baker, N.A.; Kerk, S.A.; Ky, A.; Ehlers, A.P.; Varban, O.A.; Lyssiotis, C.A.; et al. The human type 2 diabetes-specific visceral adipose tissue proteome and transcriptome in obesity. Sci. Rep. 2021, 11, 17394. [Google Scholar] [CrossRef]
- Saxena, A.; Mathur, N.; Tiwari, P.; Mathur, S.K. Whole transcriptome RNA-seq reveals key regulatory factors involved in type 2 diabetes pathology in peripheral fat of Asian Indians. Sci. Rep. 2021, 11, 10632. [Google Scholar] [CrossRef]
- Kleinstein, S.E.; McCorrison, J.; Ahmed, A.; Hasturk, H.; Van Dyke, T.E.; Freire, M. Transcriptomics of type 2 diabetic and healthy human neutrophils. BMC Immunol. 2021, 22, 37. [Google Scholar] [CrossRef]
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andréasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Kim, J.; Okamoto, H.; Ni, M.; Wei, Y.; Adler, C.; Murphy, A.J.; Yancopoulos, G.D.; Lin, C.; Gromada, J. RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab. 2016, 24, 608–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.J.; Schug, J.; Won, K.J.; Liu, C.; Naji, A.; Avrahami, D.; Golson, M.L.; Kaestner, K.H. Single-cell transcriptomics of the human endocrine pancreas. Diabetes 2016, 65, 3028–3038. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, N.; George, J.; Bolisetty, M.; Kursawe, R.; Sun, L.; Sivakamasundari, V.; Kycia, I.; Robson, P.; Stitzel, M.L. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res. 2017, 27, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Alwine, J.C.; Kemp, D.J.; Stark, G.R. Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proc. Natl. Acad. Sci. USA 1977, 74, 5350–5354. [Google Scholar] [CrossRef] [Green Version]
- Trayhurn, P. Northern blotting. Proc. Nutr. Soc. 1996, 55, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Fehr, J.E.; Trotter, G.W.; Oxford, J.T.; Hart, D.A. Comparison of Northern blot hybridization and a reverse transcriptase-polymerase chain reaction technique for measurement of mRNA expression of metalloproteinases and matrix components in articular cartilage and synovial membrane from horses with osteoarthritis. Am. J. Vet. Res. 2000, 61, 900–905. [Google Scholar] [CrossRef]
- Taniguchi, M.; Miura, K.; Iwao, H.; Yamanaka, S. Quantitative assessment of DNA microarrays-comparison with Northern blot analyses. Genomics 2001, 71, 34–39. [Google Scholar] [CrossRef]
- Raghavendra, P.; Pullaiah, T. Chapter 2: RNA-Based Applications in Diagnostic and Therapeutics for Cancer. In Advances in Cell and Molecular Diagnostics, 1st ed.; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar] [CrossRef]
- Wong, M.L.; Medrano, J.F. Real-time PCR for mRNA quantitation. Biotechniques 2005, 39, 75–85. [Google Scholar] [CrossRef]
- Perez, L.J.; Rios, L.; Trivedi, P.; D’Souza, K.; Cowie, A.; Nzirorera, C.; Webster, D.; Brunt, K.; Legare, J.F.; Hassan, A.; et al. Validation of optimal reference genes for quantitative real time PCR in muscle and adipose tissue for obesity and diabetes research. Sci. Rep. 2017, 7, 3612. [Google Scholar] [CrossRef] [Green Version]
- Clarke, J.D.; Zhu, T. Microarray analysis of the transcriptome as a stepping stone towards understanding biological systems: Practical considerations and perspectives. Plant J. 2006, 45, 630–650. [Google Scholar] [CrossRef]
- Rachinger, N.; Fischer, S.; Böhme, I.; Linck-Paulus, L.; Kuphal, S.; Kappelmann-Fenzl, M.; Bosserhoff, A.K. Loss of gene information: Discrepancies between RNA sequencing, cDNA microarray, and qRT-PCR. Int. J. Mol. Sci. 2021, 22, 9349. [Google Scholar] [CrossRef]
- Hurd, P.J.; Nelson, C.J. Advantages of next-generation sequencing versus the microarray in epigenetic research. Brief. Funct. Genom. Proteom. 2009, 8, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Slonim, D.K.; Yanai, I. Getting started in gene expression microarray analysis. PLoS Comput. Biol. 2009, 5, e1000543. [Google Scholar] [CrossRef]
- Zito, A.; Lualdi, M.; Granata, P.; Cocciadiferro, D.; Novelli, A.; Alberio, T.; Casalone, R.; Fasano, M. Gene set enrichment analysis of interaction networks weighted by node centrality. Front. Genet. 2021, 12, 577623. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA sequencing and analysis. Cold Spring Harb. Protoc. 2015, 11, 951–969. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.S.; Van Vleet, T.R.; Ciurlionis, R.; Buck, W.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Liguori, M.J. Comparison of RNA-seq and microarray gene expression platforms for the toxicogenomic evaluation of liver from short-term rat toxicity studies. Front. Genet. 2019, 9, 636. [Google Scholar] [CrossRef] [Green Version]
- Roh, S.W.; Abell, G.C.; Kim, K.H.; Nam, Y.D.; Bae, J.W. Comparing microarrays and next-generation sequencing technologies for microbial ecology research. Trends Biotechnol. 2010, 28, 291–299. [Google Scholar] [CrossRef]
- Liao, Y.; Shi, W. Read trimming is not required for mapping and quantification of RNA-seq reads at the gene level. NAR Genom Bioinform. 2020, 2, lqaa068. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Huang, Y.; Lei, J.; Luo, H.; Zhu, X. The single-cell sequencing: New developments and medical applications. Cell Biosci. 2019, 26, 53. [Google Scholar] [CrossRef] [Green Version]
- Alessandrì, L.; Arigoni, M.; Calogero, R. Differential expression analysis in single-cell transcriptomics. Methods Mol. Biol. 2019, 1979, 425–432. [Google Scholar] [CrossRef]
- Wang, T.; Li, B.; Nelson, C.E.; Nabavi, S. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinform. 2019, 20, 40. [Google Scholar] [CrossRef] [Green Version]
- Solis-Herrera, C.; Triplitt, C.; Cersosimo, E. Pathogenesis of type 2 diabetes mellitus. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of adipose tissue inflammation to the development of type 2 diabetes mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar] [CrossRef]
- Hardy, O.T.; Czech, M.P.; Corvera, S. What causes the insulin resistance underlying obesity? Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Tajima, T.; Goda, N.; Fujiki, N.; Hishiki, T.; Nishiyama, Y.; Senoo-Matsuda, N.; Shimazu, M.; Soga, T.; Yoshimura, Y.; Johnson, R.S.; et al. HIF-1alpha is necessary to support gluconeogenesis during liver regeneration. Biochem. Biophys. Res. Commun. 2009, 387, 789–794. [Google Scholar] [CrossRef]
- Norouzirad, R.; González-Muniesa, P.; Ghasemi, A. Hypoxia in obesity and diabetes: Potential therapeutic effects of hyperoxia and nitrate. Oxid. Med. Cell. Longev. 2017, 2017, 5350267. [Google Scholar] [CrossRef]
- Boden, G. Effects of free fatty acids (FFA) on glucose metabolism: Significance for insulin resistance and type 2 diabetes. Exp. Clin. Endocrinol. Diabetes 2003, 111, 121–124. [Google Scholar] [CrossRef]
- Cnop, M.; Abdulkarim, B.; Bottu, G.; Cunha, D.A.; Igoillo-Esteve, M.; Masini, M.; Turatsinze, J.V.; Griebel, T.; Villate, O.; Santin, I.; et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 2014, 63, 1978–1993. [Google Scholar] [CrossRef] [Green Version]
- Agrafioti, P.; Morin-Baxter, J.; Tanagala, K.K.K.; Dubey, S.; Sims, P.; Lalla, E.; Momen-Heravi, F. Decoding the role of macrophages in periodontitis and type 2 diabetes using single-cell RNA-sequencing. FASEB J. 2022, 36, e22136. [Google Scholar] [CrossRef]
- Aronson, D.; Edelman, E.R. Coronary artery disease and diabetes mellitus. Cardiol Clin. 2014, 32, 439–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007-2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef] [Green Version]
- Hadi, H.A.; Suwaidi, J.A. Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag. 2007, 3, 853–876. [Google Scholar]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef] [Green Version]
- Ni, T.; Wang, N.; Mao, Z.G.; Yao, M. Identification of marker genes in diabetic wounds by DNA microarray study. Genet. Mol. Res. 2013, 12, 5348–5355. [Google Scholar] [CrossRef]
- Ridiandries, A.; Tan, J.T.M.; Bursill, C.A. The role of chemokines in wound healing. Int. J. Mol. Sci. 2018, 19, 3217. [Google Scholar] [CrossRef] [Green Version]
- Jordan, A.R.; Racine, R.R.; Hennig, M.J.; Lokeshwar, V.B. The role of CD44 in disease pathophysiology and targeted treatment. Front. Immunol. 2015, 6, 182. [Google Scholar] [CrossRef]
- Liu, L.F.; Kodama, K.; Wei, K.; Tolentino, L.L.; Choi, O.; Engleman, E.G.; Butte, A.J.; McLaughlin, T. The receptor CD44 is associated with systemic insulin resistance and proinflammatory macrophages in human adipose tissue. Diabetologia 2015, 58, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Rosen, J.; Yosipovitch, G. Skin manifestations of diabetes mellitus. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- American Diabetes Association. Standards of medical care in diabetes. Diabetes Care 2004, 27 (Suppl. 1), 15–35. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Chen, W.; Chi, J.; Wang, L. Comparison of transcriptome between type 2 diabetes mellitus and impaired fasting glucose. Med. Sci. Monit. 2016, 22, 4699–4706. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, P.; Ferreira, S.R. An update on cardiovascular risk of metabolic syndrome. Arq. Bras. Endocrinol. Metabol. 2003, 47, 220–227. [Google Scholar] [CrossRef]
- Denis, G.V. Bromodomain coactivators in cancer, obesity, type 2 diabetes, and inflammation. Discov. Med. 2010, 10, 489–499. [Google Scholar]
- Abdellatif, A.M.; Oishi, H.; Itagaki, T.; Jung, Y.; Shawki, H.H.; Okita, Y.; Hasegawa, Y.; Suzuki, H.; El-Morsy, S.E.; El-Sayed, M.A.; et al. β-cell-specific Mafk overexpression impairs pancreatic endocrine cell development. PLoS ONE 2016, 11, e0150010. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Zhang, X. The profiling and role of miRNAs in diabetes mellitus. J. Diabetes Clin. Res. 2019, 1, 5–23. [Google Scholar] [CrossRef]
- Tonyan, Z.N.; Nasykhova, Y.A.; Mikhailova, A.A.; Glotov, A.S. MicroRNAs as potential biomarkers of type 2 diabetes mellitus. Russ. J. Genet. 2021, 57, 764–777. [Google Scholar] [CrossRef]
- Poy, M.N.; Eliasson, L.; Krutzfeldt, J.; Kuwajima, S.; Ma, X.; Macdonald, P.E.; Pfeffer, S.; Tuschl, T.; Rajewsky, N.; Rorsman, P.; et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 2004, 432, 226–230. [Google Scholar] [CrossRef]
- Karolina, D.S.; Tavintharan, S.; Armugam, A.; Sepramaniam, S.; Pek, S.L.; Wong, M.T.; Lim, S.C.; Sum, C.F.; Jeyaseelan, K. Circulating miRNA profiles in patients with metabolic syndrome. J. Clin. Endocrinol. Metab. 2012, 97, E2271–E2276. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Chang, X.; Yin, L.; Li, J.; Zhou, T.; Zhang, C.; Chen, X. Expression and DNA methylation status of microRNA-375 in patients with type 2 diabetes mellitus. Mol. Med. Rep. 2014, 9, 967–972. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Zhu, J.; Han, W.; Jiang, X.; Xu, M.; Zhao, Y.; Dong, Q.; Pang, Z.; Guan, Q.; Gao, L.; et al. Significance of serum microRNAs in pre-diabetes and newly diagnosed type 2 diabetes: A clinical study. Acta Diabetol. 2011, 48, 61–69. [Google Scholar] [CrossRef]
- He, Y.; Ding, Y.; Liang, B.; Lin, J.; Kim, T.K.; Yu, H.; Hang, H.; Wang, K. A systematic study of dysregulated microRNA in type 2 diabetes mellitus. Int. J. Mol. Sci. 2017, 18, 456. [Google Scholar] [CrossRef]
- Bommer, G.T.; Gerin, I.; Feng, Y.; Kaczorowski, A.J.; Kuick, R.; Love, R.E.; Zhai, Y.; Giordano, T.J.; Qin, Z.S.; Moore, B.B.; et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 2007, 17, 1298–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Xu, H.; Pan, X.; Wu, W.; Wang, H.; Yan, L.; Zhang, M.; Liu, X.; Xia, S.; Shao, Q. miR-34a and miR-125b are upregulated in peripheral blood mononuclear cells from patients with type 2 diabetes mellitus. Exp. Ther. Med. 2017, 14, 5589–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Peng, R.; Li, A.; Zhang, L.; Liu, H.; Peng, H.; Zhang, Z. Sequence variation in microRNA-34a is associated with diabetes mellitus susceptibility in a southwest Chinese Han population. Int. J. Clin. Exp. Pathol. 2018, 11, 1637–1644. [Google Scholar]
- Karolina, D.S.; Armugam, A.; Tavintharan, S.; Wong, M.T.; Lim, S.C.; Sum, C.F.; Jeyaseelan, K. MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS ONE 2011, 6, e22839. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.M.; Jeong, H.J.; Park, S.Y.; Lee, W. Induction of miR-29a by saturated fatty acids impairs insulin signaling and glucose uptake through translational repression of IRS-1 in myocytes. FEBS Lett. 2014, 588, 2170–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.M.; Chen, L.H.; Hong, M.; Chen, Y.Y.; Yang, X.R.; Tang, S.M.; Yuan, Q.F.; Chen, W.W. Serum microRNA profiling and bioinformatics analysis of patients with type 2 diabetes mellitus in a Chinese population. Mol. Med. Rep. 2017, 15, 2143–2153. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Sundquist, J.; Zöller, B.; Memon, A.A.; Palmér, K.; Sundquist, K.; Bennet, L. Determination of 14 circulating microRNAs in Swedes and Iraqis with and without diabetes mellitus type 2. PLoS ONE 2014, 9, e86792. [Google Scholar] [CrossRef]
- Gallagher, I.J.; Scheele, C.; Keller, P.; Nielsen, A.R.; Remenyi, J.; Fischer, C.P.; Roder, K.; Babraj, J.; Wahlestedt, C.; Hutvagner, G.; et al. Integration of microRNA changes in vivo identifies novel molecular features of muscle insulin resistance in type 2 diabetes. Genome Med. 2010, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Ślusarz, A.; Pulakat, L. The two faces of miR-29. J. Cardiovasc. Med. 2015, 16, 480–490. [Google Scholar] [CrossRef]
- Liang, Y.Z.; Dong, J.; Zhang, J.; Wang, S.; He, Y.; Yan, Y.X. Identification of neuroendocrine stress response-related circulating microRNAs as biomarkers for type 2 diabetes mellitus and insulin resistance. Front. Endocrinol. 2018, 9, 132. [Google Scholar] [CrossRef]
- Jia, L.; Zhang, R. Alterations in gene expression profiles in children with type 2 diabetes mellitus and its mechanisms. J. Cent. South Univ. 2018, 43, 826–834. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Donath, M.Y.; Mandrup-Poulsen, T. Role of IL-1beta in type 2 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Sun, N.; Xu, Q.; Jiang, Z.; Li, C. Comparative analysis of mRNA expression profiles in Type 1 and Type 2 diabetes mellitus. Epigenomics 2019, 11, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Ninichuk, V.; Anders, H.J. Chemokine receptor CCR1: A new target for progressive kidney disease. Am. J. Nephrol. 2005, 25, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Rojewska, E.; Zychowska, M.; Piotrowska, A.; Kreiner, G.; Nalepa, I.; Mika, J. Involvement of macrophage inflammatory protein-1 family members in the development of diabetic neuropathy and their contribution to effectiveness of morphine. Front. Immunol. 2018, 9, 494. [Google Scholar] [CrossRef] [Green Version]
- Quezada-Calvillo, R.; Sim, L.; Ao, Z.; Hamaker, B.R.; Quaroni, A.; Brayer, G.D.; Sterchi, E.E.; Robayo-Torres, C.C.; Rose, D.R.; Nichols, B.L. Luminal starch substrate “brake” on maltase-glucoamylase activity is located within the glucoamylase subunit. J. Nutr. 2008, 138, 685–692. [Google Scholar] [CrossRef] [Green Version]
- Nichols, B.L.; Quezada-Calvillo, R.; Robayo-Torres, C.C.; Ao, Z.; Hamaker, B.R.; Butte, N.F.; Marini, J.; Jahoor, F.; Sterchi, E.E. Mucosal maltase-glucoamylase plays a crucial role in starch digestion and prandial glucose homeostasis of mice. J. Nutr. 2009, 139, 684–690. [Google Scholar] [CrossRef]
- Garten, A.; Petzold, S.; Schuster, S.; Körner, A.; Kratzsch, J.; Kiess, W. Nampt and its potential role in inflammation and type 2 diabetes. Handb. Exp. Pharmacol. 2011, 203, 147–164. [Google Scholar] [CrossRef]
- Lee, H.Y.; Yea, K.; Kim, J.; Lee, B.D.; Chae, Y.C.; Kim, H.S.; Lee, D.W.; Kim, S.H.; Cho, J.H.; Jin, C.J.; et al. Epidermal growth factor increases insulin secretion and lowers blood glucose in diabetic mice. J. Cell Mol. Med. 2008, 12, 1593–1604. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wang, Z.; Liu, G.; Jin, C.; Zhang, Q.; Man, S.; Wang, Z. miR-657 promotes macrophage polarization toward M1 by targeting FAM46C in gestational diabetes mellitus. Mediat. Inflamm. 2019, 2019, 4851214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maachi, H.; Fergusson, G.; Ethier, M.; Brill, G.N.; Katz, L.S.; Honig, L.B.; Metukuri, M.R.; Scott, D.K.; Ghislain, J.; Poitout, V. HB-EGF signaling is required for glucose-induced pancreatic β-Cell proliferation in rats. Diabetes 2020, 69, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akerfeldt, M.C.; Laybutt, D.R. Inhibition of Id1 augments insulin secretion and protects against high-fat diet-induced glucose intolerance. Diabetes 2011, 60, 2506–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig-Słomczyńska, A.H.; Seweryn, M.T.; Kapusta, P.; Pitera, E.; Mantaj, U.; Cyganek, K.; Gutaj, P.; Dobrucka, Ł.; Wender-Ożegowska, E.; Małecki, M.T.; et al. The transcriptome-wide association search for genes and genetic variants which associate with BMI and gestational weight gain in women with type 1 diabetes. Mol. Med. 2021, 27, 6. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.J.; Kollara, A.; Shathasivam, P.; Ringuette, M.J. Ventricular zone expressed PH Domain Containing 1 (VEPH1): An adaptor protein capable of modulating multiple signaling transduction pathways during normal and pathological development. Cell. Commun. Signal. 2019, 17, 116. [Google Scholar] [CrossRef] [Green Version]
- Alkhatatbeh, M.J.; Enjeti, A.K.; Acharya, S.; Thorne, R.F.; Lincz, L.F. The origin of circulating CD36 in type 2 diabetes. Nutr. Diabetes 2013, 3, e59. [Google Scholar] [CrossRef]
- Jermendy, A.; Toschi, E.; Aye, T.; Koh, A.; Aguayo-Mazzucato, C.; Sharma, A.; Weir, G.C.; Sgroi, D.; Bonner-Weir, S. Rat neonatal β cells lack the specialised metabolic phenotype of mature β cells. Diabetologia 2011, 54, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Weir, G.C.; Aguayo-Mazzucato, C.; Bonner-Weir, S. β-cell dedifferentiation in diabetes is important, but what is it? Islets 2013, 5, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Okamoto, H.; Kim, J.; Ni, M.; Adler, C.; Cavino, K.; Na, E.; Murphy, A.J.; Yancopoulos, G.D.; Lin, C.; et al. Single-cell RNAseq reveals that pancreatic β-cells from very old male mice have a young gene signature. Endocrinology 2016, 157, 3431–3438. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.; Mulas, F.; Sui, Y.; Guan, T.; Miller, N.; Tan, Y.; Liu, F.; Jin, W.; Carrano, A.C.; Huising, M.O.; et al. Pseudotemporal ordering of single cells reveals metabolic control of postnatal β cell proliferation. Cell Metab. 2017, 25, 1160–1175. [Google Scholar] [CrossRef] [Green Version]
- Baron, M.; Veres, A.; Wolock, S.L.; Faust, A.L.; Gaujoux, R.; Vetere, A.; Ryu, J.H.; Wagner, B.K.; Shen-Orr, S.S.; Klein, A.M.; et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 2016, 3, 346–360. [Google Scholar] [CrossRef] [Green Version]
- Groen, N.; Leenders, F.; Mahfouz, A.; Munoz-Garcia, A.; Muraro, M.J.; de Graaf, N.; Rabelink, T.J.; Hoeben, R.; van Oudenaarden, A.; Zaldumbide, A.; et al. Single-cell transcriptomics links loss of human pancreatic β-cell identity to ER stress. Cells 2021, 10, 3585. [Google Scholar] [CrossRef]
- Teo, A.K.K.; Lim, C.S.; Cheow, L.F.; Kin, T.; Shapiro, J.A.; Kang, N.; Burkholder, W.; Lau, H.H. Single-cell analyses of human islet cells reveal de-differentiation signatures. Cell Death Discov. 2018, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- Enge, M.; Arda, H.E.; Mignardi, M.; Beausang, J.; Bottino, R.; Kim, S.K.; Quake, S.R. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell 2017, 171, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Qadir, M.M.F.; Álvarez-Cubela, S.; Klein, D.; van Dijk, J.; Muñiz-Anquela, R.; Moreno-Hernández, Y.B.; Lanzoni, G.; Sadiq, S.; Navarro-Rubio, B.; García, M.T.; et al. Single-cell resolution analysis of the human pancreatic ductal progenitor cell niche. Proc. Natl. Acad. Sci. USA 2020, 117, 10876–10887. [Google Scholar] [CrossRef]
- Domínguez-Bendala, J.; Qadir, M.M.F.; Pastori, R.L. Pancreatic progenitors: There and back again. Trends Endocrinol. Metab. 2019, 30, 4–11. [Google Scholar] [CrossRef]
- Bonnycastle, L.L.; Gildea, D.E.; Yan, T.; Narisu, N.; Swift, A.J.; Wolfsberg, T.G.; Erdos, M.R.; Collins, F.S. Single-cell transcriptomics from human pancreatic islets: Sample preparation matters. Biol. Methods Protoc. 2019, 4, bpz019. [Google Scholar] [CrossRef]
- Wang, Y.J.; Kaestner, K.H. Single-cell RNA-seq of the pancreatic islets--a promise not yet fulfilled? Cell Metab. 2019, 29, 539–544. [Google Scholar] [CrossRef] [Green Version]
- Muraro, M.J.; Dharmadhikari, G.; Grün, D.; Groen, N.; Dielen, T.; Jansen, E.; van Gurp, L.; Engelse, M.A.; Carlotti, F.; de Koning, E.J.; et al. A single-cell transcriptome atlas of the human pancreas. Cell Syst. 2016, 3, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Dorrell, C.; Schug, J.; Canaday, P.S.; Russ, H.A.; Tarlow, B.D.; Grompe, M.T.; Horton, T.; Hebrok, M.; Streeter, P.R.; Kaestner, K.H.; et al. Human islets contain four distinct subtypes of β cells. Nat. Commun. 2016, 7, 11756. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, G.D.; Gromada, J.; Sussel, L. Heterogeneity of the pancreatic β cell. Front. Genet. 2017, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Cook, J.P.; Morris, A.P. Multi-ethnic genome-wide association study identifies novel locus for type 2 diabetes susceptibility. Eur. J. Hum. Genet. 2016, 24, 1175–1180. [Google Scholar] [CrossRef] [Green Version]
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.J.; et al. The genetic architecture of type 2 diabetes. Nature 2016, 536, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, A.; Wessel, J.; Willems, S.M.; Zhao, W.; Robertson, N.R.; Chu, A.Y.; Gan, W.; Kitajima, H.; Taliun, D.; Rayner, N.W.; et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat. Genet. 2018, 50, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Taskesen, E.; van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Baron, V.; Van Obberghen, E. Mechanism of insulin action. C. R. Seances Soc. Biol. Fil. 1995, 189, 25–41. [Google Scholar]
- King, A.J. The use of animal models in diabetes research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef] [Green Version]
- Milojević, M.; Rožanc, J.; Vajda, J.; Ćurić, L.Č.; Paradiž, E.; Stožer, A.; Maver, U.; Vihar, B. In vitro disease models of the endocrine pancreas. Biomedicines 2021, 9, 1415. [Google Scholar] [CrossRef]
- Everaert, C.; Luypaert, M.; Maag, J.L.V.; Cheng, Q.X.; Dinger, M.E.; Hellemans, J.; Mestdagh, P. Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data. Sci. Rep. 2017, 7, 1559. [Google Scholar] [CrossRef] [Green Version]



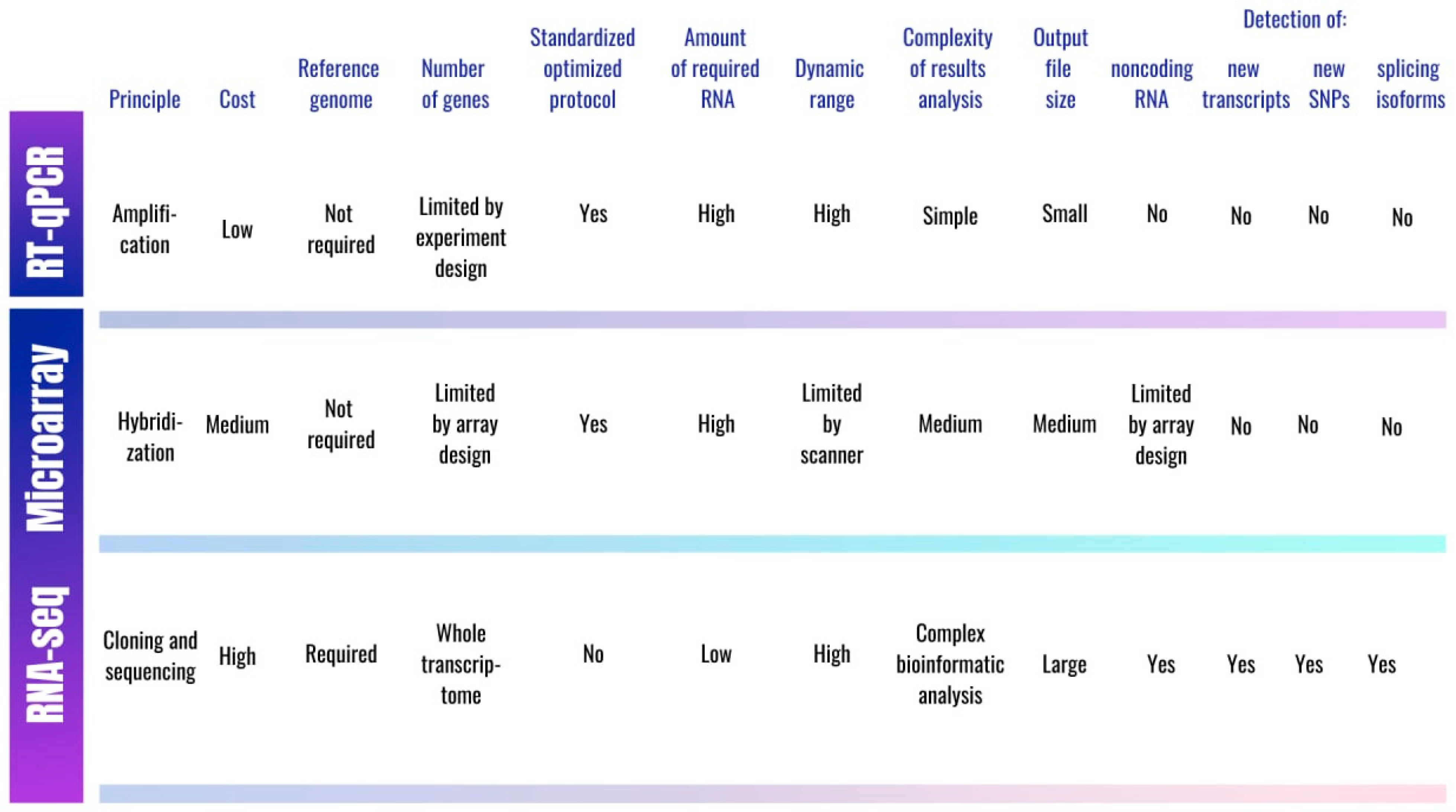{kind=link}
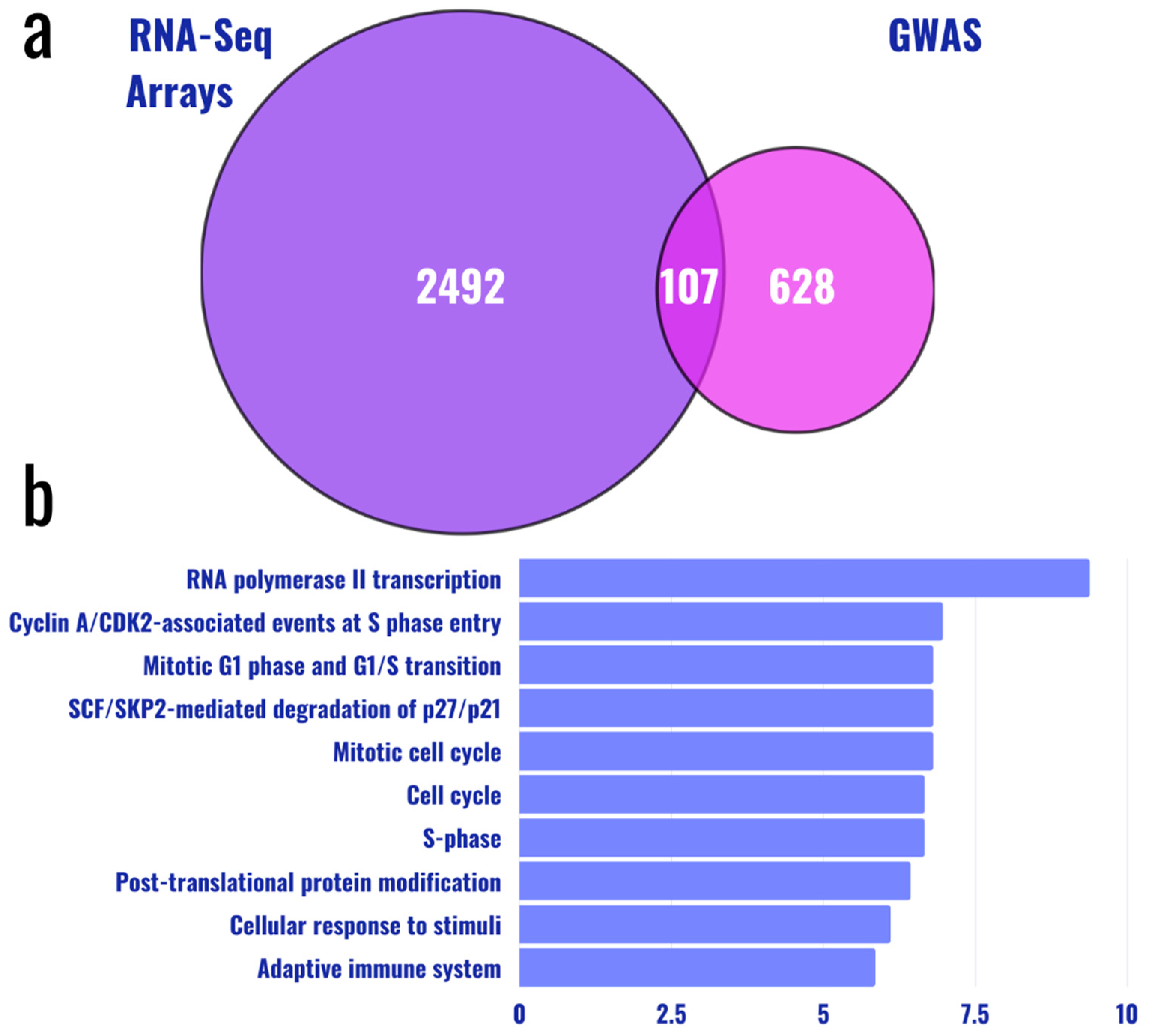{kind=link}
| Sample Type | Transcriptomic Technique | Validation | Study Group | References |
|---|---|---|---|---|
| PBMC | Microarray | qPCR | 43 newly diagnosed T1D, 12 newly diagnosed T2D, 24 HC | [16] |
| Whole blood | Microarray | qPCR | 6 patients with metabolic syndrome, 6 CAD, 8 T2D, 6 rheumatoid arthritis patients, 9 HC | [17] |
| Whole blood | Microarray | qPCR | 84 T2D, 60 HC | [18,19] |
| Human islet cells | Microarray | – | 7 non-diabetic subjects, 6 T2D donors | [20] |
| Whole blood | Microarray | – | 19 T1D, 20 T2D, 17 GDM | [21] |
| Human islet cells | Microarray | qPCR | 67 non-diabetic donors, 10 T2D donors | [22] |
| PBMC | Microarray | qPCR | 5 poorly controlled T2D, 7 well-controlled T2D, 6 normoglycemic individuals | [23] |
| PBMC | Microarray | – | 10 healthy individuals with extreme insulin resistance, 10 healthy individuals with extreme insulin sensitivity | [24] |
| Whole blood | Microarray | qPCR | 20 T2D with diabetic retinopathy, 10 T2D without diabetic retinopathy | [25] |
| Abdominal omental adipose tissues | Microarray | – | 12 T2D, 12 HC | [26] |
| Adipose tissue from thigh | Microarray | qPCR | 30 T2D, 30 HC | [27] |
| PBMC | Microarray | qPCR | 5 poorly controlled T2D with dyslipidemia and periodontitis, 7 well-controlled T2D with dyslipidemia and periodontitis, 6 normoglycemic with dyslipidemia and periodontitis, 6 healthy individuals with periodontitis, 6 HC | [28,29] |
| Whole blood | Microarray | TaqMan Low Density Array | 2 T2D, 2 HC | [30] |
| Whole blood | Microarray | – | 12 T2D, 19 HC | [31] |
| Neurone, astrocyte, and endothelial cell | Microarray | NanoString nCounter platform + Immunohistochemical validation of protein expression | 6 T2D, 6 HC | [32] |
| Skeletal muscle | RNA-Seq | – | 271 participants with glucose tolerance ranging from normal to newly diagnosed T2D | [33] |
| PBMC | RNA-Seq | – | 2 T2D, 2 CAD, 6 T2D + CAD, 7 HC | [34] |
| Skin samples | RNA-Seq | – | 74 T2D, 148 HC | [35] |
| Endothelial cells from cubital vein | RNA-Seq | – | 5 T2D, 5 HC | [36] |
| Whole blood | RNA-Seq | – | 6 T2D with thirst and fatigue, 6 HC | [37] |
| Neutrophils | RNA-Seq | qPCR | 5 newly diagnosed T2D, 5 HC | [38] |
| Visceral adipose tissue | RNA-Seq | qPCR | 10 T2D, 10 HC | [39] |
| Adipose tissue from thigh | RNA-Seq | qPCR | 5 T2D, 5 HC | [40] |
| Neutrophils | RNA-Seq | – | 11 T2D, 7 HC | [41] |
| Human islet cells | Single-cell RNA-Seq | RNA in situ hybridization | 4 T2D, 6 HC | [42] |
| Human islet cells | Single-cell RNA-Seq | – | 6 T2D, 12 HC | [43] |
| Human islet cells | Single-cell RNA-Seq | – | 1 T1D donor, 3 T2D donors, 2 children, 3 HC | [44] |
| Human islet cells | Single-cell RNA-Seq | RNA in situ hybridization | 3 T2D, 5 HC | [45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonyan, Z.N.; Nasykhova, Y.A.; Danilova, M.M.; Barbitoff, Y.A.; Changalidi, A.I.; Mikhailova, A.A.; Glotov, A.S. Overview of Transcriptomic Research on Type 2 Diabetes: Challenges and Perspectives. Genes 2022, 13, 1176. https://doi.org/10.3390/genes13071176
Tonyan ZN, Nasykhova YA, Danilova MM, Barbitoff YA, Changalidi AI, Mikhailova AA, Glotov AS. Overview of Transcriptomic Research on Type 2 Diabetes: Challenges and Perspectives. Genes. 2022; 13(7):1176. https://doi.org/10.3390/genes13071176
Chicago/Turabian StyleTonyan, Ziravard N., Yulia A. Nasykhova, Maria M. Danilova, Yury A. Barbitoff, Anton I. Changalidi, Anastasiia A. Mikhailova, and Andrey S. Glotov. 2022. "Overview of Transcriptomic Research on Type 2 Diabetes: Challenges and Perspectives" Genes 13, no. 7: 1176. https://doi.org/10.3390/genes13071176