
Description of Two New Cases of AQP1 Related Pulmonary Arterial Hypertension and Review of the Literature

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Case Presentation
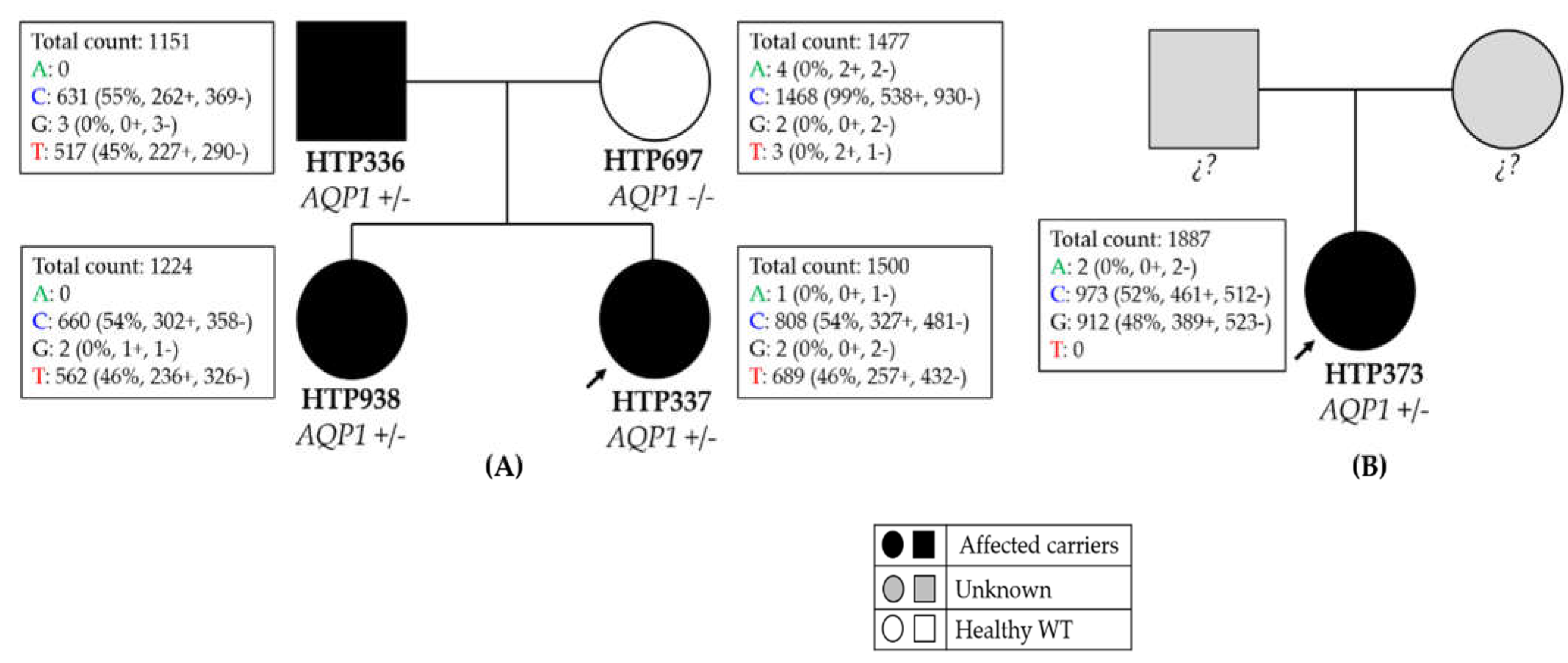
2.1.1. Family 1
2.1.2. Family 2
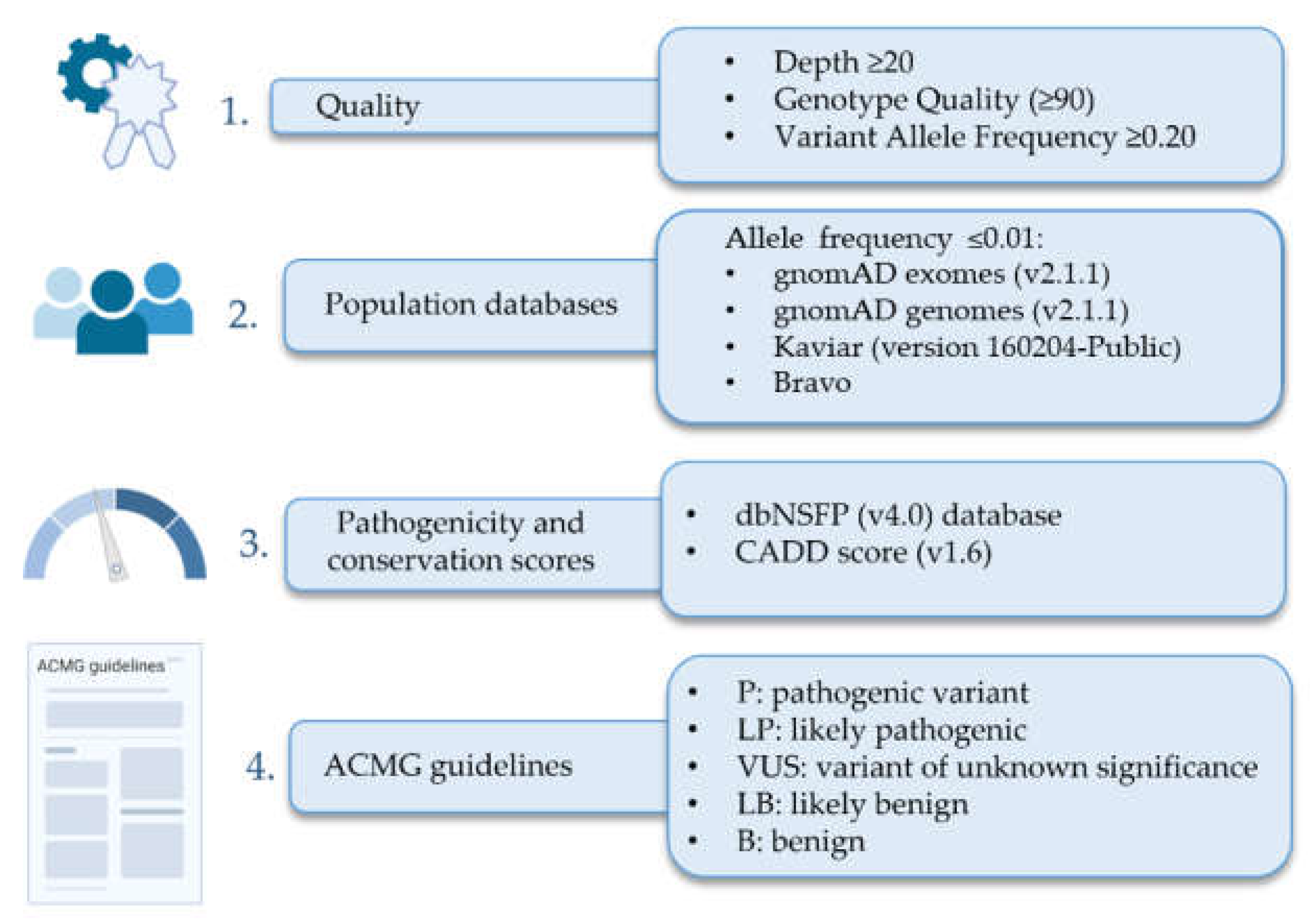
2.2. Genetic Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitbon, O.; Gomberg-Maitland, M.; Granton, J.; Lewis, M.I.; Mathai, S.C.; Rainisio, M.; Stockbridge, N.L.; Wilkins, M.R.; Zamanian, R.T.; Rubin, L.J. Clinical trial design and new therapies for pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galiè, N.; Humbert, M.; Vachiery, J.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar]
- Hemnes, A.R.; Humbert, M. Pathobiology of pulmonary arterial hypertension: Understanding the roads less travelled. Eur. Respir. Rev. 2017, 26, 170093. [Google Scholar] [CrossRef]
- Hassoun, P.M. Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 385, 2361–2376. [Google Scholar] [CrossRef]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J. Familial primary pulmonary hypertension (Gene PPH1) is caused by mutations in the bone morphogenetic protein receptor–II gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A.; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef] [Green Version]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nature 2018, 9, 1416. [Google Scholar] [CrossRef] [Green Version]
- Sui, H.; Han, B.; Lee, J.K.; Walian, P.; Jap, B.K. Structural basis of water-specific transport through the AQP1 water channel. Nature 2001, 414, 872–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadoun, S.; Papadopoulos, M.C.; Hara-Chikuma, M.; Verkman, A.S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 2005, 434, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lian, T.; Jiang, X.; Liu, S.; Li, S.; Jiang, R.; Wu, W.; Ye, J.; Cheng, C.; Du, Y. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801609. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L. ClinGen—The clinical genome resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaño, J.A.T.; Hernández-Gonzalez, I.; Gallego, N.; Pérez-Olivares, C.; Ochoa Parra, N.; Arias, P.; Granda, E.; Acebo, G.G.; Lago-Docampo, M.; Palomino-Doza, J. Customized massive parallel sequencing panel for diagnosis of pulmonary arterial hypertension. Genes 2020, 11, 1158. [Google Scholar] [CrossRef] [PubMed]
- Pérez Núñez, M.; Alonso Charterina, S.; Pérez-Olivares, C.; Revilla Ostolaza, Y.; Morales Ruiz, R.; Enguita Valls, A.B.; Tenorio, J.A.; Gallego Zazo, N.; de Pablo Gafas, A.; Lapunzina, P. Radiological findings in multidetector computed tomography (MDCT) of hereditary and sporadic pulmonary veno-occlusive disease: Certainties and uncertainties. Diagnostics 2021, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, C.; Mou, C.; Dong, Y.; Tu, Y. dbNSFP v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020, 12, 103. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Escribano-Subias, P.; Blanco, I.; López-Meseguer, M.; Lopez-Guarch, C.J.; Roman, A.; Morales, P.; Castillo-Palma, M.J.; Segovia, J.; Gómez-Sanchez, M.A.; Barberà, J.A. Survival in pulmonary hypertension in Spain: Insights from the Spanish registry. Eur. Respir. J. 2012, 40, 596–603. [Google Scholar] [CrossRef]
- Dunmore, B.J.; Jones, R.J.; Toshner, M.R.; Upton, P.D.; Morrell, N.W. Approaches to treat pulmonary arterial hypertension by targeting BMPR2: From cell membrane to nucleus. Cardiovasc. Res. 2021, 117, 2309–2325. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Lau, E.M.; Montani, D.; Jaïs, X.; Sitbon, O.; Simonneau, G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation 2014, 130, 2189–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Launay, D.; Sitbon, O.; Hachulla, E.; Mouthon, L.; Gressin, V.; Rottat, L.; Clerson, P.; Cordier, J.; Simonneau, G.; Humbert, M. Survival in systemic sclerosis-associated pulmonary arterial hypertension in the modern management era. Ann. Rheum. Dis. 2013, 72, 1940–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Launay, D.; Sobanski, V.; Hachulla, E.; Humbert, M. Pulmonary hypertension in systemic sclerosis: Different phenotypes. Eur. Respir. Rev. 2017, 26, 170056. [Google Scholar] [CrossRef] [Green Version]
- Grigoryev, D.N.; Mathai, S.C.; Fisher, M.R.; Girgis, R.E.; Zaiman, A.L.; Housten-Harris, T.; Cheadle, C.; Gao, L.; Hummers, L.K.; Champion, H.C. Identification of candidate genes in scleroderma-related pulmonary arterial hypertension. Transl. Res. 2008, 151, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Li, Y.; Yan, Y.; Shi, H.; Zou, T.; Shao, W.; Wang, Q. Identification and validation of key genes associated with systemic sclerosis-related pulmonary hypertension. Front. Genet. 2020, 816, 1–13. [Google Scholar] [CrossRef]
- Liu, M.; Liu, Q.; Pei, Y.; Gong, M.; Cui, X.; Pan, J.; Zhang, Y.; Liu, Y.; Liu, Y.; Yuan, X. Aqp-1 gene knockout attenuates hypoxic pulmonary hypertension of mice. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 48–62. [Google Scholar] [CrossRef] [Green Version]
- Schuoler, C.; Haider, T.J.; Leuenberger, C.; Vogel, J.; Ostergaard, L.; Kwapiszewska, G.; Kohler, M.; Gassmann, M.; Huber, L.C.; Brock, M. Aquaporin 1 controls the functional phenotype of pulmonary smooth muscle cells in hypoxia-induced pulmonary hypertension. Basic Res. Cardiol. 2017, 112, 30. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| This Study | Gräf et al., 2018 | Wang et al., 2019 | ||||
|---|---|---|---|---|---|---|
| Patient | Family 1 (HTP336) | Family 1 (HTP337) | Family 1 (HTP938) | Family 2 (HTP373) | N = 9 | N = 8 |
| Sex | Male | Female | Female | Female | 4 females + 5 males | NA |
| PAH diagnosis | HPAH | HPAH | HPAH | PAH-SSc, PVOD-like | 2 HAPH + 7 IPAH | 8 IPAH |
| Age at diagnosis (years) | 38 | 7 | 22 | 62 | 32.2 (25.3-46.2) | NA |
| Current age (years) | 48 | 13 | 22 | Sudden cardiac death 2 years after diagnosis | NA | NA |
| NYHA functional class | IV | II | I | III | IV (50%) III (37.5%) I (12.5%) | NA |
| MPAP (mmHg) PAWP (mmHg) PVR (Wood units) DLCO (%) CO (L/min) | 45 15 5.2 120 5.8 | 42 13 10.6 NA NA | 37 11 4.93 90 5.7 | 41 3 16.5 23 NA | 61.5 (48.0-68.2) 8 (7.5-8.0) NA 81 (78.2-91.0) 4.7 (4.3-5.3) | NA NA NA NA NA |
| 6MWT (meters) | 480 | 516 | NA | 310 | NA | NA |
| FVC FEV1 FEV1/FVC (%) | 2.39 2.20 92 | NA NA NA | NA NA NA | NA NA NA | NA NA NA | NA NA NA |
| Last known PAH therapies | Double initial therapy including intravenous epoprostenol and sildenafil | Initial double oral therapy with iPDE5 and ERA | NA | Double sequential therapy with intravenous epoprostenol and ERA | NA | NA |
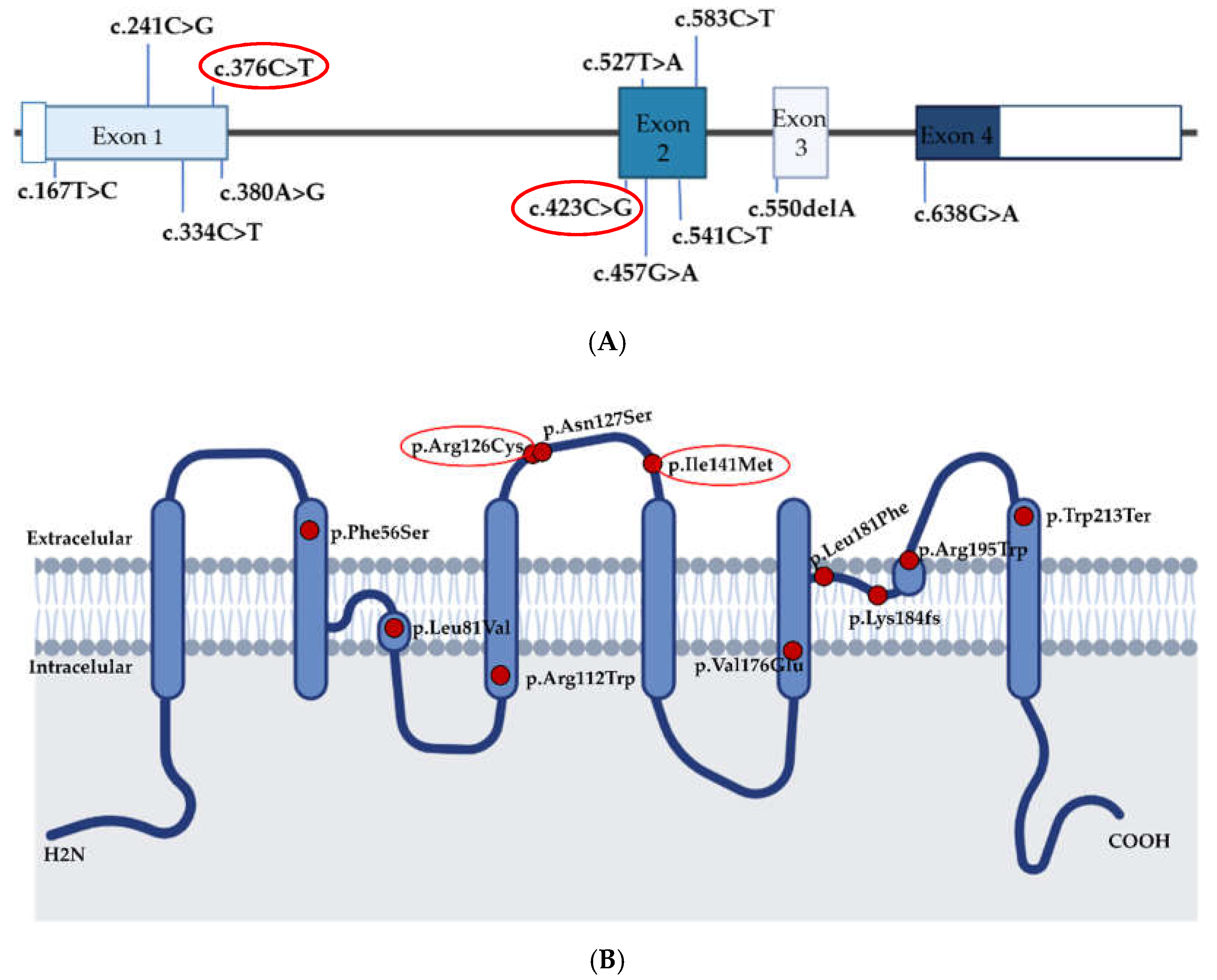
| Chr. Coordinate | PAH Patients with Variant | cDNA Position 1 | Protein Position | Variant Effect | Population Frequency 2 | DbSNFP 3 + CADD | ACMG 5 | Reference |
|---|---|---|---|---|---|---|---|---|
| Chr7:30951691 | 1 | c.167T > C | p.Phe56Ser | Missense | Absent | 16/1 + 28 | NA | [13] |
| Chr7:30961786 | 2 | c.241C > G | p.Leu81Val | Missense | Absent | 6/6 + 10 | NA | [13] |
| Chr7:30962212 | 1 | c.334C > T | p.Arg112Trp | Missense | Absent | 16/1 + 35 | NA | [13] |
| Chr7:30951900 | 5 | c.376C > T | p.Arg126Cys | Missense | Absent | 10/6 + 33 | VUS 6 | [10,13] This study |
| Chr7:30951904 | 1 | c.380A > G | p.Asn127Ser | Missense | 0.00000834 | 14/3 + 25 | NA | [13] |
| Chr7:30961719 | 1 | c.423C > G | p.Ile141Met | Missense | Absent | 6/10 + 22 | VUS | This study |
| Chr7:30961753 | 1 | c.457G > A | p.Val153Met | Missense | Absent | 15/1 + 27 | NA | [13] |
| Chr7:30961823 | 2 | c.527T>A | p.Val176Glu | Missense | Absent | 17/0 + 33 | NA | [10] |
| Chr7:30961837 | 1 | c.541C > T | p.Leu181Phe | Missense | Absent | 17/0 + 29 | NA | [10] |
| Chr7:30963232 | 1 | c.550delA | p.Lys184fsTer18 | frameshift | Absent | NA 4 | NA | [13] |
| Chr7:30962212 | 4 | c.583C > T | p.Arg195Trp | Missense | 0.0000319 | 18/0 + 35 | NA | [10] |
| Chr7:30963072 | 1 | c.638G > A | p.Trp213Ter | Nonsense | Absent | 8/1 + 43 | NA | [10] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallego-Zazo, N.; Cruz-Utrilla, A.; del Cerro, M.J.; Ochoa Parra, N.; Blanco, J.N.; Arias, P.; Lapunzina, P.; Escribano-Subias, P.; Tenorio-Castaño, J. Description of Two New Cases of AQP1 Related Pulmonary Arterial Hypertension and Review of the Literature. Genes 2022, 13, 927. https://doi.org/10.3390/genes13050927
Gallego-Zazo N, Cruz-Utrilla A, del Cerro MJ, Ochoa Parra N, Blanco JN, Arias P, Lapunzina P, Escribano-Subias P, Tenorio-Castaño J. Description of Two New Cases of AQP1 Related Pulmonary Arterial Hypertension and Review of the Literature. Genes. 2022; 13(5):927. https://doi.org/10.3390/genes13050927
Chicago/Turabian StyleGallego-Zazo, Natalia, Alejandro Cruz-Utrilla, María Jesús del Cerro, Nuria Ochoa Parra, Julián Nevado Blanco, Pedro Arias, Pablo Lapunzina, Pilar Escribano-Subias, and Jair Tenorio-Castaño. 2022. "Description of Two New Cases of AQP1 Related Pulmonary Arterial Hypertension and Review of the Literature" Genes 13, no. 5: 927. https://doi.org/10.3390/genes13050927