
A Novel Cuproptosis-Related Prognostic Gene Signature and Validation of Differential Expression in Clear Cell Renal Cell Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Multiomics Data Source and Preprocessing
2.2. Differential Expression Analysis and Validation
2.3. Gene Network and Enrichment Analysis of CRGs
2.4. Construct Prognostic Signature of Cuproptosis-Related Genes with Penalized Regression
2.5. Analysis of Correlation with Immune Infiltration
2.6. Statistical Analysis
3. Results
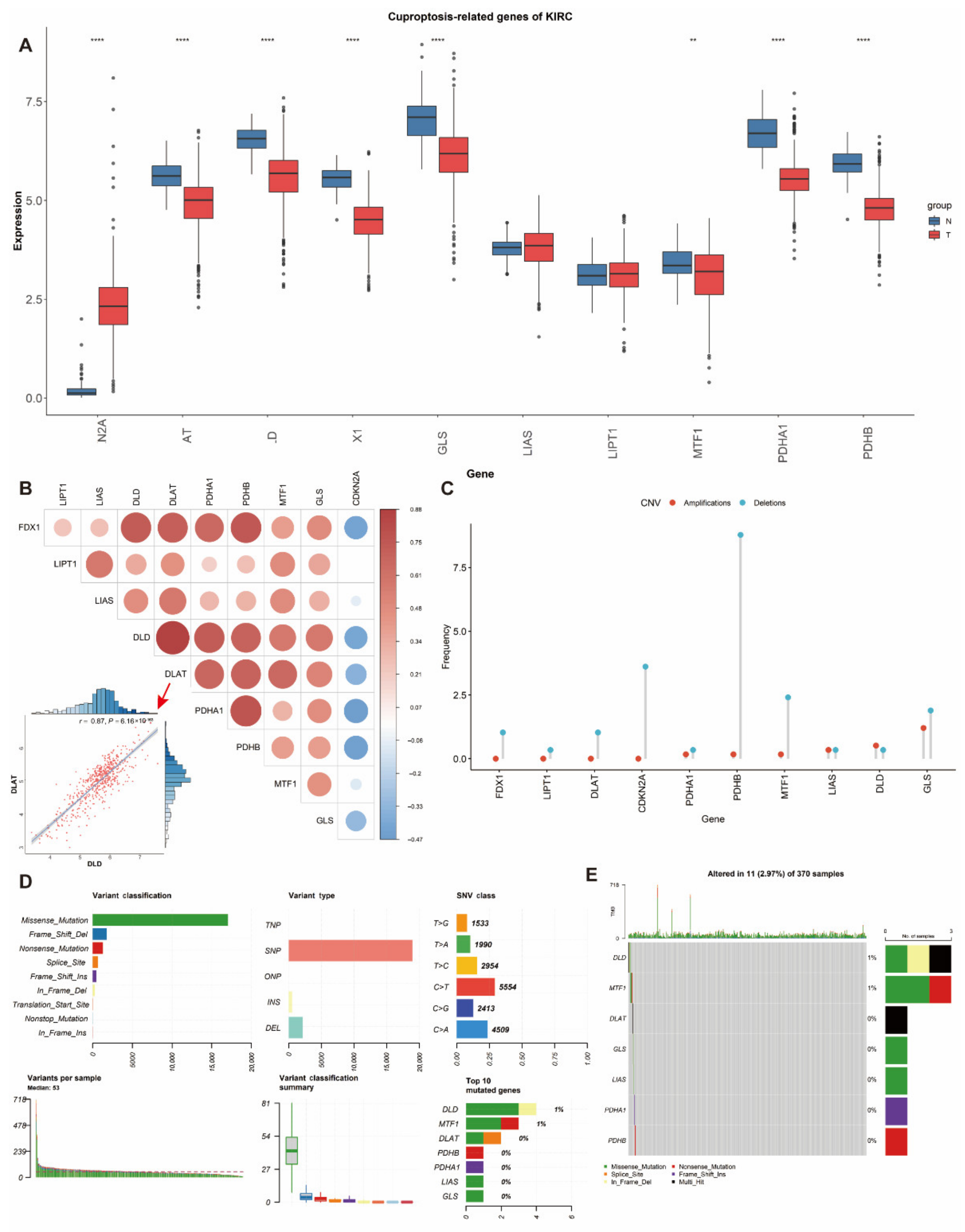
3.1. Differential Expression and Genetic Alterations of Cuproptosis-Related Genes in ccRCC
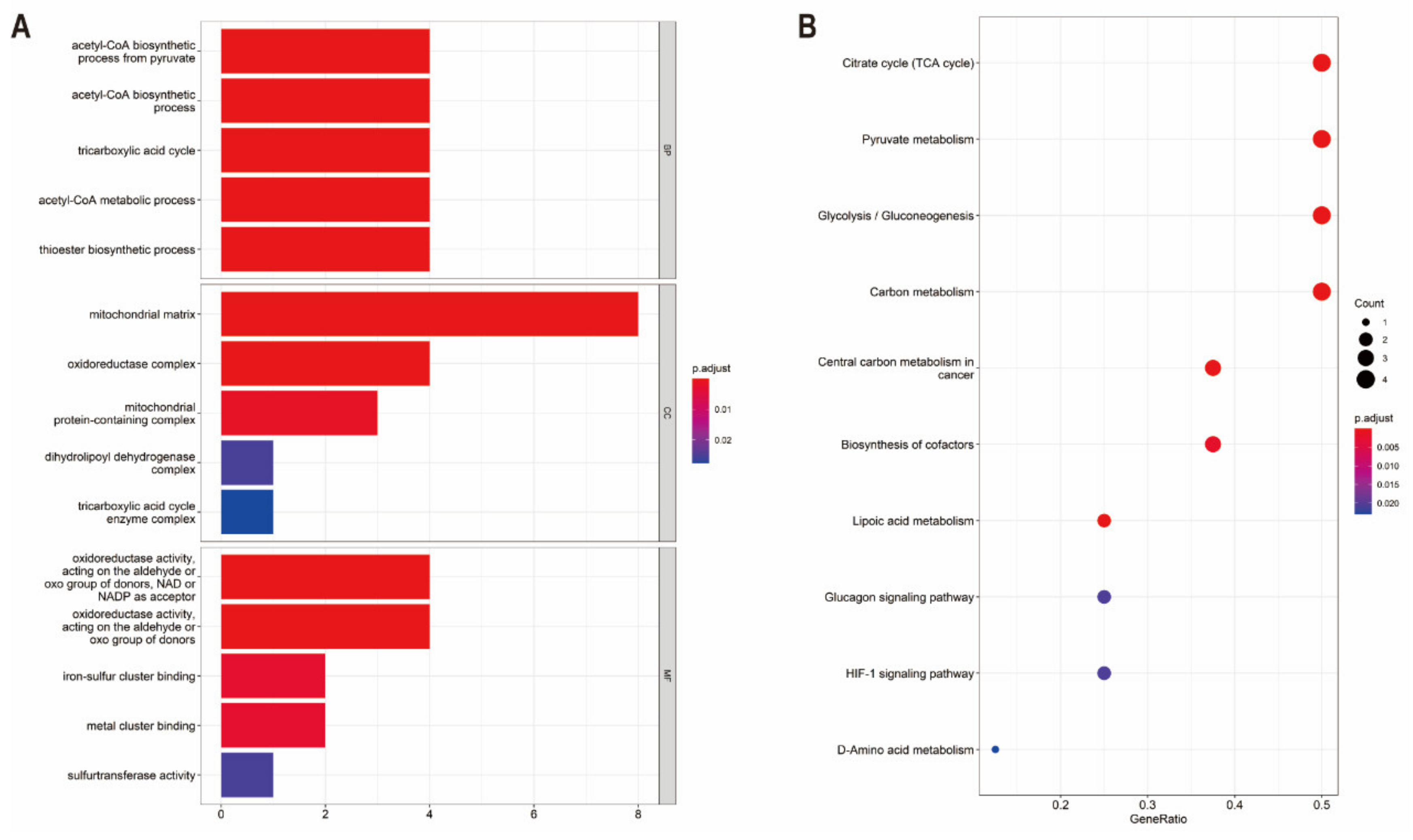
3.2. Functional Enrichment and Protein–Protein Interaction Analysis of CRGs
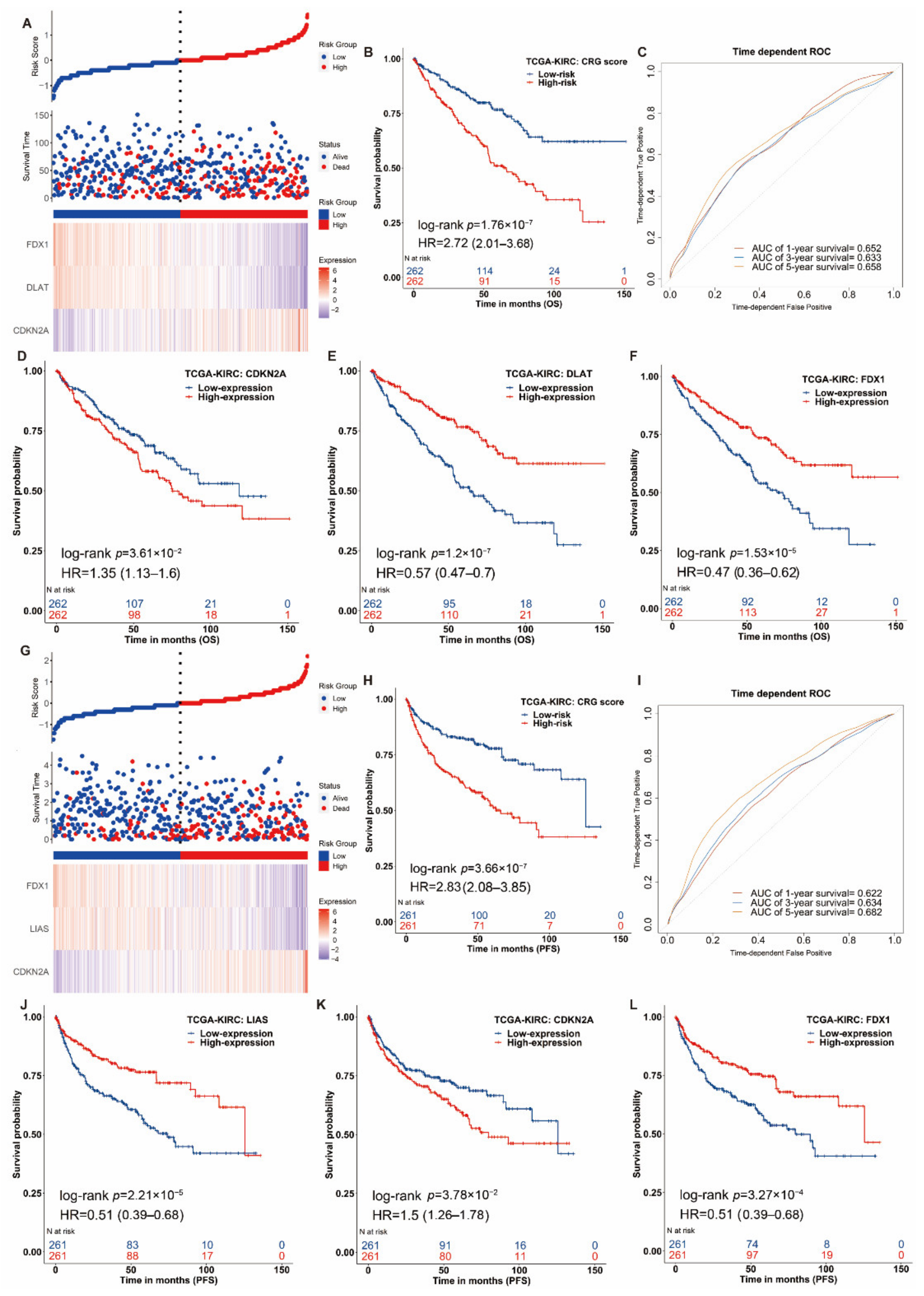
3.3. Construction of the Prognostic Signature of Cuproptosis-Related Genes in ccRCC
3.4. Nomogram Development and Validation for ccRCC
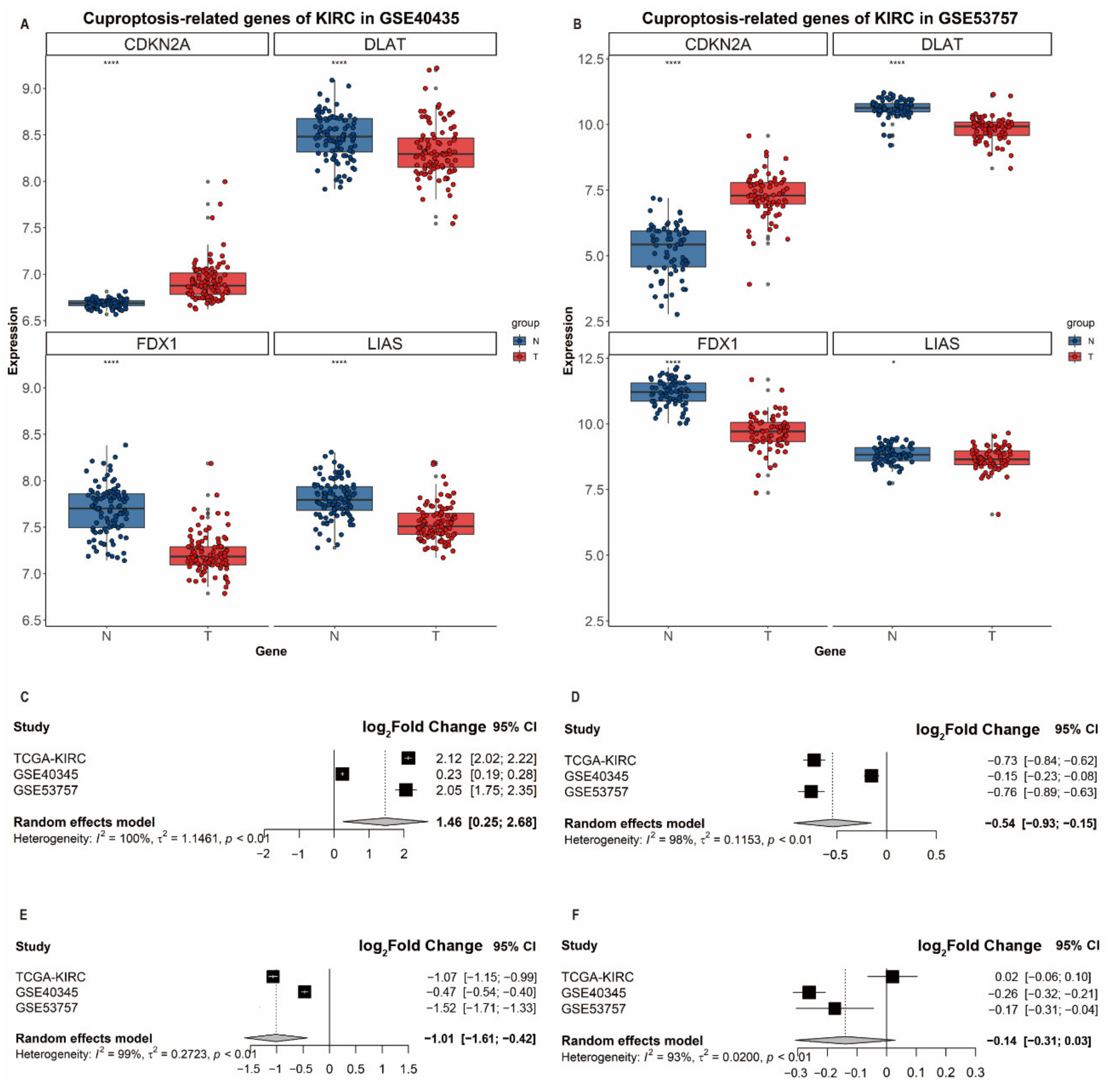
3.5. Validation of Differential Expression of CDKN2A, DLAT, FDX1 and LIAS in ccRCC
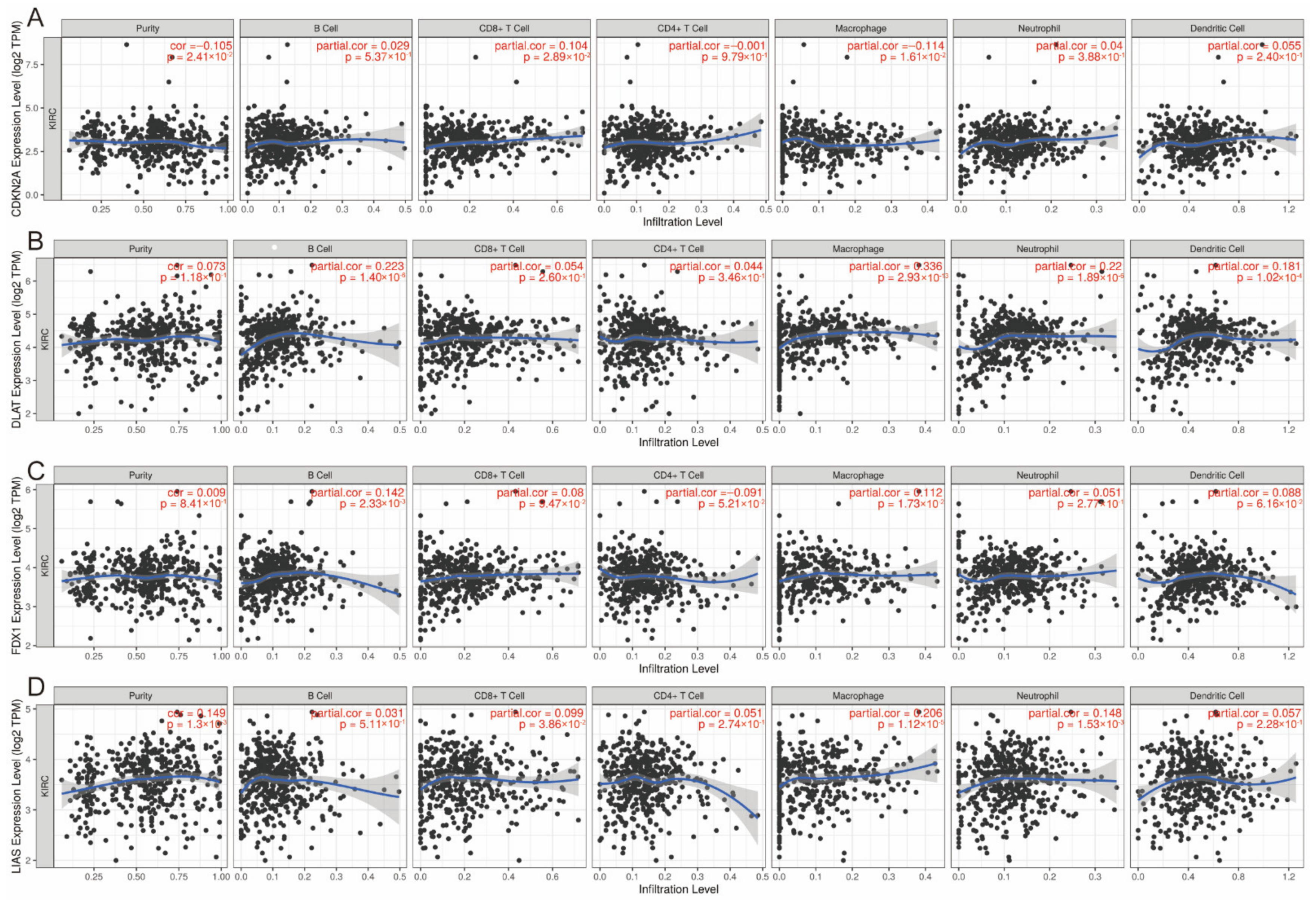
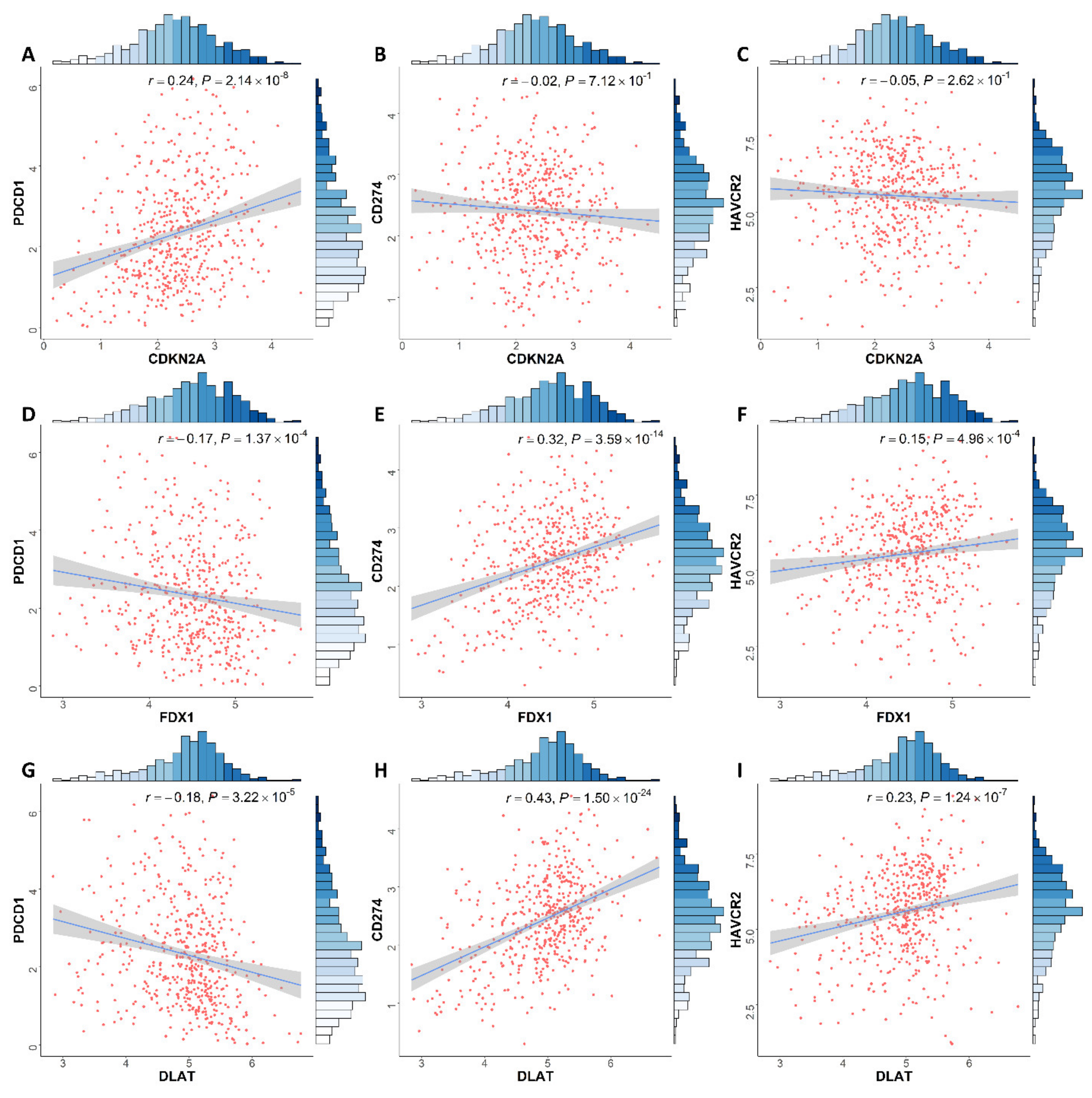
3.6. Correlation between Expression of CRGs and Immune Infiltration Levels in ccRCC
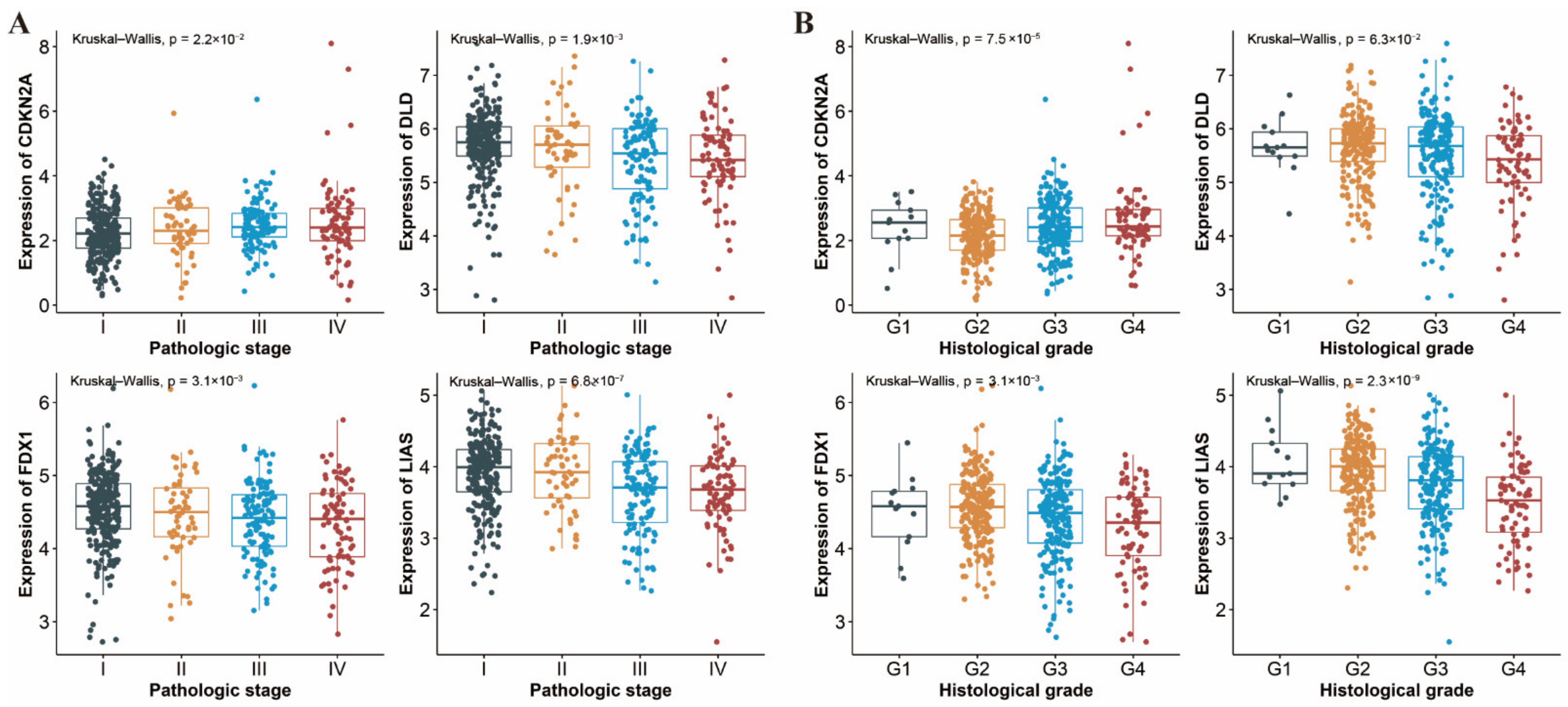
3.7. Differential Expression of CRGs in Different Pathologic Stages and Histological Grades of ccRCC
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Gao, J.; Rathmell, W.K. Renal cell carcinoma. BMJ 2014, 349, g4797. [Google Scholar] [CrossRef] [PubMed]
- Lalani, A.-K.A.; McGregor, B.A.; Albiges, L.; Choueiri, T.K.; Motzer, R.; Powles, T.; Wood, C.; Bex, A. Systemic Treatment of Metastatic Clear Cell Renal Cell Carcinoma in 2018: Current Paradigms, Use of Immunotherapy, and Future Directions. Eur. Urol. 2019, 75, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Walker, C.L.; Rathmell, W.K. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat. Rev. Nephrol. 2021, 17, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Blockhuys, S.; Celauro, E.; Hildesjö, C.; Feizi, A.; Stål, O.; Fierro-González, J.C.; Wittung-Stafshede, P. Defining the human copper proteome and analysis of its expression variation in cancers. Metallomics 2017, 9, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Ge, E.J.; Bush, A.I.; Casini, A.; Cobine, P.A.; Cross, J.R.; DeNicola, G.M.; Dou, Q.P.; Franz, K.J.; Gohil, V.M.; Gupta, S.; et al. Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. Cancer 2022, 22, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Andreux, P.; Poitry-Yamate, C.; Auwerx, J.; Hanahan, D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 19507–19512. [Google Scholar] [CrossRef] [Green Version]
- Babak, M.V.; Ahn, D. Modulation of Intracellular Copper Levels as the Mechanism of Action of Anticancer Copper Complexes: Clinical Relevance. Biomedicines 2021, 9, 852. [Google Scholar] [CrossRef]
- Brady, D.C.; Crowe, M.S.; Greenberg, D.N.; Counter, C.M. Copper Chelation Inhibits BRAF-Driven Melanomagenesis and Counters Resistance to BRAF and MEK1/2 Inhibitors. Cancer Res. 2017, 77, 6240–6252. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.I.; Gu, X.; Kiefer, R.M.; Ralle, M.; Gade, T.P.; Brady, D.C. Altered copper homeostasis underlies sensitivity of hepatocellular carcinoma to copper chelation. Metallomics 2020, 12, 1995–2008. [Google Scholar] [CrossRef]
- Chen, D.; Cui, Q.C.; Yang, H.; Dou, Q.P. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006, 66, 10425–10433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Day, S.J.; Eggermont, A.M.M.; Chiarion-Sileni, V.; Kefford, R.; Grob, J.J.; Mortier, L.; Robert, C.; Schachter, J.; Testori, A.; Mackiewicz, J.; et al. Final results of phase III SYMMETRY study: Randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Linehan, W.M.; Schmidt, L.S.; Crooks, D.R.; Wei, D.; Srinivasan, R.; Lang, M.; Ricketts, C.J. The Metabolic Basis of Kidney Cancer. Cancer Discov. 2019, 9, 1006–1021. [Google Scholar] [CrossRef] [Green Version]
- Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [CrossRef] [Green Version]
- Wettersten, H.I.; Aboud, O.A.; Lara, P.N.; Weiss, R.H. Metabolic reprogramming in clear cell renal cell carcinoma. Nat. Rev. Nephrol. 2017, 13, 410–419. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repecka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wozniak, M.B.; Le Calvez-Kelm, F.; Abedi-Ardekani, B.; Byrnes, G.; Durand, G.; Carreira, C.; Michelon, J.; Janout, V.; Holcatova, I.; Foretova, L.; et al. Integrative genome-wide gene expression profiling of clear cell renal cell carcinoma in Czech Republic and in the United States. PLoS ONE 2013, 8, e57886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Roemeling, C.A.; Radisky, D.C.; Marlow, L.A.; Cooper, S.J.; Grebe, S.K.; Anastasiadis, P.Z.; Tun, H.W.; Copland, J.A. Neuronal pentraxin 2 supports clear cell renal cell carcinoma by activating the AMPA-selective glutamate receptor-4. Cancer Res. 2014, 74, 4796–4810. [Google Scholar] [CrossRef] [Green Version]
- Franz, M.; Rodriguez, H.; Lopes, C.; Zuberi, K.; Montojo, J.; Bader, G.D.; Morris, Q. GeneMANIA update 2018. Nucleic Acids Res. 2018, 46, W60–W64. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, C.; Dong, X.; Shen, S.; Lai, L.; He, J.; You, D.; Lin, L.; Zhu, Y.; Huang, H.; et al. Independent Validation of Early-Stage Non-Small Cell Lung Cancer Prognostic Scores Incorporating Epigenetic and Transcriptional Biomarkers with Gene-Gene Interactions and Main Effects. Chest 2020, 158, 808–819. [Google Scholar] [CrossRef]
- Saldana, D.F.; Feng, Y. SIS: An R Package for Sure Independence Screening in Ultrahigh-Dimensional Statistical Models. J. Stat. Softw. 2018, 83, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Heagerty, P.J.; Zheng, Y. Survival model predictive accuracy and ROC curves. Biometrics 2005, 61, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhang, J.; Tu, H.; Liang, D.; Chang, D.W.; Ye, Y.; Wu, X. Soluble immune checkpoint-related proteins as predictors of tumor recurrence, survival, and T cell phenotypes in clear cell renal cell carcinoma patients. J. ImmunoTher. Cancer 2019, 7, 334. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, J.D. Nonparametric Statistical Methods. Technometrics 1974, 16, 477–478. [Google Scholar] [CrossRef]
- Haynes, W. Benjamini-Hochberg Method. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.-H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013; p. 78. [Google Scholar]
- Dörsam, B.; Fahrer, J. The disulfide compound α-lipoic acid and its derivatives: A novel class of anticancer agents targeting mitochondria. Cancer Lett. 2016, 371, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H.; Klein, A.P.; Hruban, R.H.; Roberts, N.J. The Role of Inherited Pathogenic CDKN2A Variants in Susceptibility to Pancreatic Cancer. Pancreas 2021, 50, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Śledzińska, P.; Bebyn, M.G.; Furtak, J.; Kowalewski, J.; Lewandowska, M.A. Prognostic and Predictive Biomarkers in Glio-mas. Int. J. Mol. Sci. 2021, 22, 10373. [Google Scholar] [CrossRef]
- Bui, T.O.; Dao, V.T.; Nguyen, V.T.; Feugeas, J.-P.; Pamoukdjian, F.; Bousquet, G. Genomics of Clear-cell Renal Cell Carci-noma: A Systematic Review and Meta-analysis. Eur. Urol. 2022, 81, 349–361. [Google Scholar] [CrossRef]
- Kahlson Martha, A.; Dixon Scott, J. Copper-induced cell death. Science 2022, 375, 1231–1232. [Google Scholar] [CrossRef]
- Whiteside, S.A.; Razvi, H.; Dave, S.; Reid, G.; Burton, J.P. The microbiome of the urinary tract—A role beyond infection. Nat. Rev. Urol. 2015, 12, 81–90. [Google Scholar] [CrossRef]
- Hyre, A.N.; Kavanagh, K.; Kock, N.D.; Donati, G.L.; Subashchandrabose, S. Copper Is a Host Effector Mobilized to Urine during Urinary Tract Infection to Impair Bacterial Colonization. Infect. Immun. 2017, 85, e01041-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.; Narayanamoorthy, S.; Gamre, S.; Majumdar, A.G.; Goswami, M.; Gami, U.; Cherian, S.; Subramanian, M. Hydroxychavicol, a key ingredient of Piper betle induces bacterial cell death by DNA damage and inhibition of cell division. Free Radic. Biol. Med. 2018, 120, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Nanni, V.; di Marco, G.; Sacchetti, G.; Canini, A.; Gismondi, A. Oregano Phytocomplex Induces Programmed Cell Death in Melanoma Lines via Mitochondria and DNA Damage. Foods 2020, 9, 1486. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Zhang, X.; Zangeneh, M.M.; Zhang, Y. Efficient biogenesis of Cu2O nanoparticles using extract of Camellia sinensis leaf: Evaluation of catalytic, cytotoxicity, antioxidant, and anti-human ovarian cancer properties. Bioorgan. Chem. 2021, 106, 104468. [Google Scholar] [CrossRef]
- Strasser, A.; Vaux, D.L. Cell Death in the Origin and Treatment of Cancer. Mol. Cell 2020, 78, 1045–1054. [Google Scholar] [CrossRef]
- Robinson, A.G.; Booth, C.M.; Eisenhauer, E.A. Progression-free survival as an end-point in solid tumours--perspectives from clinical trials and clinical practice. Eur. J. Cancer 2014, 50, 2303–2308. [Google Scholar] [CrossRef] [PubMed]
- Pasalic, D.; McGinnis, G.J.; Fuller, C.D.; Grossberg, A.J.; Verma, V.; Mainwaring, W.; Miller, A.B.; Lin, T.A.; Jethanandani, A.; Espinoza, A.F.; et al. Progression-free survival is a suboptimal predictor for overall survival among metastatic solid tumour clinical trials. Eur. J. Cancer 2020, 136, 176–185. [Google Scholar] [CrossRef]
- Fallowfield, L.J.; Fleissig, A. The value of progression-free survival to patients with advanced-stage cancer. Nat. Rev. Clin. Oncol. 2011, 9, 41–47. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bian, Z.; Fan, R.; Xie, L. A Novel Cuproptosis-Related Prognostic Gene Signature and Validation of Differential Expression in Clear Cell Renal Cell Carcinoma. Genes 2022, 13, 851. https://doi.org/10.3390/genes13050851
Bian Z, Fan R, Xie L. A Novel Cuproptosis-Related Prognostic Gene Signature and Validation of Differential Expression in Clear Cell Renal Cell Carcinoma. Genes. 2022; 13(5):851. https://doi.org/10.3390/genes13050851
Chicago/Turabian StyleBian, Zilong, Rong Fan, and Lingmin Xie. 2022. "A Novel Cuproptosis-Related Prognostic Gene Signature and Validation of Differential Expression in Clear Cell Renal Cell Carcinoma" Genes 13, no. 5: 851. https://doi.org/10.3390/genes13050851