
Activation of Esterase D by FPD5 Inhibits Growth of A549 Lung Cancer Cells via JAB1/p53 Pathway

Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Materials
2.2. Cell Culture
2.3. Western Blot Analysis
2.4. Co-Immunoprecipitation (Co-IP)
2.5. Nuclear and Cytoplasmic Extraction
2.6. Cell Staining for Immunofluorescence Microscopy
2.7. RNA Interference (RNAi)
2.8. Quantitative Real-Time PCR (qPCR)
2.9. Flow Cytometric Analysis of Cell Cycle Distribution
2.10. Quantitative Analysis and Statistical Analysis
3. Results
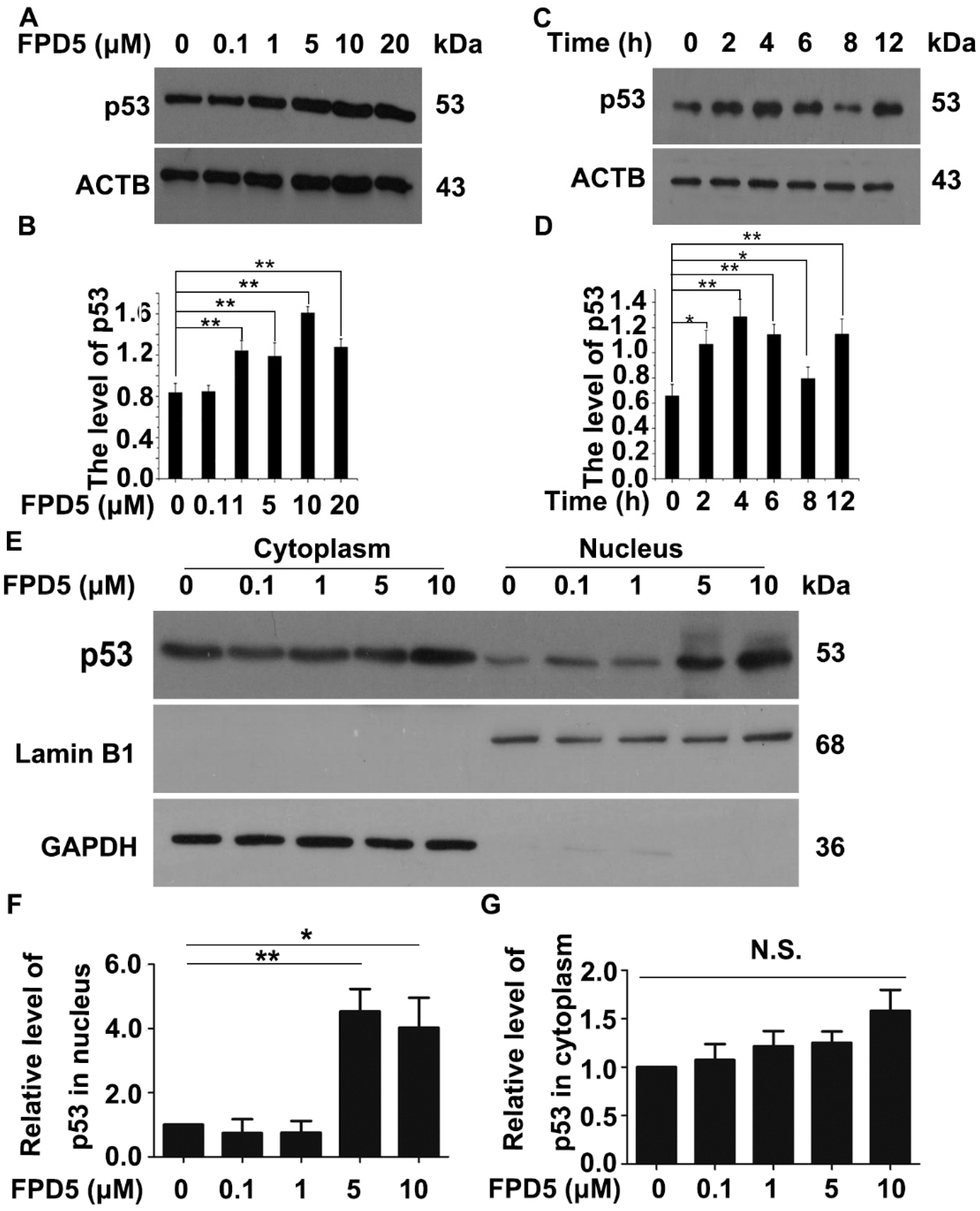
3.1. FPD5 Increased the Protein Level of p53 in the Nucleus of A549 Lung Cancer Cells
3.2. The Upregulation of p53 by FPD5 Was Associated with ESD
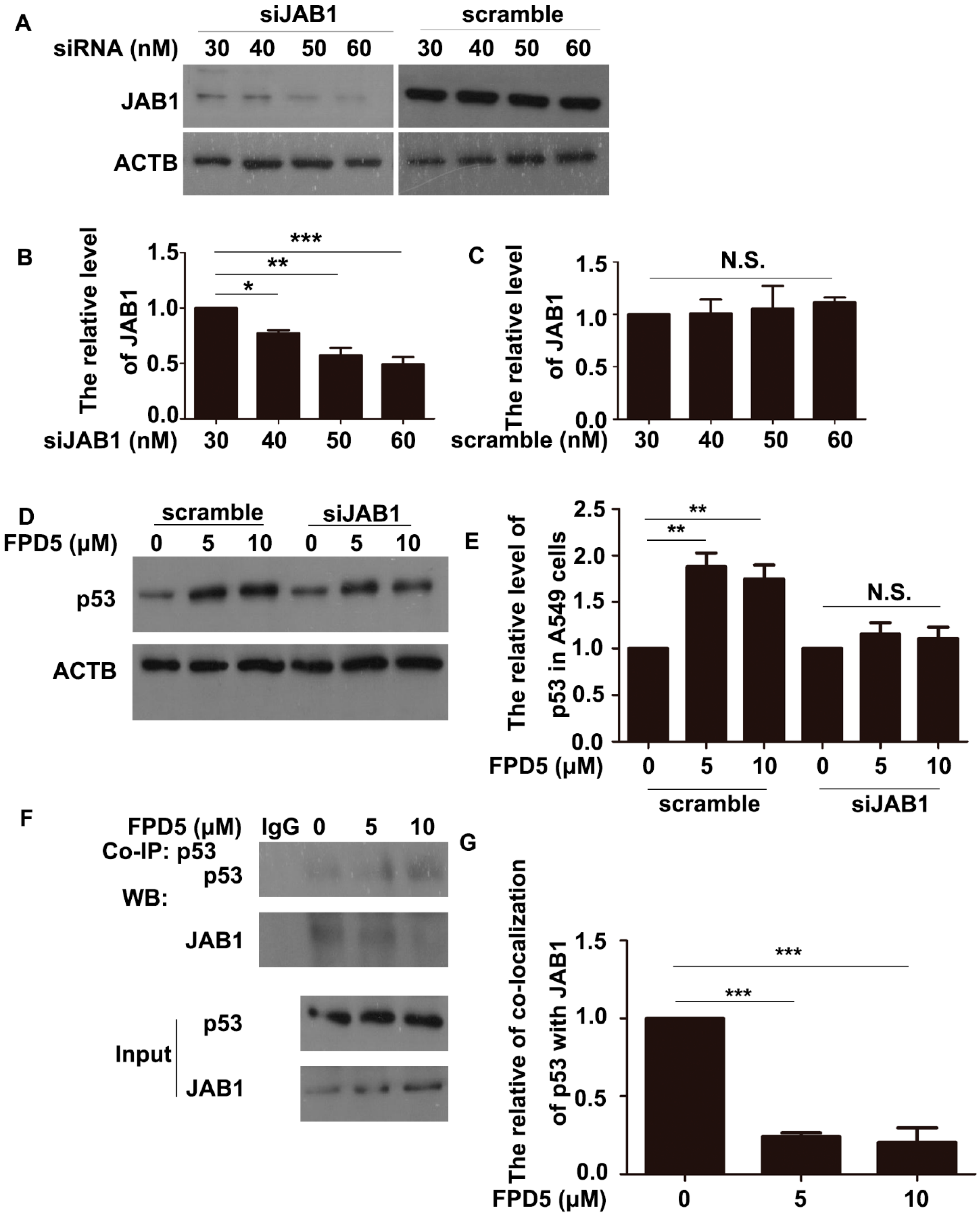
3.3. ESD Regulated p53 via JAB1 and Inhibited the Interaction between JAB1 and p53
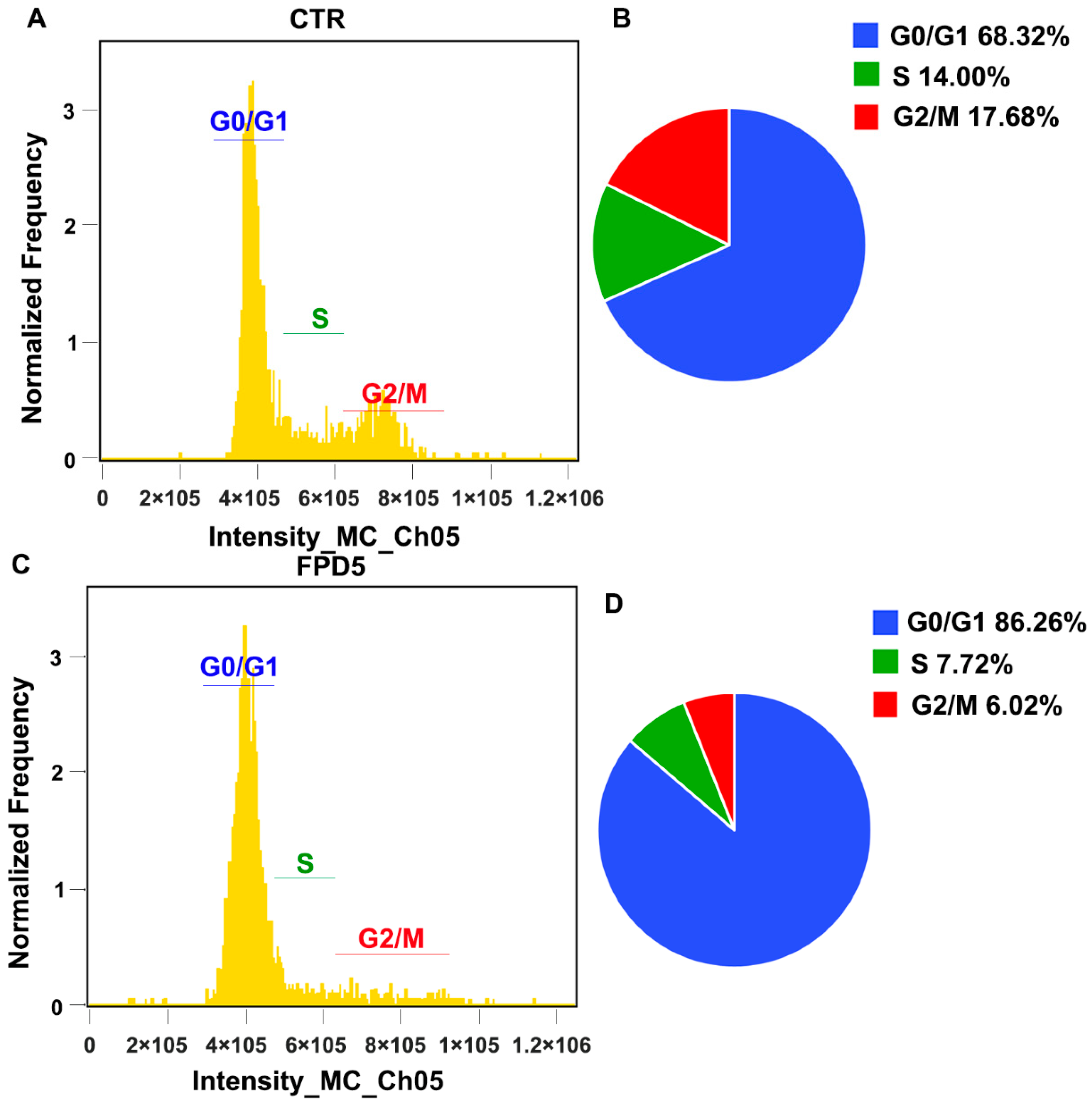
3.4. Activation of ESD Inhibited the Cell Cycle Progress of A549 Cells
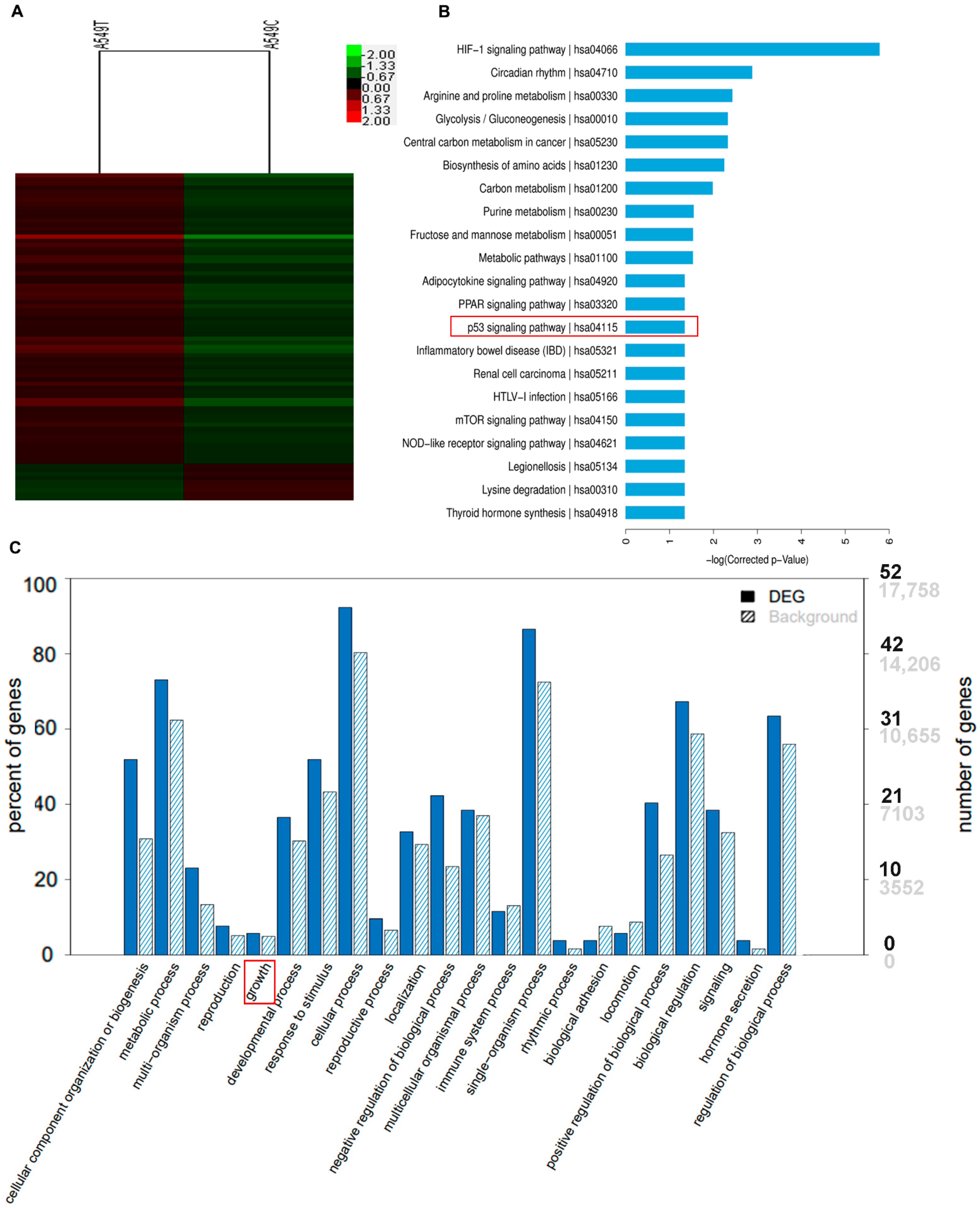
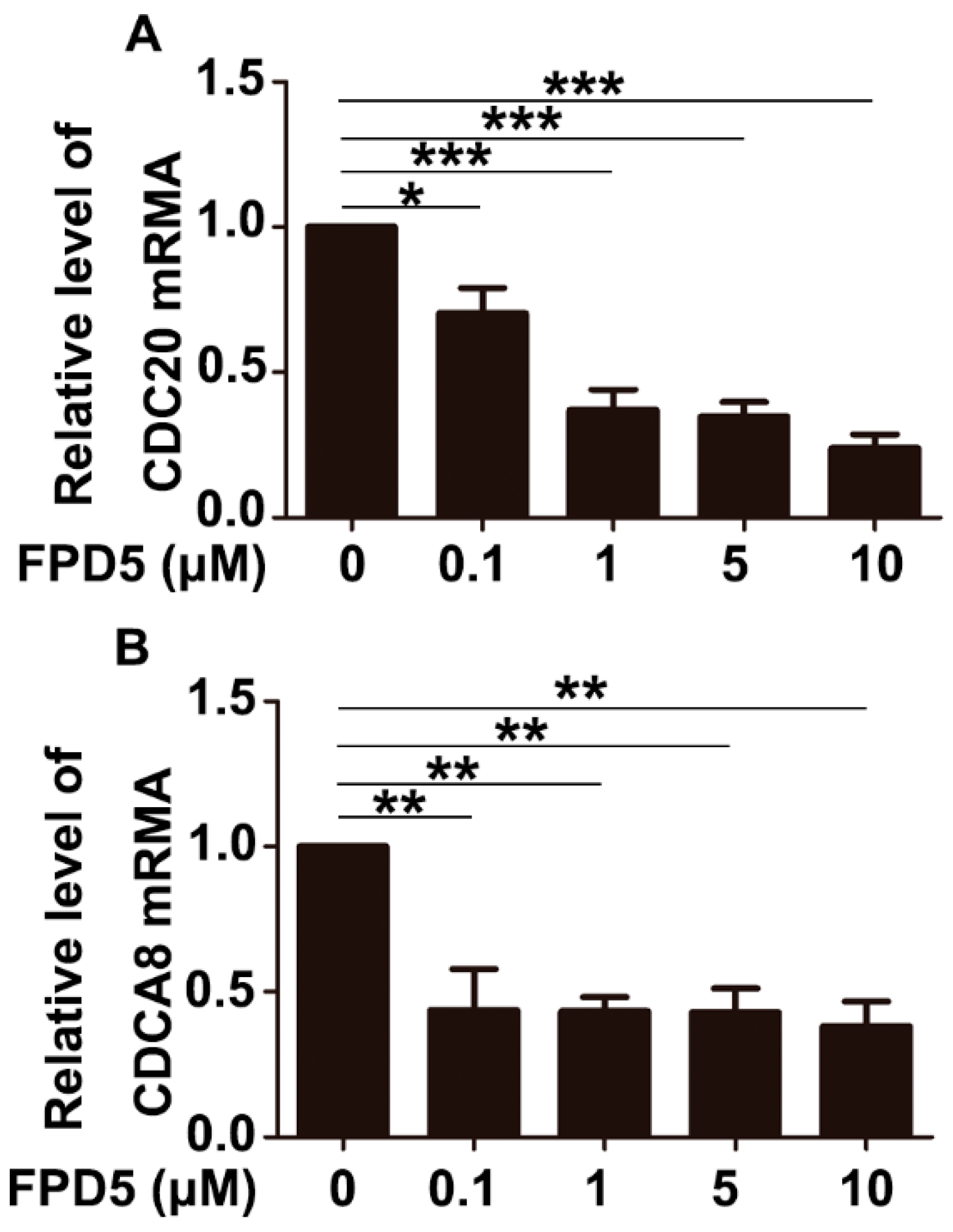
3.5. p53 Suppressed the Gene Expression of CDCA8 and CDC20
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Sparkes, R.S.; Spence, M.A.; Mohandas, T.; Shapiro, L.J.; Funderburk, S.J. Human gene mapping, genetic linkage, and clinical applications. Ann. Intern. Med. 1980, 93, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Cummins, I.; McAuley, K.; Fordham-Skelton, A.; Schwoerer, R.; Steel, P.G.; Davis, B.G.; Edwards, R. Unique regulation of the active site of the serine esterase S-formylglutathione hydrolase. J. Mol. Biol 2006, 359, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Eiberg, H.; Mohr, J. Identity of the polymorphisms for esterase D and S-formylglutathione hydrolase in red blood cells. Hum. Genet. 1986, 74, 174–175. [Google Scholar] [CrossRef]
- Apeshiotis, F.; Bender, K. Evidence that S-formylglutathione hydrolase and esterase D polymorphisms are identical. Hum. Genet. 1986, 74, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.M.; Morgan, C.; Gallie, B.; Phillips, R.A.; Squire, J. Re-evaluation of the sublocalization of esterase D and its relation to the retinoblastoma locus by in situ hybridization. Cytogenet. Cell Genet. 1987, 44, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Kit, S.; Hazen, M.; Qavi, H.; Dubbs, D.R.; Pathak, S. Integration site(s) of herpes simplex virus type 1 thymidine kinase gene and regional assignment of the gene for aminoacylase-1 in human chromosomes. Cytogenet. Cell Genet. 1980, 26, 93–103. [Google Scholar] [CrossRef]
- Aguirre, A.I.; Vicario, A.; Mazon, L.I.; de Pancorbo, M.M.; Estomba, A.; Lostao, C. Acid phosphatase, adenosine deaminase and esterase D polymorphisms in the Spanish Basque population. Hum. Hered. 1991, 41, 93–102. [Google Scholar] [CrossRef]
- Chuang, L.M.; Tai, T.Y.; Wang, T.R.; Chang, Y.C.; Chen, K.H.; Lin, R.S.; Lin, B.J. Esterase D and retinoblastoma gene loci are tightly linked to Wilson’s disease in Chinese pedigrees from Taiwan. Hum. Genet. 1991, 87, 465–468. [Google Scholar] [CrossRef]
- Wiedl, T.; Arni, S.; Roschitzki, B.; Grossmann, J.; Collaud, S.; Soltermann, A.; Hillinger, S.; Aebersold, R.; Weder, W. Activity-based proteomics: Identification of ABHD11 and ESD activities as potential biomarkers for human lung adenocarcinoma. J. Proteom. 2011, 74, 1884–1894. [Google Scholar] [CrossRef]
- Chen, X.; Lin, Z.; Su, L.; Cui, X.; Zhao, B.; Miao, J. Discovery of a fluorescigenic pyrazoline derivative targeting ubiquitin. Biochem. Biophys. Res. Commun. 2020, 528, 256–260. [Google Scholar] [CrossRef]
- Chen, X.; Yang, Y.; Su, L.; Cui, X.; Shao, J.; Liu, S.; Zhao, B.; Miao, J. Finding the mechanism of esterase D activation by a small molecule. Bioorg. Med. Chem. Lett. 2020, 30, 127150. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanapathipillai, M. Treating p53 Mutant Aggregation-Associated Cancer. Cancers 2018, 10, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W. The role of p53 gene family in reproduction. Cold Spring Harb. Perspect. Biol. 2009, 1, a001073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Wang, W.; El-Deiry, W.S. Current strategies to target p53 in cancer. Biochem. Pharmacol. 2010, 80, 724–730. [Google Scholar] [CrossRef]
- Meek, D.W. Regulation of the p53 response and its relationship to cancer. Biochem. J. 2015, 469, 325–346. [Google Scholar] [CrossRef]
- Xu, J.; Patel, N.H.; Gewirtz, D.A. Triangular Relationship between p53, Autophagy, and Chemotherapy Resistance. Int. J. Mol. Sci. 2020, 21, 8991. [Google Scholar] [CrossRef]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef] [Green Version]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Frohlich, L.F. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.W.; Oh, W.; Song, J. Jab1 as a mediator of nuclear export and cytoplasmic degradation of p53. Mol. Cells 2006, 22, 133–140. [Google Scholar] [PubMed]
- Chen, X.; Su, L.; Yang, Y.; Qv, J.; Wei, T.; Cui, X.; Shao, J.; Liu, S.; Xu, Z.; Zhao, B.; et al. A new activator of esterase D decreases blood cholesterol level through ESD/JAB1/ABCA1 pathway. J. Cell. Physiol. 2021, 236, 4750–4763. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, X.; Yao, W.; Cui, X.; Li, N.; Lin, Z.; Zhao, B.; Miao, J. Esterase D stabilizes FKBP25 to suppress mTORC1. Cell. Mol. Biol. Lett. 2021, 26, 50. [Google Scholar] [CrossRef] [PubMed]
- van der Heiden, C.; Geurtzen, A.F.; Brink, W.; de Klerk, J.B.; Beemer, F.A. Esterase D: Evaluation of a potential derived gene marker for hereditary retinoblastoma. Clin. Chim. Acta 1988, 173, 81–87. [Google Scholar] [CrossRef]
- Mueller, R.F. Retinoblastoma and esterase D. Lancet 1982, 2, 823. [Google Scholar] [CrossRef]
- Kazmierczak, M.; Luczak, M.; Lewandowski, K.; Handschuh, L.; Czyz, A.; Jarmuz, M.; Gniot, M.; Michalak, M.; Figlerowicz, M.; Komarnicki, M. Esterase D and γ 1 actin level might predict results of induction therapy in patients with acute myeloid leukemia without and with maturation. Med. Oncol. 2013, 30, 725. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhu, Z.; Cao, W.; Yang, F.; Zhang, X.; Li, D.; Zhang, K.; Li, P.; Mao, R.; Liu, X.; et al. Esterase D enhances type I interferon signal transduction to suppress foot-and-mouth disease virus replication. Mol. Immunol. 2016, 75, 112–121. [Google Scholar] [CrossRef]
- Kondo, I.; Nishigaki, I.; Yamakawa, K.; Hamaguchi, H. The esterase D polymorphism: Analysis of esterase D 7 by two-dimensional gel electrophoresis. Jinrui Idengaku Zasshi 1984, 29, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Legler, P.M.; Kumaran, D.; Swaminathan, S.; Studier, F.W.; Millard, C.B. Structural characterization and reversal of the natural organophosphate resistance of a D-type esterase, Saccharomyces cerevisiae S-formylglutathione hydrolase. Biochemistry 2008, 47, 9592–9601. [Google Scholar] [CrossRef]
- Lee, W.H.; Bookstein, R.; Wheatley, W.; Benedict, W.F.; Lee, E.Y. A null allele of esterase D is a marker for genetic events in retinoblastoma formation. Hum. Genet. 1987, 76, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Smith, A.D.; Ferraldeschi, R.; Al-Lazikani, B.; Workman, P.; de Bono, J.S. Drug discovery in advanced prostate cancer: Translating biology into therapy. Nat. Rev. Drug Discov. 2016, 15, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef]
- Oh, W.; Lee, E.W.; Sung, Y.H.; Yang, M.R.; Ghim, J.; Lee, H.W.; Song, J. Jab1 induces the cytoplasmic localization and degradation of p53 in coordination with Hdm2. J. Biol. Chem. 2006, 281, 17457–17465. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.C.; Chen, J.; Su, C.H.; Yang, H.Y.; Lee, M.H. Roles for CSN5 in control of p53/MDM2 activities. J. Cell. Biochem. 2008, 103, 1219–1230. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Title | Gene Symbol | Log2 Ratio |
|---|---|---|
| Downregulated gene | ||
| Cell division cycle associated 8 | CDCA8 | −1.50 |
| Cell division cycle 20 | CDC20 | −1.74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, W.; Yang, Y.; Chen, X.; Cui, X.; Zhou, B.; Zhao, B.; Lin, Z.; Miao, J. Activation of Esterase D by FPD5 Inhibits Growth of A549 Lung Cancer Cells via JAB1/p53 Pathway. Genes 2022, 13, 786. https://doi.org/10.3390/genes13050786
Yao W, Yang Y, Chen X, Cui X, Zhou B, Zhao B, Lin Z, Miao J. Activation of Esterase D by FPD5 Inhibits Growth of A549 Lung Cancer Cells via JAB1/p53 Pathway. Genes. 2022; 13(5):786. https://doi.org/10.3390/genes13050786
Chicago/Turabian StyleYao, Wen, Yuejun Yang, Xinpeng Chen, Xiaoling Cui, Bangzhao Zhou, Baoxiang Zhao, Zhaomin Lin, and Junying Miao. 2022. "Activation of Esterase D by FPD5 Inhibits Growth of A549 Lung Cancer Cells via JAB1/p53 Pathway" Genes 13, no. 5: 786. https://doi.org/10.3390/genes13050786