
A Novel de Novo Variant in 5′ UTR of the NIPBL Associated with Cornelia de Lange Syndrome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Clinical Evaluations
2.3. Cytogenetic and Molecular Studies
2.4. Quantitative Real-Time PCR
2.5. Plasmid Construction and Dual-Luciferase Assays
2.6. Statistical Analysis
3. Result
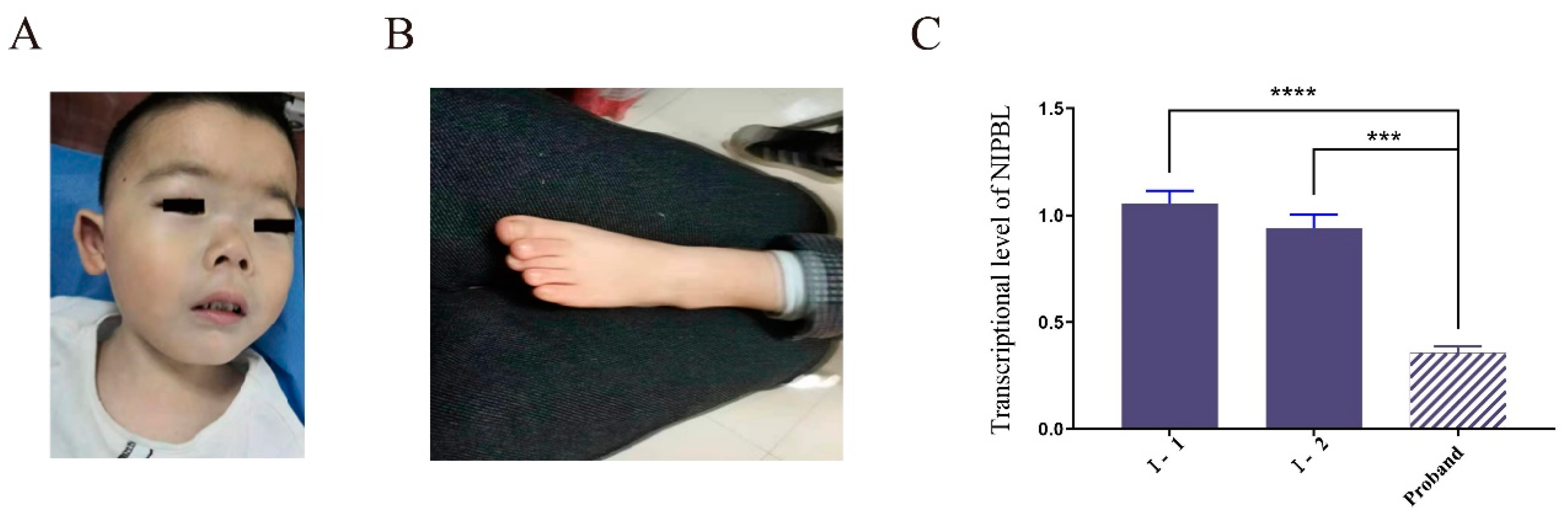
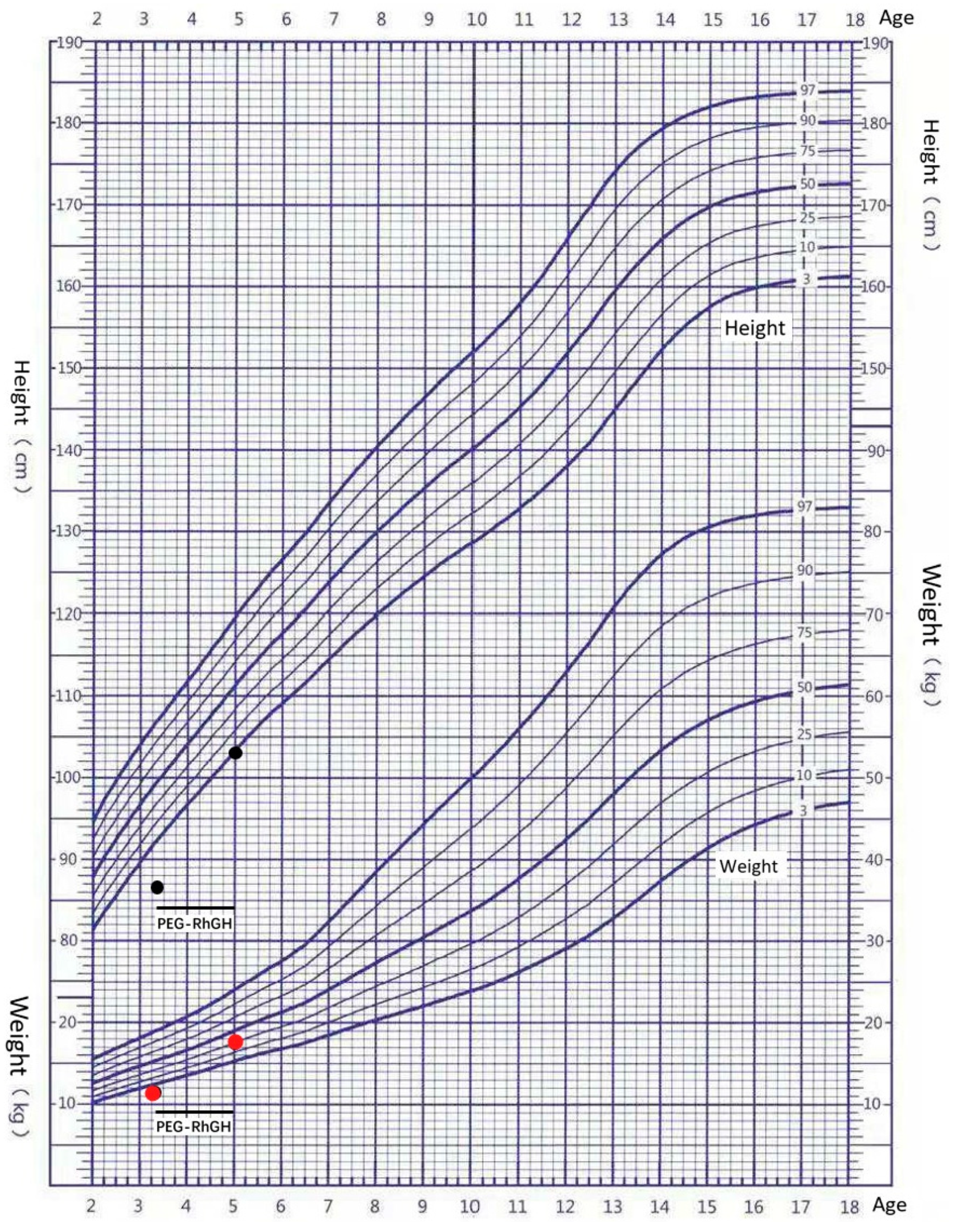
3.1. Clinical Features of the Patient
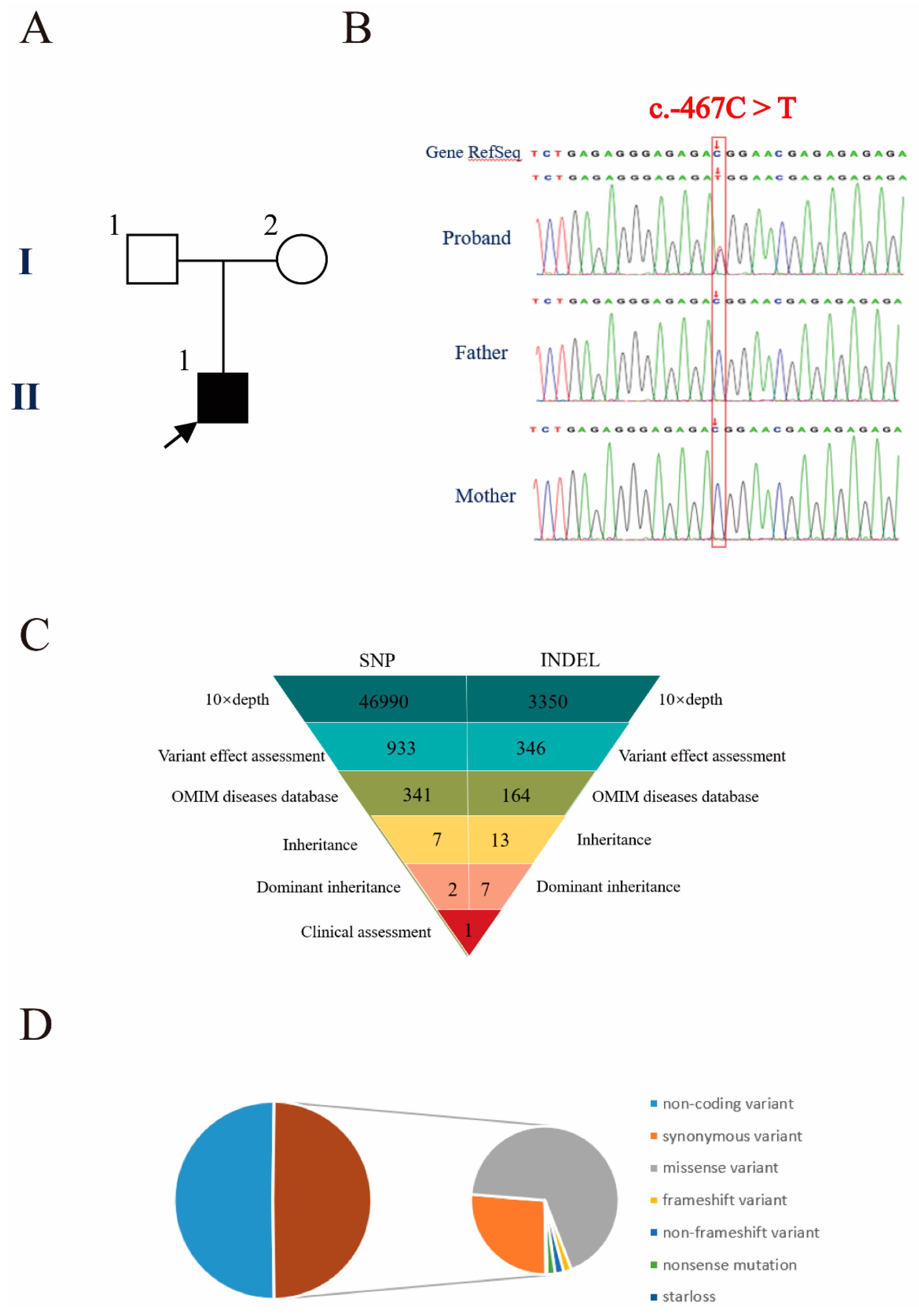
3.2. Genetic Diagnosis
3.3. Decreased Expression of the Variant Transcript
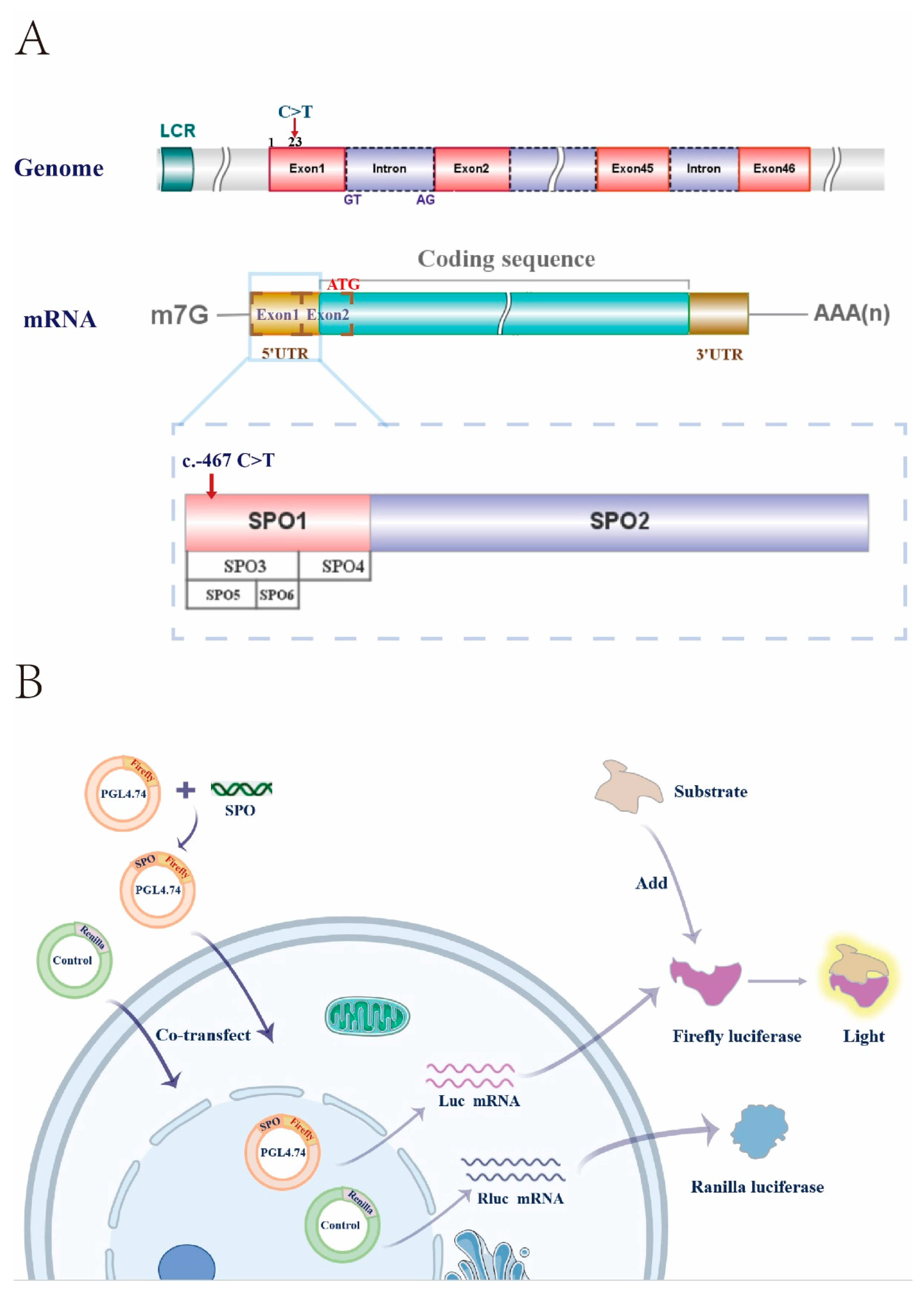
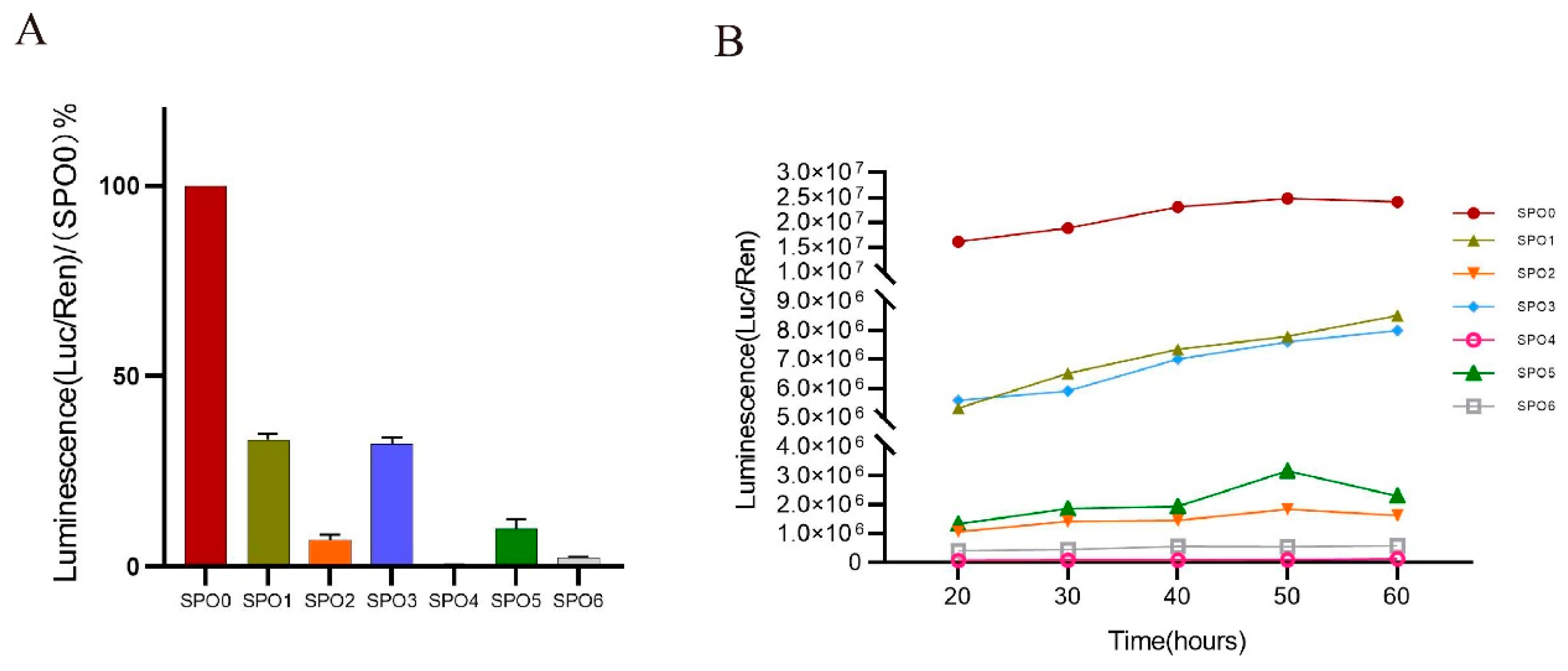
3.4. Truncations of the 5′ UTR Influence the Transcription Levels of the NIPBL
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Angelo, S.; Milena, M.; Antonella, L.; Massa, V. Cornelia de Lange Syndrome: From a Disease to a Broader Spectrum. Genes 2021, 12, 1075. [Google Scholar]
- Li, Q.; Chang, G.; Yin, L.; Li, J.; Huang, X.; Shen, Y.; Li, G.; Xu, Y.; Wang, J.; Wang, X. Clinical and molecular analysis in a cohort of Chinese children with Cornelia de Lange syndrome. Sci. Rep. 2020, 10, 21224. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, T.; Mekaru, K.; Nakada, M.; Nitta, H.; Masamoto, H.; Aoki, Y. A Case of Cornelia de Lange Syndrome: Difficulty in Prenatal Diagnosis. Case Rep. Obstet. Gynecol. 2019, 2019, 4530491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avagliano, L.; Bulfamante, G.P.; Massa, V. Cornelia de Lange syndrome: To diagnose or not to diagnose in utero? Birth Defects Res. 2017, 109, 771–777. [Google Scholar] [CrossRef]
- Li, S.; Miao, H.; Yang, H.; Wang, L.; Gong, F.; Chen, S.; Zhu, H.; Pan, H. A report of 2 cases of Cornelia de Lange syndrome (CdLS) and an analysis of clinical and genetic characteristics in a Chinese CdLS cohort. Mol. Genet. Genom. Med. 2020, 8, e1225. [Google Scholar]
- Liu, C.; Li, X.; Cui, J.; Dong, R.; Lv, Y.; Wang, D.; Zhang, H.; Li, X.; Li, Z.; Ma, J.; et al. Analysis of clinical and genetic characteristics in 10 Chinese individuals with Cornelia de Lange syndrome and literature review. Mol. Genet. Genom. Med. 2020, 8, e1471. [Google Scholar] [CrossRef]
- Ciosk, R.; Shirayama, M.; Shevchenko, A.; Tanaka, T.; Toth, A.; Shevchenko, A.; Nasmyth, K. Cohesin’s binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol. Cell 2000, 5, 243–254. [Google Scholar] [CrossRef]
- Zuin, J.; Dixon, J.R.; van der Reijden, M.I.; Ye, Z.; Kolovos, P.; Brouwer, R.W.W.; van de Corput, M.P.C.; van de Werken, H.J.G.; Knoch, T.A.; van Ijcken, W.F.J.; et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, 996–1001. [Google Scholar] [CrossRef] [Green Version]
- Sofueva, S.; Yaffe, E.; Chan, W.C.; Georgopoulou, D.; Rudan, M.V.; Mira-Bontenbal, H.; Pollard, S.; Schroth, G.P.; Tanay, A.; Hadjur, S. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J. 2013, 32, 3119–3129. [Google Scholar] [CrossRef] [Green Version]
- Seitan, V.C.; Faure, A.J.; Zhan, Y.; McCord, R.P.; Lajoie, B.R.; Ing-Simmons, E.; Lenhard, B.; Giorgetti, L.; Heard, E.; Fisher, A.G.; et al. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. 2013, 23, 2066–2077. [Google Scholar] [CrossRef] [Green Version]
- Dorsett, D. Cohesin: Genomic insights into controlling gene transcription and development. Curr. Opin. Genet. Dev. 2011, 21, 199–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muto, A.; Calof, A.L.; Lander, A.D.; Schilling, T.F. Multifactorial origins of heart and gut defects in nipbl-deficient zebrafish, a model of Cornelia de Lange Syndrome. PLoS Biol. 2011, 9, e1001181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsett, D.; Ström, L. The ancient and evolving roles of cohesin in gene expression and DNA repair. Curr. Biol. CB 2012, 22, R240–R250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garfinkle, E.A.R.; Gruber, T.A. A tale of two genes: A new connection between NIPBL and NPM1 in acute myeloid leukemia. Haematologica 2019, 104, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Mazzola, M.; Deflorian, G.; Pezzotta, A.; Ferrari, L.; Fazio, G.; Bresciani, E.; Saitta, C.; Ferrari, L.; Fumagalli, M.; Parma, M.; et al. NIPBL: A new player in myeloid cell differentiation. Haematologica 2019, 104, 1332–1341. [Google Scholar] [CrossRef] [Green Version]
- Muto, A.; Ikeda, S.; Lopez-Burks, M.E.; Kikuchi, Y.; Calof, A.L.; Lander, A.D.; Schilling, T.F. Nipbl and mediator cooperatively regulate gene expression to control limb development. PLoS Genet. 2014, 10, e1004671. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.G.; Laval, S.; Chen, F.; Rock, M.J.; Strachan, T.; Peters, H. Neural crest cell-specific inactivation of Nipbl or Mau2 during mouse development results in a late onset of craniofacial defects. Genesis 2014, 52, 687–694. [Google Scholar] [CrossRef]
- Borck, G.; Zarhrate, M.; Cluzeau, C.; Bal, E.; Bonnefont, J.-P.; Munnich, A.; Cormier-Daire, V.; Colleaux, L. Father-to-daughter transmission of Cornelia de Lange syndrome caused by a mutation in the 5′ untranslated region of the NIPBL Gene. Hum. Mutat. 2006, 27, 731–735. [Google Scholar] [CrossRef]
- Krawczynska, N.; Wierzba, J.; Wasag, B. Genetic Mosaicism in a Group of Patients With Cornelia de Lange Syndrome. Front. Pediatrics 2019, 7, 203. [Google Scholar] [CrossRef]
- Selicorni, A.; Russo, S.; Gervasini, C.; Russo, S.; Gervasini, C.; Castronovo, P.; Milani, D.; Cavalleri, F.; Bentivegna, A.; Masciadri, M.; et al. Clinical score of 62 Italian patients with Cornelia de Lange syndrome and correlations with the presence and type of NIPBL mutation. Clin. Genet. 2007, 72, 98–108. [Google Scholar] [CrossRef]
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.-M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange syndrome: First international consensus statement. Nat. Rev. Genet. 2018, 19, 649–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, M.; Poke, G.; Ferry, Q.; Williamson, K.; Aldridge, R.; Meynert, A.M.; Bengani, H.; Chan, C.Y.; Kayserili, H.; Avci, Ş.; et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J. Med. Genet. 2014, 51, 659–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huisman, S.; Mulder, P.A.; Redeker, E.; Bader, I.; Bisgaard, A.-M.; Brooks, A.; Cereda, A.; Cinca, C.; Clark, D.; Cormier-Daire, V.; et al. Phenotypes and genotypes in individuals with SMC1A variants. Am. J. Med. Genet. Part A 2017, 173, 2108–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoumans, J.; Wincent, J.; Barbaro, M.; Djureinovic, T.; Maguire, P.; Forsberg, L.; Staaf, J.; Thuresson, A.C.; Borg, Å.; Nordgren, A.; et al. Comprehensive mutational analysis of a cohort of Swedish Cornelia de Lange syndrome patients. Eur. J. Hum. Genet. EJHG 2007, 15, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.; Arora, S.; Schuster, S.L.; Corey, L.; Fitzgibbon, M.; Wladyka, C.L.; Wu, X.; Coleman, I.M.; Delrow, J.J.; Corey, E.; et al. Multiplexed functional genomic analysis of 5′ untranslated region mutations across the spectrum of prostate cancer. Nat. Commun. 2021, 12, 4217. [Google Scholar] [CrossRef]
- Schultz, I.C.; Bertoni, A.P.S.; Wink, M.R. Mesenchymal Stem Cell-Derived Extracellular Vesicles Carrying miRNA as a Potential Multi Target Therapy to COVID-19: An In Silico Analysis. Stem Cell Rev. Rep. 2021, 17, 341–356. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Chen, Q.; Yuan, K.; Zhu, J.; Fang, Y.; Yan, Q.; Wang, C. A Novel de Novo Variant in 5′ UTR of the NIPBL Associated with Cornelia de Lange Syndrome. Genes 2022, 13, 740. https://doi.org/10.3390/genes13050740
Chen Y, Chen Q, Yuan K, Zhu J, Fang Y, Yan Q, Wang C. A Novel de Novo Variant in 5′ UTR of the NIPBL Associated with Cornelia de Lange Syndrome. Genes. 2022; 13(5):740. https://doi.org/10.3390/genes13050740
Chicago/Turabian StyleChen, Yonghua, Qingqing Chen, Ke Yuan, Jianfang Zhu, Yanlan Fang, Qingfeng Yan, and Chunlin Wang. 2022. "A Novel de Novo Variant in 5′ UTR of the NIPBL Associated with Cornelia de Lange Syndrome" Genes 13, no. 5: 740. https://doi.org/10.3390/genes13050740